
Abstract
Telomere-related disorders are a clinically and genetically heterogeneous group of disorders characterized by premature telomere shortening and proliferative failure of a variety of tissues. This study reports the spectrum of telomere-related gene variants and telomere length in Nordic patients referred for genetic testing due to suspected telomere-related disorder. We performed Sanger sequencing of the genes TERT, TERC, DKC1, and TINF2 on 135 unrelated index patients and measured telomere length by qPCR on DNA from peripheral blood leukocytes. We identified pathogenic or likely pathogenic variants in 10 index patients, all of which had short telomeres compared to age-matched healthy controls. Six of the 10 variants were novel; three in TERC (n.69_74dupAGGCGC, n.122_125delGCGG, and n.407_408delinsAA) and three in TERT (p.(D684G), p.(R774*), and p.(*1133Wext*39)). The high proportion of novel variants identified in our study highlights the need for solid interpretation of new variants that may be detected. Measurement of telomere length is a useful approach for evaluating pathogenicity of genetic variants associated with telomere-related disorders.
Similar content being viewed by others
Introduction
Telomeres constitute the protective complexes of the termini of eukaryotic chromosomes. The telomeres shorten by each cell division due to incomplete replication of the lagging strand, and as a consequence, most somatic cells have limited replicative capacity [1,2,3]. However, telomere attrition can be compensated by telomerase-mediated elongation, and telomerase is expressed in highly proliferating cells including stem cells, germ cells, and activated lymphocytes. The telomerase ribonucleoprotein complex consists of an RNA component (TERC), a catalytic reverse transcriptase component (TERT), and accessory proteins of importance for complex stability and binding to the telomeres, including DKC1, NHP2, NOP10, and GAR1 [4].
Genetic variants that affect the function in genes involved in telomere maintenance result in disorders referred to as “telomeropathies” or “telomere syndromes” with premature telomere shortening and proliferative failure of a variety of tissues [3, 5]. The patients present with overlapping, highly variable phenotypes, with differences in penetrance and age of onset [6]. Different variants in a telomere related gene may cause different disorders, and individuals with the same variant may show different clinical features, ranging from very mild to severe [7]. Furthermore, families with telomeropathies often display genetic anticipation, a phenomenon which decreases age of onset and increases severity of symptoms in later generations [8]. The telomeropathies include dyskeratosis congenita (DC), which is characterized by bone marrow failure, abnormal skin pigmentation, nail dystrophy, and oral leukoplakia [9]. Other telomere-related disorders include Hoyeraal–Hreidarsson syndrome (HHS) [10], pulmonary fibrosis (PF) [11, 12], aplastic anemia (AA) [13, 14], myelodysplastic syndrome (MDS) [15], acute myeloid leukemia (AML) [16], and liver cirrhosis [17, 18].
The most commonly mutated genes involve components of the telomerase complex (TERT, TERC, and DKC1) [9, 19, 20] and the telomere-binding shelterin complex (TINF2 encoding TIN2) [7, 21]. With this study, we aim to describe the spectrum of genetic variants in patients referred to our laboratory at Umeå University hospital in Sweden for genetic testing of telomere-related genes, as part of routine health care. We further describe telomere length in these patients.
Methods
Patients
Since 2011, patients with suspicion of a telomere-related disorder have been referred to the Department of Clinical Genetics, Umeå University hospital, Sweden, for genetic testing of TERT, TERC, DKC1 and TINF2, and/or telomere length measurements, as part of routine health care. For this study, we included 135 unrelated Nordic patients where both genetic screening and telomere length had been requested between 2011 and 2016. The study was approved by the Regional ethical review board in Umeå (Dnr 2016/258-31), and the patients and/or their guardians provided informed consent.
Genetic analysis and classification of variant pathogenicity
Genomic DNA was extracted from peripheral blood leukocytes and analyzed by Sanger sequencing of all exons in the genes TERT and TERC on an ABI 3500xL Dx Genetic Analyzer (Applied Biosystems). Furthermore, 40 index patients were additionally analyzed for DKC1 and TINF2 if requested on the referral, or if TERT and TERC were negative and the telomeres were short for their age. All exons of DKC1 and exon 6 of TINF2 were analyzed, which is the only exon of this gene where pathogenic variants have been found so far.
Sequences were evaluated by Sequencer software and identified variants denoted by Human Genome Variation Society nomenclature. Assessment of the pathogenicity of all identified variants, regardless if they were novel or previously reported, was performed using Alamut software (Interactive Biosoftware, Rouen, France) in accordance with American College of Medical Genetics (ACMG) guidelines [22]. This software incorporates different programs for in silico prediction of the effect of amino acid changes and splicing variants. Additionally, it gives allele frequencies in the dbSNP and Exome Aggregation Consortium (ExAC) databases [23] and shows if a variant has been reported before in Human Gene Mutation Database (HGMD) or ClinVar. The metrics used for classification of variants included population allele frequency data, evolutionary conservation, in silico predictions, prior publications, family studies/segregation, and telomere length measurements performed in our laboratory. Variants were categorized into five classes: pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, and benign. Identified variants were considered to be pathogenic or likely pathogenic only if an effect on the protein (or the secondary structure of the molecule in the case of TERC) could be predicted, and/or if the variant was absent from control individuals, and/or if it co-segregated with affected family members, and/or if the telomere length of the index patient was short compared to controls. All variants reported in this paper (except those classified as benign or likely benign) have been submitted to the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar) and can be found under the submission name “TEL001”.
Telomere length measurement
Relative telomere length (RTL) was determined in DNA extracted from total peripheral blood white cells from patients (n = 135) and controls (n = 113), by the quantitative PCR method described by Cawthon et al. [24], with minor modifications as described in the Supplemental materials and methods. Briefly, each DNA sample was analyzed in triplicate wells in separate telomere (TEL) and single copy gene (HBG) reactions on an ABI 7900HT instrument (Applied Biosystems), at two separate occasions in 96-well plates. TEL/HBG (T/S) values were calculated by the 2−ΔCt method, where ΔCt = CtTEL−CtHBG. The RTL value was generated by dividing samples T/S value with the T/S value of a reference cell line DNA (CCRF-CEM) included in all runs. A standard curve of the reference cell line DNA was included in every run to monitor PCR efficiency. The RTL value of each sample was plotted against the individual's age in a graph. An individual’s RTL was classified as normal, within the lower normal distribution, or short, compared to 113 normal controls (age 0–83 years). RTL means were compared between groups by ANOVA with Bonferroni correction.
Results
Sequencing of telomere-related genes and telomere length measurement
We determined telomere length and screened for genetic variants in core components of the telomerase or shelterin complex in 135 index patients with suspected telomere-related disorders. The phenotypic information in the referrals was sparse in some patients and had a combination of features for other patients, but the main referral reasons are summarized in Table 1.
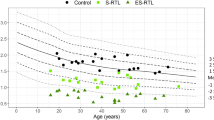
In total, we identified 10 patients with pathogenic or likely pathogenic variants, six of which were novel (Table 2). RTL was measured by quantitative PCR in all index patients and compared with healthy controls of similar age-distribution (Fig. 1a). The RTL of individuals with pathogenic or likely pathogenic variants (0.66±0.18) was significantly shorter compared to patients with no identified variant (1.36±0.40, p < 0.001) and healthy controls (1.46±0.35, p < 0.001) (Fig. 1a, b). There was no significant difference in RTL between patients with no identified variant and healthy controls (1.36 vs. 1.46, p = 0.115). Pedigrees and additional clinical features for family members with novel variants are shown in Fig. 2 and Supplemental Table 1, respectively. The genomic coordinates for the identified variants are shown in Supplemental Table 2.

Telomere lengths in patients with pathogenic or likely pathogenic variants are shorter compared with healthy controls. a Scatter plot of relative telomere length (RTL) with age in healthy controls (n = 113, gray circles) and patients with suspected telomeropathies; classified as pathogenic or likely pathogenic (n = 10, triangles), VUS (n = 2, unfilled squares), or negative (n = 123, black circles) for variants in the analyzed genes (TERC, TERT, DKC1, TINF2). Every dot corresponds to a measurement from a single individual. The line of best fit for each subgroup is shown. b Box plot analysis of age and RTL, respectively, in the healthy controls and index patients with or without pathogenic or likely pathogenic variant. The box represents the interquartile range which contains the 50% of values. The whiskers are lines that extend from the box to the highest and lowest values, excluding extreme outliers. The median is indicated as a line across the box. RTL was compared between groups by ANOVA with Bonferroni correction; ** p < 0.001

Pedigrees of families with novel variants showing co-segregation with disease. a TERC or b TERT. All identified variants, except the one in family T14, occurred in heterozygous form. The arrow indicates the index case. Circles represent females; squares represent males. Symbols with a slash indicate deceased subjects. Black symbols represent individuals with disease features; white symbols represent unaffected individuals; ? denotes that no clinical information is known. The presence or absence of a variant is indicated by plus or minus signs, respectively. The family number and genetic variant are listed above each family. Roman numericals indicate the generation. Numbers in parentheses indicate individuals for whom no DNA sample was available. AA aplastic anemia, Muc mucocutaneous features, PF pulmonary fibrosis, SCT stem cell transplantation, TCP thrombocytopenia
Novel genetic variants in TERC
In TERC, three novel variants were identified (Supplemental Fig. 1A). Two of the variants were located in the pseudoknot domain in the proposed secondary structure of the molecule, and were predicted to disrupt base pairing in the double-stranded helical regions. The first variant, n.69_74dupAGGCGC, occurred in the P2a.1 stem region. The index patient was a 40-year-old woman with MDS/AML. The pedigree indicated features of anticipation with thrombocytopenia and progression to MDS/AML at an even younger age for each generation in the family (Fig. 2a; family T4). Telomere length studies showed that the index patient (IV:17) had short telomeres. Segregation analysis demonstrated that affected relatives from which blood sample was available (III:10, III:14, and IV:1) carried the same novel variant, confirming its co-segregation with disease. The telomeres were short in individuals III:10 and III:14, whereas they were within the normal distribution for individual IV:1.
The second novel variant in the pseudoknot domain, n.122_125delGCGG, occurred in the P2b stem region. The index patient was a 46-year-old woman with MDS, who had developed thrombocytopenia during pregnancy at 38 years of age. Her brother and father had MDS and features of lung and liver disease at an age under 50 years (Fig. 2a; family T3). The variant was inherited from the affected father (II:1), whereas no DNA from the deceased brother (III:1) was available. The variant was also found to be present in an asymptomatic brother (III:3) and his son (IV:3). Telomere length measurements showed that the index patient and all relatives carrying the novel disease variant had short telomeres for their age.
The third novel variant, n.407_408delinsAA, was predicted to disrupt conserved base pairing in the P8b stem region of the CR7 domain. This variant was found in a 52-year-old man with MDS, who had affected relatives with thrombocytopenia, liver cirrhosis, and premature graying of the hair (Fig. 2a; family T13). Family studies showed that the index patient had inherited the variant from his affected mother (II:3), and that they both had short telomeres. No DNA was available from other family members.
Novel genetic variants in TERT
In TERT, three novel variants were identified (Supplemental Fig. 1B). The first variant, c.2320C>T, p.(R774*), occurred in the reverse transcriptase domain and was predicted to replace an arginine with a stop codon. The patient carrying this variant was an 11-year-old boy with thrombocytopenia, dysmorphic nails, and rough tongue (Fig. 2b; family T22). He had very short telomeres (Fig. 1; sample taken as 5 year old). No family history was known, but due to its deleterious nature, p.(R774*) was considered pathogenic.
The second variant, c.3399A>G, p.(*1133Wext*39), was identified in the C-terminal end of the gene and was predicted to replace the stop codon with an insertion of extra amino acids. The index patient was a 17-year-old man with AA and liver affection (Fig. 2b; family T5). Telomere length examination showed short telomeres. Family studies demonstrated that he had inherited the novel variant from his asymptomatic mother (II:2), whose telomeres were within the lower normal distribution. The father (II:1) did not carry the variant and had normal telomeres. The maternal uncle (II:3) had died of PF at age 40, but since no DNA was available co-segregation could not be confirmed. Due to the deleterious nature of p.(*1133Wext*39) and its absence in the general population, it was considered likely pathogenic.
The third variant, c.2051A>G, p.(D684G), changes a conserved residue in the reverse transcriptase domain. The index patient was a 32-year-old man with hypoplastic MDS transformed into leukemia, dystrophic nails, and reticular skin pigmentation (Fig. 2b; family T14). Of special notice is that he carried the variant in homozygous form, inherited from heterozygous asymptomatic parents. Furthermore, an affected sister (II:2) with tongue cancer at age 26 was homozygous for the same variant and an unaffected sister (II:3) did not carry it. The telomeres were very short in both homozygous individuals, whereas they were short in the father (I:1), within the lower normal distribution in the mother (I:2), and normal in the unaffected sister. Based on the co-segregation analysis and telomere length results, p.(D684G) was considered to be likely pathogenic and associated with autosomal recessive inheritance in this family.
To be noted, p.(D684G) was also identified in two additional index patients, but in heterozygous form; in the 11-year-old boy in family T22 described above, and in a 58-year-old man with hypoplastic MDS, lung fibrosis, liver cirrhosis, and short telomeres (Fig. 2b; family T11). Co-segregation analysis in families T11 and T22 could not be performed, and we therefore classified p.(D684G) as a VUS in these families.
Another novel VUS in TERT, c.2287-5G>A, was identified in a 19-year-old man with congenital kidney disorder who had developed thrombocytopenia and anemia (Fig. 2b; family T10). According to the referral, he had an 8-year-old half-brother with MDS. The variant was predicted to significantly decrease the strength of the natural splice consensus site and create a new splice acceptor site, which would cause an in-frame insertion of one amino acid. However, we classified this splice variant as a VUS since the index patient had normal telomeres and no co-segregation studies could be performed.
Previously reported genetic variants
In four patients, we identified variants that had previously been reported and that we classified as pathogenic according to ACMG guidelines. In a 64-year-old woman with anemia, PF, and multiple forms of cancer, the variant TERC n.107G>T in the P3 region of the pseudoknot was found [25]. The woman had short telomeres and unknown family history. In a 45-year-old woman with anemia, thrombocytopenia and MDS, the variant TERT c.1892G>A, p.(R631Q) was identified [26,27,28]. The woman had short telomeres and an affected son and sister who also carried the variant. In an 18-year-old man with thrombocytopenia and dysmorphic nails, the variant DKC1 c.203A>G, p.(H68R) was identified [29]. The man had short telomeres and unknown family history. In a 9-year-old girl with AA and dysmorphic nails, the variant TINF2 c.845G>A, p.(R282H) was found [7, 21]. The girl had very short telomeres and no family history. Analysis of the parents demonstrated that the variant had occurred de novo.
Likely benign variants
Several likely benign variants were identified, including the TERT variants p.(A279T) in seven patients, p.(A1062T) in five patients, and p.(H412Y) in three patients, and the TERC variant n.228G>A in one patient. These variants were considered to have no clinical significance since they are prevalent in the general population; p.(A279T) has been identified in 5.0% of European chromosomes by the ExAC database, p.(A1062T) in 2.1%, and p.(H412Y) in 1.5%. The TERC variant n.228G>A in this study was identified in a man of African origin. This variant has been reported in 2.8% of African chromosomes by ExAC.
Discussion
Novel variants of TERC and TERT associated with telomeropathies
In this study, we identified six novel and four previously reported pathogenic or likely pathogenic variants in telomere related genes among 135 Nordic patients with suspicion of telomere-related disorders. All novel variants were located in highly conserved regions, and were absent or extremely rare among controls, and/or co-segregated with disease in the family, supporting their pathogenicity.
In TERC, three of the four identified variants disrupt the conserved base pairing in the pseudoknot, which is the region of the molecule where most pathogenic variants occur. The pseudoknot region binds to TERT and is essential for telomerase activity [30]. The novel variants n.69_74dupAGGCGC and n.122_125delGCGG occurred in the double-stranded helical regions P2a.1 and P2b. Other variants that affect function in these conserved stems, such as n.67G>A and n.72C>G in P2a.1, and n.95_96delGC in P2b, have been reported in patients with DC or AA [13, 25]. The authors reported significantly short telomeres of these patients, as well as almost completely abolished telomerase activity, demonstrating the importance of these stem regions for TERC function. The third identified pseudoknot variant, n.107G>T in the P3 stem region, has previously been reported in a patient with AA who had inherited the variant from an asymptomatic father [25]. This patient had short telomeres compared to controls and the telomerase activity in vitro was less than 1% of wild type [25]. The fourth identified variant, the novel n.407_408delinsAA, occurred in the CR7 domain. CR7 is a highly conserved base-pairing region that is essential for stability and accumulation of TERC within the cell [31]. Another variant that affects function in the same region, n.408C>G, has been shown to co-segregate in a family with DC [20]. In a functional assay, n.408C>G greatly reduced telomerase activity, whereas a compensatory variant targeting the corresponding base rescued the activity [32], demonstrating the importance of this region.
In TERT, the identified variants were spread throughout the gene, which is in accordance with previous reports. The variant p.(R631Q) has previously been reported in families with DC, PF, and MDS/AML, and shown to completely abolish telomerase activity [26,27,28]. The novel variant p.(R774*) in the reverse transcriptase domain is predicted to result in a loss-of-function. Loss-of-function variants are considered to affect function as they will result in haploinsufficiency, which is a well-established disease mechanism with telomerase variants [19]. Another loss-of-function variant in the same domain, p.(R889*), has been reported in a patient with AA [33]. The novel variant p.(*1133Wext*39) was classified as likely pathogenic due to its deleterious nature and absence in the general population. Another variant in the same C-terminal end of TERT (a 177 bp-deletion referred to as “E1116fsX1127”) has been reported in a family with PF and shown to greatly reduce telomerase activity, demonstrating the importance of this region for normal function of the protein [12]. The novel variant p.(D684G) was identified in three different families; in homozygous form in the index patient of family T14, and in heterozygous form in families T11 and T22. This variant is observed in 0.0136% in the ExAC database, which is higher than expected for an autosomal dominant disorder. In the literature, a few patients with autosomal recessive TERT variants have been described [34,35,36], supporting this mechanism of inheritance. Based on the co-segregation analysis and telomere length results, we considered p.(D684G) to be a likely pathogenic variant associated with autosomal recessive inheritance in family T14. In family T22, the index patient carried the additional variant p.(R774*), which explain the patient's symptoms and short telomeres. In family T11, the index patient carried only p.(D684G), although it is possible that he may have another disease variant in a gene not analyzed.
In DKC1, the identified variant p.(H68R) has previously been reported in a patient with DC [29]. Although no segregation analysis was reported by the authors, their patient had short telomeres and a positive family history. Two other variants affecting the same amino acid residue have been reported; p.(H68Q) in a sporadic case with features of both classic DC and HHS [37], and p.(H68Y) in a child with HHS who had a symptomatic brother carrying the same variant [25], supporting the pathogenicity of p.(H68R).
The identified TINF2 variant p.(R282H) has been reported as one of the most common variants among pediatric DC patients [7, 21]. Patients with TINF2 variants that affect function have very short telomeres with early age of onset and severe manifestations of the disease [7], which was also the case for the index patient in this study. Neither parent was found to carry p.(R282H), which is consistent with the observation that the majority of TINF2 variants arise de novo [7].
Telomere length analysis is important for determining pathogenicity of novel variants
The symptoms, clinical findings, and telomere-related findings in this study show a large diversity between the patients. A pathogenic or likely pathogenic variant was identified in 7.4% (10/135) of our index patients. This can be compared with a study where next-generation sequencing detected variants predicted to affect function in 5.1% (5/98) of AA patients and in 13.6% (15/110) of MDS patients [38]. In another study, 5% (10/200) of patients with bone marrow failure had a TERT or TERC variant that affect function [39]. However, comparison of studies is complicated due to different number of genes analyzed and different diagnoses of the included patients. Of the 12 patients in our study with an identified pathogenic or likely pathogenic variant or VUS, eight had a family history of a related phenotype, indicating that a disease variant is more likely to be found in those patients having a positive family history. However, investigation of a telomere-related disorder has to be considered even in the absence of a family history.
In most of the analyzed patients, no pathogenic or likely pathogenic variant was identified. There is a possibility that some of these individuals in fact do not have a telomere-related disorder, or that any variants are missed due to technical limitations or location in other genes not included in this study. This is especially true for the patients who were found to have short telomeres and/or a positive family history. At our laboratory, we have chosen to analyze only the genes TERT, TERC, DKC1, and TINF2. However, variants that affect function have been identified in other telomere-related genes, such as NHP2 and NOP10 in autosomal recessive DC [40, 41], RTEL1 in HHS [42], PARN in severe DC [43], TCAB1 in DC [44], and CTC1 in Coats Plus [45]. With large scale next-generation sequencing-based approaches, the number of variants in new disease genes will continue to increase.
It can be difficult to determine the pathogenicity of an identified novel variant in a family with a telomere-related disorder due to genetic heterogeneity and overlapping phenotypes. For example, patients with TERT variants may express PF, AA, and liver cirrhosis at different times throughout life [5], whereas others present with these symptoms at the same time, or not at all. Furthermore, due to genetic anticipation, it is common to have members of younger generations presenting earlier in life compared with older generations within a family [8]. Clinical presentation correlates with telomere length, where the majority of the most severely affected individuals display the shortest telomere lengths [2]. However, there is not a strict relationship between telomere length and severity of symptoms at an individual level [25, 46]. In this study, all but one of the patients and affected relatives with a pathogenic or likely pathogenic variant had short telomeres. The exception was individual IV:1 in family T4. Of the asymptomatic relatives, individuals III:3 and IV:3 in family T3, and individual I:1 in family T14 had short telomeres, whereas individual II:2 in family T5 and individual I:2 in family T14 had telomeres within the lower normal distribution compared to controls of similar age-distribution.
Telomere length analysis is an important tool when evaluating novel variants, as short telomeres supports pathogenicity. This is especially important for families where co-segregation analysis is not possible to perform. Since the qPCR method requires a standardized and optimized setup, and the number of patients referred for blood telomere-length analysis due to suspected telomere-related disorder is currently low, national or regional centers for telomere length measurements will be preferred. In this study, we demonstrated co-segregation of the novel variant with disease and/or short telomeres in families T3, T4, T13, and T14, thus confirming pathogenicity. For the other families co-segregation could not be demonstrated, either due to a lack of family history or unavailable samples from deceased relatives. Due to the deleterious nature of the variant and short telomeres in the index patient, we were able to classify the novel variants in families T5 and T22 as likely pathogenic and pathogenic, respectively.
In conclusion, this study increases the publicly known genetic variants associated with the telomeropathies. Identification of inherited telomeropathies is of great importance for appropriate medical care of the patient, as it influences choice of treatment and follow-up routines.
References
Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005;26:867–74.
Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704.
Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193–8.
Egan ED, Collins K. Specificity and stoichiometry of subunit interactions in the human telomerase holoenzyme assembled in vivo. Mol Cell Biol. 2010;30:2775–86.
Holohan B, Wright WE, Shay JW. Cell biology of disease: telomeropathies: an emerging spectrum disorder. J Cell Biol. 2014;205:289–99.
Armanios M. Syndromes of telomere shortening. Annu Rev Genom Hum Genet. 2009;10:45–61.
Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–3600.
Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36:447–9.
Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19:32–38.
Knight SW, Heiss NS, Vulliamy TJ, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999;107:335–9.
Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–26.
Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7.
Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–70.
Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413–24.
Xin ZT, Beauchamp AD, Calado RT, et al. Functional characterization of natural telomerase mutations found in patients with hematologic disorders. Blood. 2007;109:524–32.
Calado RT, Regal JA, Hills M, et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc Natl Acad Sci USA. 2009;106:1187–92.
Calado RT, Regal JA, Kleiner DE, et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS One. 2009;4:e7926.
Hartmann D, Srivastava U, Thaler M, et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology. 2011;53:1608–17.
Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA. 2005;102:15960–4.
Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–5.
Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82:501–9.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47.
Vulliamy TJ, Kirwan MJ, Beswick R, et al. Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS One. 2011;6:e24383.
Basel-Vanagaite L, Dokal I, Tamary H. et al. Expanding the clinical phenotype of autosomal dominant dyskeratosis congenita caused by TERT mutations. Haematologica. 2008;93:943–4.
Diaz de Leon A, Cronkhite JT, Katzenstein AL, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE. 2010;5:e10680.
Kirwan M, Vulliamy T, Marrone A, et al. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum Mutat. 2009;30:1567–73.
Carrillo J, Martinez P, Solera J, et al. High resolution melting analysis for the identification of novel mutations in DKC1 and TERT genes in patients with dyskeratosis congenita. Blood Cells Mol Dis. 2012;49:140–6.
Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100:503–14.
Martin-Rivera L, Blasco MA. Identification of functional domains and dominant negative mutations in vertebrate telomerase RNA using an in vivo reconstitution system. J Biol Chem. 2001;276:5856–65.
Ly H, Schertzer M, Jastaniah W, et al. Identification and functional characterization of 2 variant alleles of the telomerase RNA template gene (TERC) in a patient with dyskeratosis congenita. Blood. 2005;106:1246–52.
Calado RT, Regal JA, Kajigaya S, Young NS. Erosion of telomeric single-stranded overhang in patients with aplastic anaemia carrying telomerase complex mutations. Eur J Clin Invest. 2009;39:1025–32.
Marrone A, Walne A, Tamary H, et al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 2007;110:4198–205.
Du HY, Pumbo E, Manley P, et al. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111:1128–30.
Stockklausner C, Raffel S, Klermund J, et al. A novel autosomal recessive TERT T1129P mutation in a dyskeratosis congenita family leads to cellular senescence and loss of CD34+ hematopoietic stem cells not reversible by mTOR-inhibition. Aging (Albany NY). 2015;7:911–27.
Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–5.
Keel SB, Scott A, Sanchez-Bonilla M, et al. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica. 2016;101:1343–50.
Du HY, Pumbo E, Ivanovich J, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009;113:309–16.
Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA. 2008;105:8073–8.
Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16:1619–29.
Le Guen T, Jullien L, Touzot F, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22:3239–49.
Tummala H, Walne A, Collopy L, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125:2151–60.
Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011;25:11–16.
Anderson BH, Kasher PR, Mayer J, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44:338–42.
Ueda Y, Calado RT, Norberg A, et al. A mutation in the H/ACA box of telomerase RNA component gene (TERC) in a young patient with myelodysplastic syndrome. BMC Med Genet. 2014;15:68.
Acknowledgements
We would like to thank all patients and relatives who have participated in the study, and the clinicians who referred samples to our registry. We also thank laboratory assistant Susann Haraldsson for telomere length measurements. Financial support was provided through regional agreement between Umeå University and Västerbotten County Council on cooperation in the field of Medicine, Odontology and Health. SD and MH are supported by grants from the Medical Faculty of Umeå University, the Swedish Childhood Cancer Foundation, Lion's Cancer Research Foundation, Umeå University, the Kempe foundations, and Uppsala-Umeå Comprehensive Cancer Consortium.
Author contributions
Study design: AN, AR, GR, SD, and MH. Data collection: AN, KRJ, LK, YK, PB, JSM, OL, AA, and AOK. Data analysis: AN, AR, PL, and SD. Data interpretation: AN, AR, GR, SD, and MH. Drafting manuscript: AN. All authors revised the manuscript content and approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Norberg, A., Rosén, A., Raaschou-Jensen, K. et al. Novel variants in Nordic patients referred for genetic testing of telomere-related disorders. Eur J Hum Genet 26, 858–867 (2018). https://doi.org/10.1038/s41431-018-0112-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0112-8
This article is cited by
-
Genetics of human telomere biology disorders
Nature Reviews Genetics (2023)
-
DNA methylation variations and epigenetic aging in telomere biology disorders
Scientific Reports (2023)
-
FinnGen provides genetic insights from a well-phenotyped isolated population
Nature (2023)
-
The telomerase gene polymorphisms, but not telomere length, increase susceptibility to primary glomerulonephritis/end stage renal diseases in females
Journal of Translational Medicine (2020)
-
Biallelic mutations in WRAP53 result in dysfunctional telomeres, Cajal bodies and DNA repair, thereby causing Hoyeraal–Hreidarsson syndrome
Cell Death & Disease (2020)
