
Abstract
Au–Kline syndrome (AKS, OMIM 616580) is a multiple malformation syndrome, first reported in 2015, associated with intellectual disability. AKS has been associated with de novo loss-of-function variants in HNRNPK (heterogeneous ribonucleoprotein K), and to date, only four of these patients have been described in the literature. Recently, an additional patient with a missense variant in HNRNPK was also reported. These patients have striking facial dysmorphic features, including long palpebral fissures, ptosis, deeply grooved tongue, broad nose, and down-turned mouth. Patients frequently also have skeletal and connective tissue anomalies, craniosynostosis, congenital heart malformations, and renal anomalies. In this report, we describe six new patients and review the clinical information on all reported AKS patients, further delineating the phenotype of AKS. There are now a total of 9 patients with de novo loss-of-function variants in HNRNPK, one individual with a de novo missense variant in addition to 3 patients with de novo deletions of 9q21.32 that encompass HNRNPK. While there is considerable overlap between AKS and Kabuki syndrome (KS), these additional patients demonstrate that AKS does have a distinct facial gestalt and phenotype that can be differentiated from KS. This growing AKS patient cohort also informs an emerging approach to management and health surveillance for these patients.
Similar content being viewed by others


Introduction
Au–Kline syndrome (AKS, OMIM 616580) is a recently described multiple congenital malformation syndrome associated with intellectual disability. AKS is caused by loss-of-function variants in the gene HNRNPK, which encodes the heterogeneous nuclear ribonucleoprotein K (hn RNP K). Thus far, only four patients with documented loss-of-function variants in HNRNPK and two patients with microdeletions encompassing HNRNPK have been reported in the literature [1,2,3,4,5].
The initial reports of patients with AKS suggested that this syndrome has a recognizable craniofacial gestalt that includes long palpebral fissures, ptosis, a broad nasal bridge, and a characteristic down-turned mouth. Similarities between the facial features of AKS and Kabuki syndrome (KS) were identified, as a clinical diagnosis of KS was suggested for three of these initial patients [1,2,3]. The earlier descriptions of AKS patients also revealed a wide range of congenital anomalies, including congenital heart disease, genitourinary abnormalities, and skeletal abnormalities, some of which are also observed in KS. All patients also had moderate-to-severe intellectual disability, associated with hypotonia that was often profound.
Through international collaboration, we have identified five additional patients with loss-of-function variants in HNRNPK, as well as another with a 264-Kb microdeletion encompassing HNRNPK. This report of six new patients further reinforces that AKS has a recognizable facial gestalt that overlaps with, but is distinct from, KS. The growing cohort of AKS patients also provide further understanding of the emerging phenotype of AKS, including the prenatal presentation and the spectrum of malformations that can be associated with this condition. These new data inform health surveillance measures that should be considered for these patients and highlight areas that require further study.
Methods
The five new patients with loss-of-function variants in HNRNPK were diagnosed via trio-based whole-exome sequencing (WES) performed at clinical laboratories. Patient samples were initially sent to commercial gene testing companies: GeneDx, Ambry, or ARUP. The patient with the microdeletion was identified via clinical oligoarray comparative genomic hybridization (180K Cytosure ISCA v2). All patients were consented for clinical testing under their local institutional protocol. The HNRNPK variants identified in the patients discussed in this report have been submitted to ClinVar (SCV000709679.1 - SCV000709684.1 for the new patients reported, and accession numbers SCV000223813.2, SCV000223814.2 from our previous publication [1]). Consent for publication of photographs has been obtained.
Results
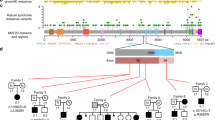
All five patients identified through clinical WES (Patients 5–9) had de novo loss-of-function variants in HNRNPK: stop, frameshift, or canonical splice variants (see Fig. 1). Patient 10 had a de novo 264-Kb microdeletion encompassing 9q21.32: arr[hg19]9q21.32 (86,328,837_86,592,487)x1 dn. This deletion is smaller than the previously described deletions [4, 5] and disrupts HNRNPK in addition to three additional genes and a microRNA with no known human disease association (GKAP1, C9ORF64, KIF27, MIR7-1).

Schematic representation of disease-causing HNRNPK mutations on the NM_002140.4 transcript. Introns (lines), coding exons (in black), and non-coding exons (in white) are represented. Published loss-of-function mutations (below) and missense mutation (above) and new mutations (above) are indicated. The scale bar represents 100 base pairs
The six new AKS patients, along with the patients previously described in the literature, have shared facial features. Long palpebral fissures, shallow orbits, ptosis, a broad nasal bridge, hypoplastic alae nasi, and a downturned mouth previously described as an “M-shaped Cupid’s bow” [3] are commonly observed (Table 1, Fig. 2). Missing teeth have been reported in two patients, and malocclusion is present in at least three. Craniosynostosis affecting the sagittal suture and/or metopic ridging has been observed in one third of the patients (Table 1). Lambdoid suture synostosis was confirmed in only Patient 1. There is also a tendency for AKS patients to appear mildly coarse, and the face tends to be long (Fig. 2).

Photographs of AKS patients. a Patient 1 at 19 years of age. b Patient 1 demonstrates narrow palate and deeply grooved tongue. c Patient 2 at 9 years of age. Note metopic ridging. d Patient 3 at 11 years. e–g Patient 8 as a newborn, age 16 months and age 4 years 5 months. h Patient 6. Note “M” shaped upper lip and deep groove to tongue. i Patient 7 at age 7. j Patient 7 demonstrates deep groove to tongue and mildly bifid tip. k, l Patient 9 at 6 months. Note excess nuchal skin. m, n Patient 10 at 4 weeks. o Patient 10 at 1 year 9 months. p Patient 10 at 8 years
Many AKS patients have ear anomalies but these are quite variable, and both sensorineural and/or conductive hearing loss is possible (Table 1, Fig. 2). Branchial defects presenting as cystic remnants have been observed in two patients. Vision abnormalities have been variable and optic nerve abnormalities have been described in at least three patients (Table 1). Patient 9 has horizontal nystagmus.
Congenital heart malformations are present in 10/12 patients (Table 2), and ventricular septal defects are most frequently observed. Ascending aortic dilation has been observed in one patient. Genitourinary anomalies are extremely common (Table 2), particularly hydronephrosis with vesicoureteral reflux (VUR), which can be severe. Patient 9 presented prenatally with prune belly sequence, with cystic kidneys associated with large cystic hygroma, pleural effusions, enlarged bladder, and club feet. He had bilateral grade 5 VUR, obstructive uropathy requiring vesicostomy, and developed chronic kidney disease. At 15 months of age, he acquired urosepsis and developed multiorgan failure and he passed away shortly thereafter. Patient 7 has had multiple urinary tract infections. He had repair of uteropelvic junction obstruction, required reflux surgery, and subsequent appendicovesicostomy and reconstruction/reimplantation surgery. He requires urinary catheterization every 4 h through a stoma and is on oxybutynin for bladder spasms. Neurogenic bladder is also present in Patient 2.
Gastrointestinal (GI) malformations are rarely described for AKS (Table 2) but motility issues appear to be common, with constipation in half of patients, frequently associated with chronic diarrhea and gastroesophageal reflux disease. Patient 2 has required long-term gastrostomy-tube feeding due to delayed gastric emptying and was diagnosed with chronic intestinal pseudoobstruction, and Patient 7 required nasogastric tube feeding in infancy.
All of the known AKS patients have global developmental delay, and older patients have been assessed to have moderate-to-severe intellectual disability (Table 2). Language acquisition is markedly delayed, and many patients use alternative communication methods. All of the known AKS patients are hypotonic with delayed gross motor development. Several patients have depressed or absent reflexes. Walking is typically markedly delayed (Table 2); however, most patients continue to improve with their gross motor skills well into late childhood and adolescence.
Myopathy or muscle disease has been considered on the differential diagnosis for several patients. Muscle biopsy in Patient 7 revealed mitochondrial complex I deficiency. The patient described by Pua et al. also had a muscle biopsy, which showed diffuse and uniform fiber atrophy suggestive of chronic neurogenic change, but no mitochondrial respiratory chain testing was reported [4]. Muscle biopsy has not been pursued for the other AKS patients. Patient 7 is on a mitochondrial treatment ‘cocktail’ with subjective improvements in energy and strength. Patient 8, who was observed to have decreased muscle bulk and weakness, has had subjective improvement on creatine supplementation. High pain tolerance was reported in at least four patients, one of whom also has unusual flushing of the skin, and another of whom has decreased sweating. Patient 2 has had a formal diagnosis of dysautonomia with absent sweating, high pain tolerance, tachycardia, and GI symptoms. The other patients in this cohort have not had formal evaluation for dysautonomia.
Brain magnetic resonance imaging (MRI) has demonstrated variable findings in the AKS cohort (Table 2). Hypoplasia or agenesis of the corpus callosum appears more common (Table 2); however, Chiari malformation was observed in Patient 7. Patient 2 developed absence seizures at age 11 years, which are well controlled on a single medication.
Skeletal findings are common (Table 2). Vertebral segmentation abnormalities include hemivertebrae, butterfly vertebrae, fusion anomalies, and extra lumbar vertebrae [1, 3]. Hypermobility is variably reported. Patient 1 has developed bilateral elbow contractures over time. Decreased bone density was identified in Patient 2, who presented with fracture, but bone density has not been routinely evaluated in the other AKS patients. Growth can also be abnormal, with reduced height, weight, and/or head circumference over time in some patients despite normal birth parameters (Table 1).
Transient hypothyroidism was present in Patient 2, and Patient 4 was reported to have thyroiditis. Patient 9 was diagnosed with hypothyroidism with low T4 and slight elevation of thyroid-stimulating hormone with neonatal cholestasis that resolved after treatment with levothyroxine. Hormone abnormalities have otherwise not been observed consistently in this cohort of patients.
Respiratory and/or sleep abnormalities have not been typically observed in AKS patients so far. However, Patient 1 was recently found to have nocturnal hypoventilation requiring bi-level positive airway pressure support. His sleep-related respiratory insufficiency and difficulties with secretions were only identified post-operatively at 19 years of age, after partial glossectomy to address open anterior bite. Polysomnography done at age 4 years in Patient 7 was normal. However, polysomnography has not been routinely done on AKS patients, and most have not reported breathing or sleep abnormalities.
The prenatal presentation of AKS is notable. Five patients presented antenatally with increased nuchal translucency, including one patient who developed hydrops and another who had a cystic hygroma. Hydronephrosis is also a common prenatal finding, present in five patients in addition to the patient with cystic kidneys and prune belly sequence (Patient 9). An umbilical cord containing two vessels was observed in three patients. Congenital heart disease was only reported for one patient prenatally. Agenesis of the corpus callosum was observed prenatally in Patient 5. Choroid plexus cysts, hyperechoic bowel, and ventriculomegaly have also been detected on ultrasound in single patients; therefore, it is not clear if these are common associations.
Discussion
Four patients with loss-of-function variants in HNRNPK and two patients with deletions involving this gene have been previously reported in the literature [1,2,3,4,5]. We present an additional six patients and further delineate the phenotype of AKS. These patients were rapidly identified through clinical WES (or reanalysis of WES data) since the publication of the original patients; therefore, we suspect AKS is not as rare as initially expected. The common phenotype between patients with HNRNPK truncating variants and the newly described microdeletion patient further supports that AKS is due to haploinsufficiency of the HNRNPK gene. Loss-of-function intolerance of HNRNPK is supported by a high pLI score of 1.0 [ExAC database, accessed March 2018].
As this paper was being finalized for submission, we became aware of a report of an individual with a de novo missense variant, NM_002140.4(HNRNPK):c.464T>C (p.(Leu155Pro)) [6]. This individual has a craniofacial gestalt suggestive of AKS, ureteropelvic junction stenosis, ureteral stenosis, and bilateral hydronephrosis. While the clinical information on this patient is limited, the similarities to AKS support that this missense change may result in significant loss of function. It remains to be seen whether other de novo missense variants in HNRNPK will be associated with an AKS phenotype or other neurodevelopmental phenotypes.
As observed from previous studies, some AKS patients demonstrate considerable overlap with KS; four patients received testing for Kabuki-related genes KMT2D and KDM6A prior to diagnosis of AKS [1,2,3]. However, the majority of patients we report in this study did not present strikingly as KS. No KS testing was considered or pursued by clinical geneticists in six patients. Genes for transforming growth factor β (TGFβ)-related conditions, including Loeys–Dietz syndrome (TGFBR1, TGFBR2) and Shprintzen–Goldberg syndrome were frequently pursued [1, 2], likely influenced by skeletal findings and craniosynostosis. Interestingly, Noonan syndrome had been considered in Patients 1, 8 and 9 due to suggestive facial dysmorphism, increased nuchal fold, generalized edema, congenital heart disease in Patient 8, and presence of cystic hygroma prenatally in Patient 9. Other diagnoses that were frequently investigated included Prader–Willi syndrome due to marked hypotonia and cryptorchidism, congenital disorders of glycosylation, mutations in FLNA and FLNB, and Pallister–Killian syndrome. Therefore, while there is significant intersection in the features of KS and AKS, AKS is a distinct condition and should also be considered in the differential for other multiple malformation syndromes. This cohort also further demonstrates the increasingly recognizable facial gestalt of AKS, which is frequently associated with long palpebral fissures, ptosis, a characteristic nose with a broad bridge and often hypoplastic alae nasi, a distinctive down-turned mouth, and a long face with an often coarse appearance.
Shared features that overlap with KS include long palpebral fissures and ear malformations, in addition to congenital malformations such as heart defects and renal anomalies. Minimal lower lateral lid eversion was observed in Patients 2, 3, 7, and 10 but has either been very mild or not present in the other patients, in contrast to KS. Some findings that are more common in AKS, such as craniosynostosis and tongue abnormalities, have only been rarely described for KS. Deeply grooved or bifid tongue was described in the first report of AKS as a striking feature [1]; however, differences in the tongue and palate are now noted in only half the patients (Table 1). Interestingly, in a well-defined clinical cohort of 48 KS patients, Armstrong et al. identified that 6% patients had craniosynostosis and 2% had a deeply fissured tongue [7]. At least five other patients with craniosynostosis and a clinical diagnosis of KS have been described in the literature [8]. However, these reports were before molecular diagnosis was readily available, and it is possible that some patients clinically diagnosed with KS actually were affected with alternative syndromes, which may have included AKS. Only one report of KS with craniosynostosis, affecting the right coronal and sagittal sutures, has been published with molecular confirmation of a mutation in the KMT2D gene. However, this patient also had a 3.2 Mb deletion of 10q22.3q23.1[9].
Other findings that are common to KS, such as fetal pads, lip nodules, and blue sclerae, have not been observed in AKS. Although recurrent infections were noted for the patient described by Pua et al. and in Patient 4 [3, 4], most patients do not have obvious immunodeficiency. Endocrine abnormalities are not typically observed in AKS, other than hypothyroidism in three patients. Additionally, AKS patients appear to have more severe global developmental delay and intellectual disability than is typically expected for KS. However, the potential neurodevelopmental outcome range for AKS is difficult to predict given the limited number of patients and possibility of ascertainment bias for more severe presentations in this early cohort.
The HNRNPK gene has been implicated in regulating both tumor-suppressor and oncogenic pathways [10]. Recurrent somatic 9q21.32 deletions encompassing HNRNPK have been observed in acute myeloid leukemias [10, 11]. Gallardo et al. demonstrated in a mouse model of HNRNPK haploinsufficiency that Hnrnpk+/− mice had reduced survival that was in part due to a predisposition to hematologic malignancy [12]. These mice also had reduced growth. Therefore, humans haploinsufficient for HNRNPK may potentially be at increased risk for malignancy. However, no individuals with AKS have been affected with cancer to date. Furthermore, despite the hematologic abnormalities observed in Hnrnpk+/− mice, which include myeloproliferation and thrombocytosis, no blood abnormalities have been observed so far in AKS. Patients 1–10 have had normal complete blood counts. The oldest known patient is currently only 19 years of age and further natural history studies are required.
Early lethality has been rare in patients with AKS. The microdeletion patient described by Pua et al. [4]. had several prolonged hospitalizations associated with respiratory distress requiring ventilatory support and died at 14 months of age with extensive aspiration pneumonia. Patient 9 also died at 15 months of age in the context of pre-existing renal disease. While more data are necessary, it appears that genitourinary system contributes disproportionately to morbidity and possibly mortality in AKS.
The degree to which other health problems are associated with AKS remains unclear. Although the connection between AKS and congenital heart disease is now obvious, the actual risk for aortic dilation remains ambiguous as it has only been described in Patient 2, who also has a family history of bicuspid aortic valve and aortic dilation present in his mother. The risk for breathing abnormalities such as nocturnal hypoventilation is also uncertain. However, given Patient 1’s experience with prolonged ventilator support after recovery from anesthesia, extra caution around anesthesia for AKS patients may be warranted. Additionally, although patients with AKS most likely have a central nervous system cause for their often profound hypotonia, given the abnormal muscle biopsy results of two patients, it is possible that this condition may also involve some degree of mitochondrial dysfunction and/or myopathy. Dysautonomia is also emerging as an important health complication for these patients that has been inadequately characterized.
The nature of growth abnormalities in AKS is also uncertain. Some patients appear to demonstrate growth restriction over time, particularly for height (to less that third percentile for Patients 1, 5, 8, 9, 10), and occasionally for head circumference, whereas other patients appear to maintain their growth velocity (Table 1). The presence of scoliosis may account for some degree of height loss, but short stature is also present in patients without significant scoliosis (Patients 6, 9, and 10). Postnatal microcephaly was also observed in multiple patients (Table 1) [4, 5]. The presence of reduced head circumference is not necessarily associated with craniosynostosis (Patients 8–10). Growth restriction does not seem to correlate obviously with the incidence of GI malformation or dysmotility.
Although it is difficult to determine health surveillance guidelines based on only a few patients, we recommend some investigations based on our preliminary understanding of AKS. Echocardiogram, renal ultrasound, MRI of the brain and spine, skeletal evaluation for spine and hip abnormalities, ophthalmology assessment, and hearing assessment should be done at baseline to screen for the common features now known to be associated with AKS. Repeating an echocardiogram periodically may be warranted given an ambiguous association with aortic dilation. Infants should be monitored for craniosynostosis. Further urinary tract work-up should be carried out if indicated clinically. ENT evaluation is likely beneficial given association with branchial defects, palatal defects, and jaw and bite abnormalities. Polysomnography and upper GI contrast studies could be considered based on symptoms. Muscle evaluations such as biopsy, electromyography, and nerve conduction studies are not necessary if the diagnosis of AKS is established but could be useful in further delineating an AKS muscle phenotype, if suggestive symptoms arise. In regards to prenatal diagnosis, many of the AKS-associated findings may lead to prenatal array testing, so it may be possible currently to detect some AKS cases due to deletion of 9q21.32 in utero.
Germline HNRNPK haploinsufficiency obviously exhibits pleiotropic effects, impacting multiple organ systems. AKS should be considered in the differential diagnosis for a patient presenting with multiple congenital malformations and intellectual disability. Special consideration should be given to Kabuki-like or Noonan-like patients presenting with craniosynostosis, more marked intellectual impairment, or atypical dysmorphology. AKS should also be considered in patients presenting with striking skeletal and connective tissue abnormalities who also have significant dysmorphism particularly in the context of intellectual disability. Further study in larger numbers of patients will be required to further clarify the impact of many of the health issues associated with AKS in order to better inform management for these patients.
References
Au PYB, You J, Caluseriu O, Schwartzentruber J, Majewski J, Bernier FP, et al. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum Mutat. 2015;36:1009–14.
Lange L, Pagnamenta AT, Lise S, Clasper S, Stewart H, Akha ES, et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin Genet. 2016;90:258–62.
Dentici ML, Barresi S, Niceta M, Pantaleoni F, Pizzi S, Dallapiccola B, et al. Clinical spectrum of Kabuki-like syndrome caused by HNRNPK haploinsufficiency. Clin Genet. 2017;93:1–7.
Pua HH, Krishnamurthi S, Farrell J, Margeta M, Ursell PC, Powers M, et al. Novel interstitial 2.6 Mb deletion on 9q21 associated with multiple congenital anomalies. Am J Med Genet A. 2014;164:237–42.
Hancarova M, Puchmajerova A, Drabova J, Karaskova E, Vlckova M, Sedlacek Z. Deletions of 9q21.3 including NTRK2 are associated with severe phenotype. Am J Med Genet A. 2015;167:264–7.
Miyake N, Inaba M, Mizuno S, Shiina M, Imagawa E, Miyatake S, et al. A case of atypical Kabuki syndrome arising from a novel missense variant in HNRNPK. Clin Genet. 2017;92:554–5.
Armstrong L, El Moneim AA, Aleck K, Aughton DJ, Baumann C, Braddock SR, et al. Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am J Med Genet. 2005;132A:265–72.
Martinez-Lage JF, Felipe-Murcia M, Navarro EG, Almagro MJ, Lopez-Guerrero AL, Perez-Espejo MA. Craniosynostosis in Kabuki syndrome. J Neurosurg Pediatr. 2010;6:198–201.
Topa A, Samuelsson L, Lovmar L, Stenman G, Kolby L. On the significance of craniosynostosis in a case of Kabuki syndrome with a concomitant KMT2D mutation and 3.2 Mbp de novo 10q22.3q23.1 deletion. Am J Med Genet A. 2017;173:2219–25.
Gallardo M, Hornbaker MJ, Zhang X, Hu P, Bueso-Ramos C, Post SM. Aberrant hnRNP K expression: all roads lead to cancer. Cell Cycle. 2016;15:1552–7.
Peniket A, Wainscoat J, Side L, Daly S, Kusec R, Buck G, et al. Del (9q) AML: clinical and cytological characteristics and prognostic implications. Br J Haematol. 2005;129:210–20.
Gallardo M, Lee HJ, Zhang X, Bueso-Ramos C, Pageon LR, McArthur M, et al. HnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015;28:486–99.
Acknowledgements
We would like to thank the patients and their families for their participation and support. We would also like to acknowledge the help and support of Marina Kerr, Lukas Lange, Alistair Pagnamenta, Jenny Taylor, and David Keays. We would also like to thank Taila Hartley (Clinical Coordinator) and Chandree Beaulieu (Project Manager) at the Children’s Hospital of Eastern Ontario Research Institute for their contribution to the infrastructure of Care4Rare. Finally, we would like to thank Care4Rare Consortium: ‘Enhanced Care for Rare Genetic Diseases in Canada’; Gene Discovery Steering Committee—Kym Boycott (lead; University of Ottawa), Alex MacKenzie (co-lead; University of Ottawa), Jacek Majewski (McGill University), Michael Brudno (University of Toronto), Dennis Bulman (University of Ottawa), David Dyment (University of Ottawa).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Au, P.Y.B., Goedhart, C., Ferguson, M. et al. Phenotypic spectrum of Au–Kline syndrome: a report of six new cases and review of the literature. Eur J Hum Genet 26, 1272–1281 (2018). https://doi.org/10.1038/s41431-018-0187-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0187-2
This article is cited by
-
Comment on Gustavson syndrome is caused by an in-frame deletion in RBMX associated with potentially disturbed SH3 domain interactions
European Journal of Human Genetics (2024)
-
Hnrnpk is essential for embryonic limb bud development as a transcription activator and a collaborator of insulator protein Ctcf
Cell Death & Differentiation (2023)
-
Hnrnpk maintains chondrocytes survival and function during growth plate development via regulating Hif1α-glycolysis axis
Cell Death & Disease (2022)
-
Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders
Genome Medicine (2021)
