
Abstract
Hirschsprung disease (HSCR) is a complex birth defect characterized by the lack of ganglion cells along a variable length of the distal intestine. A large proportion of HSCR patients remain genetically unexplained. We applied whole-genome sequencing (WGS) on 9 trios where the probands are sporadically affected with the most severe form of the disorder and harbor no coding sequence variants affecting the function of known HSCR genes. We found de novo protein-altering variants in three intolerant to change genes—CCT2, VASH1, and CYP26A1—for which a plausible link with the enteric nervous system (ENS) exists. De novo single-nucleotide and indel variants were present in introns and non-coding neighboring regions of ENS-related genes, including NRG1 and ERBB4. Joint analysis with those inherited rare variants found under recessive and/or digenic models revealed both patient-unique and shared genetic features where rare variants were found to be enriched in the extracellular matrix–receptor (ECM–receptor) pathway (p = 3.4 × 10−11). Delineation of the genetic profile of each patient might help finding common grounds that could lead to the discovery of shared molecules that could be used as drug targets for the currently ongoing cell therapy effort which aims at providing an alternative to the surgical treatment.
Similar content being viewed by others

Introduction
Hirschsprung disease (HSCR) is a complex, congenital genetic disorder characterized by the absence of enteric neurons along a variable length of the distal intestine. This is attributed to a failure in the migration of the enteric neuron precursors, the neural crest cells (NCCs). Patients present with bowel obstruction and the disease may be fatal unless it is surgically treated. The severity of the HSCR phenotype is determined by the length of the colon affected i.e. total colonic aganglionosis (TCA; 5% of the patients); long-segment aganglionosis (L-HSCR; 15% of the patients; aganglionosis spans from the rectum beyond the sigmoideum); short-segment aganglionosis (S-HSCR; 80% of the patients; aganglionic segment does not extend beyond the upper sigmoid). HSCR is a relatively rare disorder whose incidence varies significantly across populations (1.4/5,000 live-births in Chinese vs. 1/5,000 live-births in Caucasians) [1,2,3]. It is often accompanied (30% patients) by other neurological and/or developmental disorders, developmental delay, well characterized syndromes and/or chromosomal abnormalities, of which Down syndrome is the commonest [1]. HSCR most commonly presents sporadically although it can be familial (5–20% of the patients) and manifests with low, sex-dependent penetrance and phenotypic variability.
Our own and others’ data indicate that only a fraction of the HSCR patients (15% of the sporadic and 50% of the familial cases) are explained by rare variants in coding sequences (CDS) of genes involved in signaling pathways that govern the development of the enteric nervous system (ENS), including, RET, EDNRB, EDN3, SOX10, NRG1, NRG3, SEMA3D among others [1, 2, 4,5,6]. We have also shown that common variants in RET, NRG1 and/or SEMA3D—the latter only in Caucasians- are strongly associated with the commonest form of HSCR (male, S-HSCR, sporadic) [2, 6,7,8] while rare CDS function-altering variants are more likely to be identified in the most severe and less frequent form (female, L-HSCR and familial cases) [2]. Copy number variations (CNVs) have also been linked with the HSCR phenotype. Our previous CNV analysis suggested a role for NRG3 in HSCR etiology and provided insights into the contribution of structural variants in both syndromic and non-syndromic HSCR [5]. The relevance of CNVs in HSCR is underscored by the non-random association of HSCR with syndromes involving chromosome abnormalities. Incidentally, the two major HSCR genes, RET and EDNRB, were discovered through the detection of interstitial deletions in their respective loci.
In spite of the vast screening of HSCR patients for CDS variants in candidate genes or for large structural variations, a large proportion of HSCR patients, including those with the most severe phenotype, remain unexplained. This implies that other genetic lesions anywhere in the genome (including non CDS; NCDS) exist, and, given our previous observations and the severity and rarity of the disorder, these variants are likely to be rare.
In this study, we have applied WGS on nine trios where the probands (i) had L-HSCR or TCA, (ii) harbored no rare CDS variants affecting the function of RET and other known HSCR genes and, (iii) were born to unaffected parents. Given that this is a pilot study and the first WGS in L-HSCR or TCA, our aim is to capture different types of genetic variations and get a glimpse of what the genetic architecture of the most severe HSCR phenotype may be. The genetic profile generated for each patient revealed unique and shared genetic features that, if exploited and replicated, could lead to findings of shared molecules that could be used as drug targets for the currently ongoing cell therapy efforts (exemplified in Fattahi et al. [9]) which aims at providing an alternative to the only existing treatment i.e. the surgical removal of the affected intestine- which is not without devastating sequels for the patients.
Materials and Methods
Patients and controls
A total of 27 samples (9 sporadic HSCR patients affected with either TCA or L-HSCR together with their unaffected parents) were included in the study. A previous screening for CDS variants in RET indicated that none of patients had CDS variants affecting the function in this major HSCR gene. Besides availability, these 9 trios were selected for the study because they did not harbor any rare CDS function-altering variant in known HSCR genes in spite of these being more likely to account for patients with L-HSCR. Thus, we argued that if rare function-altering variants underlie the disorder, these should either map to CDS of yet-to-know genes or to regulatory NCDS which are only accessible through WGS. The characteristics of the patients are described in Table S1. Data from 493 Chinese healthy individuals participating in an in-house WGS sequencing project were added to the calling set and were used as “local controls”. The study was approved by the institutional review board of The University of Hong Kong together with the Hospital Authority (IRB: UW 06-349 T/1374). Blood samples were drawn from all participants after obtaining informed consent (parental consent in newborns and children below age 7) and experiments were carried out in “accordance” with the approved guidelines.
Whole-genome sequencing and variation detection
Genomic DNA extracted from blood was assessed for quality by PicoGreen and gel electrophoresis. Library preparation was done using The Illumina TruSeq Nano DNA HT Sample Prep Kit (Catalog#: FC-121-4003) and then sequenced at 30-fold (30×) coverage using Illumina HiSeqX Ten (150-bp paired-end sequencing) by Macrogen Inc. through The University of Hong Kong, Centre for Genomic Sciences (HKU, CGS).
Burrows-Wheeler Aligner (BWA, v.0.7.12) [10] was used to align sequence reads to the human reference genome (GRCh37/hg19) and GATK v.3.3.0 [11] was employed for pre-processing sequence reads and calling single-nucleotide variants (SNVs) and small insertion and deletions (indels). KGGSeq (v1.0 + ) [12] was used for annotation. Metrics for sequence and variant calling quality are detailed in Table S2.
To attain maximum accuracy, CNVs were called using 4 different and complementary software: CNVnator [13], Seeksv [14], DELLY [15] and LUMPY [16]. Default parameters were used in all software except for CNVnator, where the bin size used to partition the genome was set to 50 bp. Only those CNVs >50 bp called by at least 3 software and were supported by at least 10 soft-clip reads were selected for downstream analysis. CNVs with >50% of length overlapping with centromere or short repeat regions were excluded. We used BEDTools [17] to calculate the overlap of CNVs among individuals. Two CNVs were considered as shared when the reciprocal overlap was >50%. Annotation of CNVs was done by ANNOVAR [18].
De novo CDS and NCDS SNV/indels
For identification of de novo SNV/indels, we used KGGSeq and GATK PhaseByTransmission with the following criteria: (i) minor allele frequency (MAF) in public databases <0.01% (1000 Genomes, NHLBI Exome Sequencing Project and Exome Aggregation Consortium databases); (ii) variant call rate >80% across all samples; (iii) biallelic; (iv) proportion of reads supporting the alternative allele for heterozygous genotype >25% for the proband and <5% for both parents; (v) the alternative allele should not appear in the control dataset; (vi) sequencing depth >20× at the variant for the proband; (vii) GATK PhaseByTransmission probability(TP) >20 (in Phred scale). De novo SNVs were phased using DeNovoGear software to assess which parental chromosome harbored the de novo allele. All de novo CDS and selected NCDS SNV/indels were validated by Sanger sequencing.
Data can be accessed on: https://databases.lovd.nl/shared/individuals (Patient IDs 133232, 133234, 133265, 133266, 133267, 133268, 134081).
Inherited CDS SNV/indels
For the selection of homozygous or compound heterozygous (CH) CDS variants, the following criteria were used: (i) MAF<1% in public databases and 493 in-house controls; (ii) biallelic; (iii) no homozygous genotypes at that site observed in the controls for homozygous CDS variants; (iv) no compound heterozygote in the same gene present in pseudocontrols (parental untransmitted alleles) for CH CDS variants and (v) expressed in vagal NCCs. For all those genes known or reported to be involved in the development of the ENS in either human or mouse (117 ENS genes; Table S3), we used the following two criteria: (i) MAF in public databases <1% except for RET (MAF < 5%; where multiple low-frequency missense variants were reported to be highly associated with HSCR) and (ii) biallelic. Thus, the main filtering criteria for non-synonymous and loss-of-function (LoF) variants was MAF (of both public and in-house controls) as current in silico pathogenicity prediction software are discrepant.
We also assumed the possibility of the phenotype being caused by summation effect of variants in the relevant genes, regardless of the variant deleteriousness. The Residual Variation Intolerance Score (RVIS) which measures the intolerance of a gene to variation is provided for all genes with non-synonymous or LoF de novo variants. Likewise, scores that measure the effect of the variant on the protein such as Sorting Tolerant From Intolerant (SIFT), Polymorphism Phenotyping (Polyphen2_HDIV) and Combined Annotation Dependent Depletion (CADD) scores are also provided for de novo variants. CADD also provides scores for those variants in NCDS (Table S4).
De novo CNVs
De novo CNVs had to meet the following two criteria: (i) present in patients only and (ii) absent in the CNV database constructed for all parents. CNVs documented in the database of genomic variation (DGV) [19] were removed as DGV records CNVs present in samples of non-affected individuals. Likewise, CNVs that overlapped with our in-house controls CNV dataset (493 individuals) were also removed.
Functional overlap among genes
To uncover the “di/oligo-genic model” where variants in different interacting genes co-exist in the patient through de novo or parental inheritance. We identified functional overlap among genes using STRING [20] (v10.0. Including 2031 organisms, 9.6 million proteins and 1380 million interactions) and GeneMania [21] (Indexing 2,277 association networks containing 597,392,998 interactions mapped to 163,599 genes from 9 organisms). Mutated genes encoding interacting partners to any of the seed genes identified or to any of the 117 ENS-known candidate genes were included in this model (seed genes: genes with de novo CDS, homozygous and/or CH SNVs/indels).
Results
The WGS approach was applied to 9 HSCR patients and their unaffected parents who, in addition to being affected with the most severe form of the disease, carried no known CDS variants affecting the function in the main HSCR genes, namely RET, EDNRB, EDN3, SOX10, NRG1 and PHOX2B (patient HK97C was affected with Central hypoventilation syndrome; Table S1) as indicated by a prior Sanger sequencing screening.
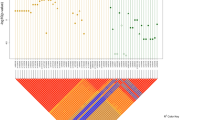
Approximately 96.95% of the genomes were covered by at least 5 reads and around 88.47% by more than 20 reads (Table S2). As for the exonic sequences, 92.55% were covered by at least 20 reads. An average of 3.4 million SNV/indels and 1692 CNVs per individual were identified by WGS (Table S2; Fig. 1).

Workflow for detection and selection of candidate HSCR susceptibility variants
Given that all patients were born to unaffected parents, we considered (i) the effect of damaging de novo germ-line variants and tested (ii) an autosomal-recessive mode of inheritance including homozygosis, CH (different variants in the same gene, one paternally and one maternally inherited) and (iii) a “di/oligo-genic model” where variants in different interacting genes co-exist in the patient through de novo or parental inheritance. The sequence analysis was done under rigorous and stringent parameters, including a composite method to ensure the CNV detection veracity.
De novo variants
First, we focused our analysis on de novo SNVs, indels (<50 bp), and CNVs (>50 bp), including only high quality calls. For de novo SNVs and indels, the MAF was set at <0.0001. We identified a total of 531 high confident de novo events including 480 SNVs and 51 indels, no de novo CNV was found (Fig. 1; Table S5). To assess which parental chromosome harbored the de novo alternative allele, read-back phasing were performed using the DeNovoGear software, where phasing was possible for 209 variants. Among these successfully phased de novo SNV/indels, 179 were on the paternal chromosome and 36 were on the maternal chromosome, which is consistent with the fact that de novo variants are predominantly of paternal origin [22].
De novo coding SNVs/indels
There was an average of 0.78 de novo exonic SNVs/indels per genome—which is in line with reported rates [23] (Table S5). All of the seven coding de novo SNV/indels selected for validation were verified (Table 1, Table S4).
Three genes—CCT2, VASH1, and CYP26A1—with de novo variants were shortlisted. According to RVIS, these genes are among the 10–25% most change-intolerant genes (Table S4 column W). A direct and plausible link with the ENS could be found for CYP26A1. CYP26A1 encodes a retinoic acid (RA)-metabolizing enzyme that regulates the RA availability in the developing gut. RA is essential for the morphogenesis and function of the ENS, therefore, it is tempting to speculate that genetic variants that change the activity of the CYP26A1 enzyme could profoundly influence the developing bowel wall and ENS. RA deficiency is thought to alter neuronal versus glial differentiation leading to gut aganglionosis [24, 25]. The protein encoded by CCT2 (for the assembly of the basal body (BBSome) involved in the formation of primary cilia and it causes Bardet-Biedl syndrome, which may include HSCR in its phenotypic spectrum. VASH1 encodes a protein binding molecule that inhibits migration, proliferation and network formation by endothelial cells as well as angiogenesis. No link with ENS could be deduced at present.
De novo SNVs/indels in non-coding sequences
We then investigated de novo SNVs/indels in non-coding sequences (NCDS introns and boundaries) of (a) genes known to be implicated in ENS development and/or HSCR disease (ENS-related genes; Table S3); (b) genes with de novo, homozygous and/or compound heterozygous variants uncovered in the present study (see below; Tables S4,S6-S9) and in five genes recently reported to play a role in HSCR [26, 27], namely DENND3, NCLN, NUP98, TBATA, and VCL.
A total of 524 small de novo variants (SNVs/indels) were identified in NCDS including 35 small deletions, 16 small insertions and 473 SNVs (Table S5; Fig. 1). Four patients had NCDS de novo SNVs/indels in intronic or neighboring regions of ENS-related genes (Table 1, S4). Patients VH106C, VH108C and HK164C, carried NRG1 intronic de novo deletions at different sites. In addition, HK164C harbored a de novo SNV downstream of the transcription factor ZEB2 (known to cause Mowat–Wilson syndrome which includes HSCR in its phenotypic spectrum) and VH106C harbored de novo intronic deletions in the gene encoding the NRG1 receptor, ERBB4, and in SEMA3D. The fourth patient, HK180C, had a de novo intronic SNV in DCC, which encodes a critical receptor for netrins, molecules that mediate perpendicular migration of the vagal enteric neurons precursors toward the mucosa [28]. Importantly, the enteric submucosal ganglia are absent in transgenic mice lacking Dcc. DCC has recently been involved in two neurodevelopmental disorders, isolated agenesis of corpus callosum and developmental split-brain syndrome [29, 30]. These, together with HSCR, are also part of the phenotypic spectrum of Mowat–Wilson syndrome. HSCR has also been reported concomitantly with isolated agenesis of corpus callosum [31].
Homozygosis and compound heterozygosis in coding sequences
Next, we tested an autosomal-recessive mode of inheritance and identified 18 genes with homozygous CDS variants (Table S6), none of which have been linked with the ENS thus far. Yet, among those, PLAT (Plasminogen Activator, Tissue Type) is known to facilitate neuronal migration during neuronal development, including ENS [32]. As expected, patient HK164C, born to consanguineous parents, was significantly enriched for homozygous variants.
Likewise, none of the 19 genes with CH rare variants belong to the ENS-related genes (Table S7). However RADIL (Ras-Associating And Dilute Domain-Containing Protein) is required for cell adhesion and migration of neural crest precursors during development and importantly, knockdown of radil in zebrafish results in multiple defects in NC-derived lineages, including pigment cells and enteric neurons [33], features reminiscent of the human Shah-Waardenburg Syndrome where HSCR is a mandatory feature.
Genes recurrently mutated
We then looked for genes recurrently mutated among those with de novo (Table S4) homozygous (Table S6), CH (Table S7) variants—altogether called seed genes hereafter—as well as in ENS-related genes (Table S3). Thus, patients were scrutinized for additional variants (inherited SNVs/indels/CNVs) in those seed and Table S3 genes. A total of 14 genes had different rare variants (SNVs/indels/CNVs) in more than one patient (Table S8). Noticeably, we identified NRG1 with de novo SNVs in 3 patients and ZEB2 harbored both de novo and inherited variants. In the remaining 12 recurrently mutated genes, the variants were all inherited. Among these, it is worth highlighting GLI2 (ENS-gene, in 2 patients), RET (ENS-gene, in 3 patients, not in known pathogenic positions) and LAMA5 (non-ENS-gene, in 4 patients). Two patients had the same inherited variant in PLXNB1 but from different paternal source, and likewise with NEK1, where 2 out of 3 patients had the same variants.
Functional overlap among genes
We also considered a di/oligo-genic recessive model where variants in two or more interacting genes co-exist in a patient. Mutated genes encoding interacting partners to any of the seed genes identified or to any of the 117 ENS-known candidate genes were included in this model (Table S9 where co-existing mutated interacting genes -according to the STRING database v.10.0- are bold and highlighted in gray; Fig. 2).

Genetic profile and the interacting network of two HSCR patients. a VH106C and b HK164C
To consider whether our findings fit into any pathological process, and/or share a niche in spite of the genetic heterogeneity observed, we proceeded with a careful examination of the genetic profile of each patient and performed gene/pathway-set enrichment analyses. Each patient had variants in at least two interacting and biologically plausible genes (Table S9) clearly suggesting a joint effect was necessary for the phenotype to occur. Joint analysis of the genetic profiles indicated that the main pathway shared by these HSCR patients was the extracellular matrix–receptor interaction pathway (ECM–receptor) as this network has significantly more interactions in the patients than expected (KEGG pathways; p = 3.4 × 10−11). Understandably, the migration along the gut of the enteric neurons precursors involves the control of cell adhesion to ECM a dynamic environment that affect the migratory behavior of the enteric neuron precursors [34]. In fact, a collagen VI-dependent pathogenic mechanism for Hirschsprung disease has been described [35]. Note for example that patient HK164C has rare SNVs in 7 interacting genes (ITGB5, COL6A3, LAMB2, ITGA2 COL1A2, SORBS1, and COL6A2; Fig. 2b) that form part of the ECM–receptor interactions. It is then conceivable that the summation or synergy among the variants in these seven interacting genes lead to HSCR in this patient. Likewise with other patients.
Discussion
Multiple lines of evidence demonstrate genetic heterogeneity and complexity in HSCR. A plethora of different genomic variants of all types and frequencies throughout the genome seem to contribute to the disorder. Consequently, HSCR is also clinically and phenotypically heterogeneous, ranging from isolated to syndromic forms with marked phenotypic variability even within a family. To comprehend this wide-range of genetic variants that might contribute to HSCR risk, we conducted a pilot study where WGS was carried out on 9 trios for the first time. These nine patients were selected because they were born to no affected parents (sporadic) and were affected with the most severe and rarest form of the disease, hence, we assumed that the involvement of rare variants was most likely and that the genetic complexity would be less than that of the more common HSCR form. In addition, the patients included were all devoid of unique CDS variants in the main gene, RET.
From the data presented here, no known variants affecting the function of genes associated with known dominant monogenic forms of the disorders were found, in spite of the hundreds of putatively function-altering variants harbored by each individual. It is clear that in the absence of RETde novo function-altering variants in more than one gene or genomic region are needed for the disease to occur. It is also evident that given the rarity and heterogeneity of the disorder, establishing disease causality for any given gene is extremely difficult. Delineation of the genetic profile of each patient might help the search for possible common grounds among these patients and draw plausible explanations for the phenotype.
A shared feature is that all nine patients have rare variants in at least two biologically plausible interacting genes (Table S9). Take patient VH106C for example, with variants in many of the HSCR genes discovered thus far, including the gene encoding SEMA3A and its receptor, PLXNB1, as well as in the genes encoding NRG1, its receptor ERBB4, and its regulator, NOTCH1 [36] (Fig. 2a). Similar pattern can be observed in the majority of patients (Table S9).
Another shared pattern that emerged would be the relevance of genes encoding proteins components of the ECM–receptor pathway which is consistent with the fact that the colonization of the gut by enteric neuron precursors is regulated by specific molecular signals from both within the neural crest and the intestinal environment. ECM molecules not only provide a physical surface for enteric neuron precursor’s migration but also signaling molecules that regulate the whole gut colonization process [37]. Likewise, the enteric neuron precursors also affect the ECM composition. Indeed, the genetic complexity observed in HSCR can be conceptually understood in the light of the molecular and cellular events that take place during the ENS development. Thus, in the same way that we understand that there is cross regulation among proteins involved in enteric neurogenesis, the HSCR phenotype may result from the summation or synergetic effect of variants in genes encoding such proteins (variants that are unlikely to cause disease on their own). Besides, the various disease mechanisms (defects in proliferation, migration, neuron vs. glia differentiation) that have been proposed for HSCR are not mutually exclusive. These can co-exist in the same patient and/or the disease mechanism may differ from patient to patient as many events can go wrong during ENS development, and hence, the present genetic complexity. Noteworthy, a new functional relationship among genes involved in ENS has recently been described whereby Ednrb is not only regulated by Sox10 but also by Zeb2 and interestingly, mimics the oligogenic nature of the human disorder. Mice with combined mutations in Zeb2 and Edn3 (double mutants) had more severe enteric anomalies compared to mice with mutations in either gene alone [38].
A third feature and perhaps the most remarkable finding is the plausible genetic overlap between HSCR and schizophrenia (SZ), autism spectrum disorders (ASD) and with syndromes—including several ciliopathies—that may have HSCR among their phenotypic spectrum. These included Joubert syndrome [39] (C5orf42), Bardet-Biedl syndrome [40] (CCT2), 3 M Syndrome (CUL7), Wolf-Hirschhorn syndrome [41] (FGFRL1), Agenesis of corpus callosum [29, 30] (DCC) and Mowat–Wilson syndrome [42] (ZEB2) among others (Table S9). Except for patient HK97C, who had been diagnosed with congenital central hypoventilation syndrome (CCHS)—yet PHOX2B was not mutated—none of the other patients were affected with the syndromes underlain by the mutated genes above. We had already reported the association of NRG1, a schizophrenia associated gene, with HSCR as well as the detection of ERBB4 deletions [5] (NRG1 receptor; also associated with schizophrenia) in HSCR patients. Here, we have also detected variants in genes involved in SZ and/or ASD including NRG1, NRG3, ERBB4, SEMA3A, PLXNB1 [43], DOCK8 [44] (de novo genic SNV), again reinforcing the idea that pathological alterations affecting pathway(s) shared by more than one disorder may indeed underlie apparently unrelated diseases. This is further supported by the frequently observed association of intestinal dysmotility with psychiatric disorders and many other neurodevelopmental and congenital disorders. By understanding the pleiotropy and the intersecting pathway(s), one can optimize the search for other causal variants underlying HSCR.
We are aware that the data presented here is that of a small sample size, and that, as stated above, establishing causality in HSCR may require the screening of a large number of patients to statistically associate genes with the disease and/or functional analysis of almost all genes identified. The latter is indeed also a daunting task. It is clear that genetic heterogeneity in rare disorders is having an impact on the scientific community when it comes to link or associate genes with diseases as prohibitive large sample sizes an equally prohibitive battery of functional tests are required to achieve statistical significance and elucidate the disease mechanism for each gene(s). Yet, as stated, suggestive evidence pointing to a gene or pathway could be really valuable in future investigations on patients with the same phenotype [45, 46].
Availability of data and materials
The datasets generated and analyzed for the current study are not publicly available due to individual privacy but are available from the corresponding author on reasonable request.
References
Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14.
Emison ES, Garcia-Barcelo M, Grice EA, et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet. 2010;87:60–74.
Garcia-Barcelo MM, Sham MH, Lui VC, et al. Chinese patients with sporadic Hirschsprung’s disease are predominantly represented by a single RET haplotype. J Med Genet. 2003;40:e122.
Tang CS, Ngan ES, Tang WK et al. Mutations in the NRG1 gene are associated with Hirschsprung disease. Hum Genet. 2011;131:67–76.
Tang CS, Cheng G, So MT, et al. Genome-wide copy number analysis uncovers a new HSCR gene: NRG3. PLoS Genet. 2012;8:e1002687.
Jiang Q, Arnold S, Heanue T, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to hirschsprung disease liability. Am J Hum Genet. 2015;96:581–96.
Garcia-Barcelo MM, Tang CS, Ngan ES, et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung’s disease. Proc Natl Acad Sci USA. 2009;106:2694–9.
Emison ES, McCallion AS, Kashuk CS, et al. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. 2005;434:857–63.
Fattahi F, Steinbeck JA, Kriks S, et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature. 2016;531:105–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Li MX, Li J, Li MLJ et al. Robust and rapid algorithms facilitate large-scale whole genome sequencing downstream analysis in an integrative framework. Nucleic Acids Res. 2017; 45 e75.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84.
Liang Y, Qiu K, Liao B, et al. Seeksv: an accurate tool for somatic structural variation and virus integration detection. Bioinformatics. 2017;33:184–91.
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339.
Layer RM, Chiang C, Quinlan AR, Hall IM: LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014; 15 R84.
Quinlan AR. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:11 12 11-34.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–66.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–D992.
Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368.
Vlasblom J, Zuberi K, Rodriguez H, et al. Novel function discovery with GeneMANIA: a new integrated resource for gene function prediction in Escherichia coli. Bioinformatics. 2015;31:306–10.
Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:241.
Gilissen C, Hehir-Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Sato Y, Heuckeroth RO. Retinoic acid regulates murine enteric nervous system precursor proliferation, enhances neuronal precursor differentiation, and reduces neurite growth in vitro. Dev Biol. 2008;320:185–98.
Simkin JE, Zhang D, Rollo BN, Newgreen DF. Retinoic acid upregulates ret and induces chain migration and population expansion in vagal neural crest cells to colonise the embryonic gut. PLoS ONE. 2013;8:e64077.
Gui H, Schriemer D, Cheng WW, et al. Whole exome sequencing coupled with unbiased functional analysis reveals new Hirschsprung disease genes. Genome Biol. 2017;18:48.
Lai FP, Lau ST, Wong JK et al. Correction of Hirschsprung-associated Mutations in Human Induced Pluripotent Stem Cells, via CRISPR/Cas9, Restores Neural Crest Cell Function. Gastroenterology 2017;153:139–153.
Gershon MD, Ratcliffe EM. Developmental biology of the enteric nervous system: pathogenesis of Hirschsprung’s disease and other congenital dysmotilities. Semin Pediatr Surg. 2004;13:224–35.
Jamuar SS, Schmitz-Abe K, D’Gama AM, et al. Biallelic mutations in human DCC cause developmental split-brain syndrome. Nat Genet. 2017;49:606–12.
Marsh AP, Heron D, Edwards TJ, et al. Mutations in DCC cause isolated agenesis of the corpus callosum with incomplete penetrance. Nat Genet. 2017;49:511–4.
Sayed M, al-Alaiyan S. Agenesis of corpus callosum, hypertrophic pyloric stenosis and Hirschsprung disease: coincidence or common etiology? Neuropediatrics. 1996;27:204–6.
Pietri T, Eder O, Blanche M, Thiery JP, Dufour S. The human tissue plasminogen activator-Cre mouse: a new tool for targeting specifically neural crest cells and their derivatives in vivo. Dev Biol. 2003;259:176–87.
Smolen GA, Schott BJ, Stewart RA, et al. A Rap GTPase interactor, RADIL, mediates migration of neural crest precursors. Genes Dev. 2007;21:2131–6.
Sasselli V, Pachnis V, Burns AJ. The enteric nervous system. Dev Biol. 2012;366:64–73.
Soret R, Mennetrey M, Bergeron KF, et al. A collagen VI-dependent pathogenic mechanism for Hirschsprung’s disease. J Clin Invest. 2015;125:4483–96.
Zhang K, Wong P, Zhang L, et al. A Notch1-neuregulin1 autocrine signaling loop contributes to melanoma growth. Oncogene. 2012;31:4609–18.
Nagy N, Goldstein AM: Enteric nervous system development: a crest cell’s journey from neural tube to colon. Semin Cell Dev Biol. 2017;66:94–106.
Watanabe Y, Stanchina L, Lecerf L, et al. Differentiation of mouse enteric nervous system progenitor cells is controlled by endothelin 3 and requires regulation of Ednrb by SOX10 and ZEB2. Gastroenterology. 2017;152:1139–50. e1134
Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20.
Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26:1039–56.
Wieczorek D, Krause M, Majewski F, et al. Unexpected high frequency of de novo unbalanced translocations in patients with Wolf-Hirschhorn syndrome (WHS). J Med Genet. 2000;37:798–804.
Garavelli L, Mainardi PC. Mowat-Wilson syndrome. Orphanet J Rare Dis. 2007;2:42.
Konopka G, Friedrich T, Davis-Turak J, et al. Human-specific transcriptional networks in the brain. Neuron. 2012;75:601–17.
Shi L. Dock protein family in brain development and neurological disease. Commun Integr Biol. 2013;6:e26839.
Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–50.
MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–76.
Acknowledgements
We are grateful to the numerous patients, their families and referring physicians that have participated in these studies in our laboratories, and the numerous members of our laboratories for their valuable contributions over many years.
Funding
This work was supported by: Theme-based Research scheme T12C-714/14-R to P.T., Human Medical Research Fund (HMRF) 02131866 and 01121516 to M.M.G.B and 0451966 to C.S.T. and Seed Fund for Basic Research of the University of Hong Kong 201606159005 to C.S.T.
Author information
Authors and Affiliations
Contributions
M.-M.G.-B., P.C.S., and P.K.T. conceived the experiments. C.S.T., X.Z. and M.-M.G.-B. wrote the article. P.C.S. and S.S.C. contributed to critical revision of the article. C.S.T. and X.Z. did the statistical analysis and data interpretation. W.Y.L. and J.S.H. assisted the analysis. M.T.S. did the genetic sample preparation and Sanger sequencing. E.S.W.N. provided the expression data. P.K.T., M.Y. and N.D.N. recruit and clinically characterized the patients.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the institutional review board of The University of Hong Kong together with the Hospital Authority (IRB: UW 06-349T/1374).
Informed consent
All participant information was anonymized and unique ID codes were presented in this manuscript. Publication of the de-identified results from all consenting participants was approved.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Tang, C.S., Zhuang, X., Lam, WY. et al. Uncovering the genetic lesions underlying the most severe form of Hirschsprung disease by whole-genome sequencing. Eur J Hum Genet 26, 818–826 (2018). https://doi.org/10.1038/s41431-018-0129-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0129-z
This article is cited by
-
Genetics of Hirschsprung’s disease
Pediatric Surgery International (2023)
-
Stem cell-based therapy for hirschsprung disease, do we have the guts to treat?
Gene Therapy (2022)
-
“Too much guts and not enough brains”: (epi)genetic mechanisms and future therapies of Hirschsprung disease — a review
Clinical Epigenetics (2019)
