
Abstract
We previously showed that a common genetic variant leads to a remarkably increased risk of type 2 diabetes (T2D) in the small and historically isolated Greenlandic population. Motivated by this, we aimed at discovering novel genetic determinants for glycated hemoglobin (HbA1C) and at estimating the effect of known HbA1C-associated loci in the Greenlandic population. We analyzed genotype data from 4049 Greenlanders generated using the Illumina Cardio-Metabochip. We performed the discovery association analysis by an additive linear mixed model. To estimate the effect of known HbA1C-associated loci, we modeled the effect in the European and Inuit ancestry proportions of the Greenlandic genome (EAPGG and IAPGG, respectively). After correcting for multiple testing, we found no novel significant associations. When we investigated loci known to associate with HbA1C levels, we found that the lead variant in the GCK locus associated significantly with HbA1C levels in the IAPGG (\(P_{IAPGG} = 4.8 \times 10^{ - 6},\,\beta _{IAPGG} = 0.13\,{\mathrm{SD}}\)). Furthermore, for 10 of 15 known HbA1C loci, the effects in IAPGG were similar to the previously reported effects. Interestingly, the ANK1 locus showed a statistically significant ancestral population differential effect, with opposing directions of effect in the two ancestral populations. In conclusion, we found only 1 of the 15 known HbA1C loci to be significantly associated with HbA1C levels in the IAPGG and that two-thirds of the loci showed similar effects in Inuit as previously found in European and East Asian populations. Our results shed light on the genetic effects across ethnicities.
Similar content being viewed by others


Introduction
The Greenlandic population comprises about ~57,000 individuals of whom the majority (>80%) has a degree of European ancestry [1]. On average, a Greenlander has 25% European ancestry [1], thus, the Greenlandic genome has both an Inuit ancestry proportion (Inuit Ancestry Proportion of the Greenlandic Genome (IAPGG)) and a European ancestry proportion (European Ancestry Proportion of the Greenlandic Genome (EAPGG)). The Greenlandic population is closely related, both genetically and culturally, to the Inuit population (Iñupiat) in Canada and Alaska, which in turn is culturally related to the Yup’ik people of Alaska and Siberia [2]. In contrast, the Greenlandic population is genetically far from any large population, has been small for many thousand years, and has been isolated in an inhospitable climate, making this population genetically interesting.
In the past 25 years, the Greenlandic population has experienced a dramatic increase in type 2 diabetes (T2D) prevalence [3,4,5]. We previously described a common loss-of-function variant in TBC1D4 with a high isolated impact on postprandial circulating glucose and insulin levels and risk of T2D in the Greenlandic population [6]. In a recessive model, the variant increased the 2-h (h) plasma glucose levels during an oral glucose tolerance test (OGTT) by 4 mmol/l and increased the risk of T2D with an odds ratio of 10.3. The effect sizes shown for the TBC1D4 variant are several fold higher than previously observed in genome-wide association studies (GWAS). Interestingly, the loss-of-function TBC1D4 variant decreased fasting plasma glucose level by 0.18 mmol/l6.
T2D can be diagnosed from measurement of plasma glucose at fasting and at 2 h during an OGTT or by measurements of glycated hemoglobin (HbA1C) levels [7]. The HbA1C level reflects the average level of plasma glucose for the individual during the past 6–12 weeks [8]. From 2011, the World Health Organization (WHO) recommended the use of standardized measurement of HbA1C levels to diagnose T2D with a cut-off point of 6.5% (48 mmol/mol) [7]. HbA1C has also been shown to be a risk factor for cardiovascular diseases, even at non-diabetic levels [9].
GWAS of HbA1C levels in the European and Asian populations with up to 31,000 individuals have identified a total of 18 genomic loci significantly associated with HbA1C in either or both populations [10, 11] (Supplementary Table 1). The majority of these loci do not associate with plasma glucose levels or T2D [10, 11], conversely six loci (ANK1, GCK, G6PC2, MTNR1B, CDKAL1, and FADS1-FADS2) have been shown to associate with fasting glucose levels or T2D [12,13,14].
Motivated by the highly increased risk of T2D imposed by the common variant in the TBC1D4 in the Greenlandic population [6], we aimed to discover novel genetic associations with HbA1C in this population. Furthermore, we aimed to investigate genomic loci already known to associate with HbA1C levels in European and East Asian populations by estimating the effect of the reported lead single-nucleotide polymorphisms (SNPs) in the Inuit ancestry proportion of the genomes of the Greenlandic population, IAPGG.
Materials and methods
Study samples
Data originated from two separate cohorts, “Inuit Health in Transition” (IHIT) [3] and B99 [15]. The IHIT cohort (N = 3115) was collected as a part of a health survey of the Greenlandic population during 2005–2010. Participants were invited from 12 regions, consisting of 21 selected towns and villages. The B99 cohort (N = 1401) was recruited during 1999–2001 as a part of a general population health survey. There was an overlap of 295 individuals between the two cohorts, who were assigned to the B99 cohort. We removed participants previously diagnosed with T2D (N = 67), as well as individuals whose genetic data did not pass quality control (see details below) making the final number of individuals in the meta-analysis 4049 (IHIT: N = 2730; B99: N = 1319).
In both Greenlandic cohorts, the HbA1C levels were measured using ion exchange high-performance liquid chromatography, in a Na-heparinized (5 μl) capillary in Ebendorf tube with 1 ml EDTA and potassium cyanide solution (0.25 mmol/l). In the IHIT cohort, this was measured by Tosoh G7 [16], whereas in the B99 cohort the measurement was done using BioRad VARIANTTM [17]. HbA1C was defined as the ratio of HbA1C to the total hemoglobin and measured in accordance to the The International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) reference method [18]. Basic information for each cohort is given in Supplementary Table 2.
The studies have received ethical approval from the Commission for Scientific Research in Greenland (project 2011–13, ref. no. 2011–056978 and project 2013–13, ref. no. 2013–090702) and were conducted in accordance with the ethical standards of the Helsinki II Declaration. Participants gave their written consent after being informed about the study both orally and in writing
We also included the Danish cohort Inter99 in our analysis of ancestral-specific effect to be representative of the European population. The Inter99 cohort (N = 6127) is a population-based randomized controlled trial, investigating the effect on cardiovascular diseases, and diabetes of a non-pharmacological intervention [19]. After removing participants with known diabetes (N = 121), the number of individuals added to the ancestral-specific effect analysis was 6006, bringing the final number in the ancestral-specific effect analyses to 10,055 individuals.
Genotyping
We genotyped all participants with the Illumina Cardio-Metabochip [20] (MetaboChip) using the HiScan system (Illumina). Illumina GenomeStudio software was used for the genotype calling. The MetaboChip consists of 196,725 SNPs selected for genetic studies on cardiovascular, metabolic, and anthropometric traits.
We removed all SNPs with a call rate below 0.99 before analysis, but kept all other SNPs regardless of minor allele frequency (MAF). Furthermore, we removed SNPs that failed a Hardy–Weinberg equilibrium test with a P-value below 10−5 in either of the two cohorts. We removed duplicate samples and excluded samples with mis-specified sex. Finally, we removed samples that had a call rate below 98% for a subset of SNPs with a MAF above 1%. A total of 426 samples and 84,448 SNPs were removed through these steps leaving 112,277 SNPs in 4049 individuals for analysis.
Loci known to associate with HbA1C level
From the literature, we found 21 SNPs across 18 loci, shown to associate genome-wide significantly with HbA1C levels in European or East Asian populations (Supplementary Table 1) [10, 11]. For three of these loci, two different lead SNPs were reported and for these we chose the SNP with the lowest reported P-value. We defined a locus as the ±240 kilo-base-pairs (kbp) region surrounding the reported lead SNP. For 7 of 18 genomic loci, the reported lead SNP was available in the quality filtered MetaboChip data, whereas we for 8 of the 18 loci analyzed a proxy SNP in high (R2 > 0.9) linkage disequilibrium (LD) in the population in which they were identified (Supplementary Table 3). For the remaining three loci (TMEM79, MYO9B,and TMPRSS6), the lead SNP was unavailable in the quality filtered MetaboChip data and we were unable to find a suitable proxy. Therefore, these three loci were omitted from our analyses. For the sake of clarity, we name these 15 loci as “known HbA1C loci” as a shorthand version for loci known to associate with HbA1C levels in European and/or East Asian populations.
Statistical analysis
The population structure of the Greenlandic population has been shown to be composed of an ancestral Inuit population with recent gene flow from the European population [1, 6] and as the genotyped individuals constitute almost 10% of the adult population, >1000 close relationships (parent–offspring or siblings) were identified [6]. To control the false-positive rate in the discovery analyses, we therefore performed association testing between each SNP and HbA1C levels using an additive linear mixed model as implemented in GEMMA [21]. This model takes the relatedness between individuals, as well as admixture into account by using a general similarity matrix (GSM). We further adjusted the analysis for age and sex by adding these as covariates in the model. As the distribution of HbA1C prior to transformation was not normal in either cohort (SkewnessIHIT = 3.70, KurtosisIHIT = 62.71, SkewnessB99 = 4.44, KurtosisB99 = 53.93) (see supplementary figure 2 and 3), we transformed the HbA1C measurements to a standard normal distribution using a rank-based inverse normal transformation on the unadjusted variable. We used GEMMA’s maximum likelihood estimator method to calculate P-values. Data from the two cohorts were analyzed separately and the results were subsequently combined in a fixed-effect meta-analysis. We reported the P-value for the two-tailed test on the combined effect. The inflation of P-values was calculated as \(\lambda = \frac{{{\mathrm{median}}\left( {{\mathbf{\chi }}_1^2\left( {1 - {\mathrm{P}}} \right)} \right)}}{{{\mathrm{median}}\left( {{\mathbf{\chi }}_1^2(0.5)} \right)}}\), where \({\mathrm{\chi }}_1^2\) is the chi-squared distribution with one degree of freedom and P is the observed P-values in the discovery analysis.
We also looked for novel associations in the known loci by using the same linear mixed model but with the genotype of the reported lead SNP for the locus as an additional covariate (conditional model) in addition to the unconditional model, and reported the results from both models alongside each other.
When comparing the effect of the lead SNPs found in European and East Asian populations with the Greenlandic cohort, the effect of interest for the current study is in the IAPGG. We therefore used asaMap [22], a novel statistical method that can perform association testing on the ancestral parts of an admixed population. First, we combined both Greenlandic cohorts, IHIT and B99, with the Danish Inter99 cohort [19], to strengthen the statistical power of asaMap and ADMIXTURE. Second, we used a rank-based inverse normal transformation separately in each cohort before combining the data. Third, to determine the proportions of Inuit ancestry for each individual in each cohort needed for the asaMap analyses, we used the software program, ADMIXTURE [23] with k = 2, implying that we assume two ancestral populations (Inuit and European) in admixed Greenlandic genome. Finally, using asaMap we ran an additive linear regression with age, sex, cohort and the first 10 principal components as covariates to adjust for cohort bias, population, and relatedness structure in lieu of a GSM. We tested three hypotheses: (1) The existence of an ancestral-specific effect (asaMap M1vM4) of the variant on HbA1C levels in the admixed Greenlandic population, (2) the variant’s effect only in IAPGG (asaMap M1vM2: βEAPGG=0), and (3) the variant’s effect only in the Inter99 cohort combined with the European ancestry proportion of the Greenlandic genome (Inter99 + EAPGG) (asaMap M1vM3: βIAPGG = 0). The applied models have previously been described in more detail [22]. We assumed that only the minor allele could have ancestry-specific effects, whereas the most frequent allele has the same effect regardless of ancestry. Therefore, the minor allele was always used as the effect allele in all of the asaMap analyses.
The inflation of P-values for the ancestral-specific effect model was calculated for all SNPs with a call rate of at least 99% and a MAF above 5% across all three cohorts. The resulting lambda (λ=0.89) was calculated as before \(\lambda = \frac{{{\mathrm{median}}\left( {{\mathrm{\chi }}_1^2\left( {1 - {\mathrm{P}}} \right)} \right)}}{{{\mathrm{median}}\left( {{\mathrm{\chi }}_1^2\left( {0.5} \right)} \right)}}\) (Supplementary Figure 1). We attribute the low lambda to the fact that many SNPs are not polymorphic in both the Inuit and the European ancestral populations.
To be able to compare the effect sizes from our analyses to the effect sizes reported in the literature, we translated the effects of known loci from percentage units to standard deviation (SD) units, as used in our study. For this, we used the 5783 non-diabetic participants of the Danish Inter99 cohort [19] where the SD of HbA1C was 0.40%.
We calculated the statistical significance thresholds using the conservative Bonferroni correction, which was calculated as \(P = \frac{{0.05}}{{N_{SNP}}}\), where NSNP is the number of SNPs. We applied different significance thresholds depending on the hypothesis tested, to reflect the different number of test in each model. In the discovery analysis, we used the standard GWAS significance threshold of P < 5×10−8. For the analyses of novel additional association signals in known loci were corrected for the total number of SNPs (2612) across all known loci \(\left( {P = \frac{{0.05}}{{2,612}} = 1.9 \times 10^{ - 5}} \right)\). The threshold chosen for the GEMMA and asaMAP analyses of the lead SNPs were corrected for the number of lead SNPs (15) tested \(\left( {P = \frac{{0.05}}{{15}} = 3.3 \times 10^{ - 3}} \right)\).
We calculated the statistical power to detect association signals at different significance thresholds given a specific MAF and effect size, by running a linear additive model on a simulated data with a normal distribution with the same variation as the combined HbA1C levels in the two Greenlandic cohorts. We then observed how often the model crossed a given significance level. We tested each of the three thresholds (genome-wide, loci-wide and lead SNP-wide) and for each effect size and MAF, we ran the simulation 100 times. To calculate statistical power for the ancestry-specific analyses, we used the asaMap model instead of a linear model, and added the real admixture proportions from the combined cohorts of Inter99, B99, and IHIT.
Results
We first performed a discovery analysis, in which we sought to detect novel associations with HbA1C in the Greenlandic population when applying an additive linear mixed model. We tested 112,277 SNPs in 4049 individuals and observed no inflation of the test statistic (λ=0.97). We found no SNPs reaching statistical genome-wide significance (P < 5 × 10−8) (Fig. 1). The strongest signal was for rs1146347_G (\({\mathrm{\beta }} = - 0.145\,{\mathrm{SD}},\,P = 1.5 \times 10^{ - 6}\)), which is located in an intergenic region with ~1 Mbp (mega base pairs) to the nearest genes (SPAG17 and TBX15; up- and downstream, respectively), none of which are known to associate with HbA1C levels or other glycemic or hematological traits. None of the most associated variants was known from the literature to associate with HbA1C levels (Supplementary Table 4) or other glycemic traits. None of these variants was significantly associated with HbA1C levels in the Danish cohort Inter99 consisting of European individuals (data not shown).

Association analyses of 112,277 markers on the MetaboChip with HbA1C levels in 4,049 Greenlanders. Left: Manhattan plot showing the –log10(P) for each marker against their position, with each chromosome colored alternatively blue and green. The red line represents the genome-wide significant level (P<5×10−8). Right: QQ plot showing the –log10(P) from the discovery analysis against the expected distribution. (Color figure online)
Next, we looked for novel association signals in loci known to be associated with HbA1C; however, we did not find any novel statistically loci-wide significant signals in neither in tests conditioned on the lead SNP in the locus nor in unconditional tests (Supplementary Table 5).
Finally, we investigated lead SNPs in known HbA1C loci. When using a linear mixed model, we found that 2 of the 15 lead SNPs associated statistically significantly with HbA1C levels in the Greenlandic cohorts (GCK rs730497_A: \({\mathrm{\beta }} = 0.079\,{\mathrm{SD}},\,P = 0.0013\), MTNR1B rs1387153_T: \({\mathrm{\beta }} = 0.081\,{\mathrm{SD}},\,P = 0.00036\)) (Supplementary Table 6). When further testing for ancestry-specific effects using the asaMap software, we found one locus with a statistically significant ancestral-specific effect (ANK1: \(P_{AnSE} = 0.00051\)). Additionally, we found one locus that associated significantly with HbA1C levels in IAPGG (GCK rs730497_A: \({\mathrm{\beta }} = 0.13\,{\mathrm{SD}},\,P_{IAPGG} = 4.8 \times 10^{ - 6}\)). In comparison, 9 of the 15 loci associated significantly (P < 3.3 × 10−3) in the European part of the asaMap analysis (Inter99 + EAPGG). Five of the six loci that did not associate in Europeans where originally reported in East Asian populations.
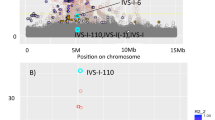
When comparing the 95% confidence interval (CI) of the effects reported in European (CEU) or East Asian (EAS) populations with the CI of association in the IAPGG, we observed that for 10 of the 15 known HbA1C loci, the 95% CI of association with HbA1C in IAPGG overlapped the 95% CI reported in CEU or EAS (Fig. 2). Of the five loci with non-overlapping CIs, two loci (ANK1, ATP11A/TUBGCP3) showed effect in the opposite direction in IAPGG compared with the effect found in CEU, however, only the ANK1 locus was nominally significant in IAPGG (PIAPGG = 0.023) (Fig. 2, Table 1).

Comparison of the reported effects of HbA1C-associated lead SNPs with the effects found Inuit proportion of the Greenlandic genome (IAPGG) and the combination of Danish Inter99 cohort and the European proportion of the Greenlandic genome (Inter99 + EAPGG). The plot shows the effect size, with 95% confidence intervals (CI), found in European (CEU) or East Asian (EAS), IAPGG and Inter99 + EAPGG for each known HbA1C locus. The effect size, beta, was measured in standard deviations (SD). Loci with a SE above one were omitted in this plot for easier viewing. The SNP for each locus is either the reported lead SNP or a proxy on the chip (Table 1)
We also investigated the lead SNP (rs7330796_T) in TBC1D4 and its association with HbA1C levels, but it showed no ancestral-specific effect (P = 0.97) nor was it significantly associated with HbA1C levels in either IAPGG or EAPGG (P = 0.7 and P = 0.75, respectively) under an additive model.
Discussion
We investigated the genetic determinants of HbA1C in the small and historically isolated Greenlandic population and found no novel association signals in the discovery analysis. Two known HbA1C-associated loci, GCK and MTNR1B, were significantly associated with HbA1C in the Greenlandic population. Furthermore, in analysis of the Inuit ancestral proportion of the Greenlandic genome (IAPGG), we found the GCK locus to associate significantly with HbA1C levels. In addition, we found that 10 of 15 known loci showed evidence of comparable effects in IAPGG when compared with the effects found in CEU or EAS populations. In contrast, two loci (ANK1 and ATP11A/TUBGCP3) showed indications of a different effect in IAPGG than reported in the literature (Fig. 2). The most likely explanation for these findings is that the tested variants in these two loci are not the causal variants and LD patterns differ between the two ancestral populations. This is especially an issue when proxy SNPs are used for the analysis. However, it is also possible the multiple association signals are present at these loci (ANK1and ATP11A/TUBGCP3) although we find no evidence for this in the current data. Lastly, three loci (HFE, HK1,and 9q31.2) had very low estimated effect allele frequencies (<0.01%) and are likely monomorphic in IAPGG.
In the discovery analysis, we did not find any statistically significant associations. This analysis was obviously limited by low statistical power (see Supplementary Table 7) for variants with moderate effects and by the non-genome-wide coverage of the MetaboChip. None of the genes in the region of the strongest signal had previously been shown to associate with HbA1C levels or metabolic traits or had any known biological relations to glucose metabolism. The top association signal is located on chromosome 1 between SPAG17 and TBX15. We recently detected a signal for selection in the region of TBX15 [24], however, the two SNPs (rs1146347 and rs4659153) are separated by ~800 kb and are not in LD in Greenlanders (R2=0.0005, D′=0.031).
In the analyses of the ancestral-specific effect of Inuit and European ancestry in the Greenlandic genome, we found one locus, GCK, which associated significantly in IAPGG. GCK encodes glucokinase (a member of the hexokinase family) and is primarily expressed in the liver and pancreas. Glucokinase is an enzyme that facilitates phosphorylation of glucose to glucose-6-phosphate, the first step in the glucose metabolism pathway [25]. The association with HbA1C is thus not surprising and with a larger effect allele frequency in IAPGG this locus may have a larger overall impact on the population, although the effect size per allele is similar to what was previously found in European populations. This finding is supported by previous reports showing a significantly higher level of HbA1C for a given level of fasting plasma glucose and 2-h plasma glucose among Greenlanders compared with Danes [26]. In contrast, the lack of a strong association in the European subset of the current study (Inter99 + EAPGG) is surprising. When investigating the GCK locus in details in Inter99, we find that the strongest association with HbA1C is carried by the rarer allele rs117316370 \(\left(MAF = 0.02,\,P = 0.0058,\,\beta = - 0.21\,{\mathrm{SD}}\right)\), which is not in strong LD (R2=0.093) with our chosen proxy SNP (rs730497) that does not associate with HbA1C for Inter99 (\(P = 0.29,\,\beta = 0.029\,{\mathrm{SD}}\)). We speculate that this might be the cause of the surprising lack of association in the subset Inter99 + EAPGG.
We found that that the ANK1 locus had the strongest difference in ancestral-specific effects among the examined known HbA1C loci. ANK1 was identified as an HbA1C-associated locus in Europeans [11], and in IAPGG we find a nominally significant association of ANK1 rs6474359_C in the opposite direction as reported in Europeans. This may indicate that the identified lead variant is not causal and that LD between the lead and causal variants differs among Inuit and European populations. Of interest, an opposite direction of effect than in Europeans of rs6474359_C was also observed in a study of the East Asian population [10]. The metaboChip does not cover the ANK1 locus densely as only four additional SNPs within 240 kb of lead SNP rs6474359 were available for analysis. None of these had a significant ancestral-specific effect nor did any associate more strongly than rs6474359 in either ancestral population (see Supplementary Table 8). A recent study found that the T2D risk allele at rs516946 leads to an increased expression of short ANK1 isoforms in skeletal muscle [27]. The T2D-associated rs516946 variant was not in strong LD (R2 < 0.2) with any amino-altering variants in the study nor with the HbA1C-associated rs6474359 in Europeans (R2 = 0.0076, using LD-link [28]). The authors speculate that the association with T2D may be driven by expression of the short isoforms of ANK1. Protein products of ANK1 have been shown to interact with insulin receptor substrate 1, through pathways enriched in skeletal muscle [29]. However, more functional studies are needed to understand how the short isoforms influence this interaction. Together this points to that the lead SNP is not the causal SNP in the ANK1 locus.
In the GEMMA meta-analyses of known HbA1C loci, the MTNR1B locus was significantly associated with HbA1C, whereas in our ancestry-specific analyses the association was nominally significant in IAPGG and statistically significant in the European proportion of the data (Inter99 + EAPGG). It has previously been suggested that the MTNR1B locus has a European-specific association with T2D [30]. Similarly, a GWAS of East Asians did not show association between SNPs in MTNR1B and HbA1C [10]. Of interest, we found almost identical effect sizes of MTNR1B rs1387153_T on HbA1C in IAPGG and Inter99 + EAPGG and showed no indication of ancestral-specific effects, which suggests that the association is not unique to Europeans. In addition, data on fasting plasma glucose in Inuit have shown an association between variants in MTNR1B and increased fasting plasma glucose level [31], which is in line with the current findings.
In conclusion, motivated by the previous finding of a high-effect T2D-associated variant in the Greenlandic population, we investigated the genetic determinants of HbA1C levels in this population. We did not detect any novel loci associated with HbA1C levels in the Greenlandic population when using data from the MetaboChip. However, we found that one known HbA1C-associated locus, GCK, associated with HbA1C in the Inuit ancestral proportion of the Greenlandic population. Secondly, we also found that the ANK1 locus shows a significant ancestral-specific effect indicating that the lead variant may not be the causal variant in the locus. Finally, for 10 of 15 known HbA1C-associated loci the effect was comparable between IAPGG and the reported effects in CEU or EAS. Our results shed light on genetic effects across populations and ethnicities and can contribute alongside other populations in future trans-ethnic meta-analyses.
Data availability
The data are available through European Genome-phenome Archive (EGA) at the url: https://ega-archive.org/studies/EGAS00001002641.
References
Moltke I, Fumagalli M, Korneliussen TS, et al. Uncovering the genetic history of the present-day Greenlandic population. Am J Hum Genet. 2015;96:54–69.
Sturtevant WC, Damas D. Handbook of North American Indians vol 5. The Arctic. In: Sturtevant WC, Damas D, editors. Handbook of North American Indians vol 5. The Arctic. Washington, D.C.: Smithsonian Institution; 1984. p. 845.
Jørgensen ME, Borch-Johnsen K, Stolk R, Bjerregaard P. Fat distribution and glucose intolerance among Greenland Inuit. Diabetes Care. 2013;36:2988–94.
Jørgensen ME, Bjeregaard P, Borch-Johnsen K, et al. Diabetes and impaired glucose tolerance among the Inuit population of Greenland. Diabetes Care. 2002;25:1766–71.
Jørgensen ME, Borch-Johnsen K, Witte DR, Bjerregaard P. Diabetes in Greenland and its relationship with urbanization. Diabet Med. 2012;29:755–60.
Moltke I, Grarup N, Jørgensen ME, et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature. 2014;512:190–3.
Report A, Consultation WHO. Use of glycated haemoglobin (HbA1c) in the diagnosis of diabetes mellitus. Diabetes Res Clin Pract. 2011;93:299–309.
Nathan DM, Turgeon H, Regan S. Relationship between glycated haemoglobin levels and mean glucose levels over time. Diabetologia. 2007;50:2239–44.
Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med. 2010;362:800–11.
Chen P, Takeuchi F, Lee J-Y, et al. Multiple nonglycemic genomic loci are newly associated with blood level of glycated hemoglobin in East Asians. Diabetes. 2014;63:2551–62.
Soranzo N, Sanna S, Wheeler E, et al. Common variants at 10 genomic loci influence hemoglobin A1C levels via glycemic and nonglycemic pathways. Diabetes. 2010;59:3229–39.
Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–16.
Scott Ra, Lagou V, Welch RP, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005.
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39:770–5.
Bjerregaard P, Curtis T, Borch-Johnsen K, et al. Inuit health in Greenland: a population survey of life style and disease in Greenland and among Inuit living in Denmark. Int J Circumpolar Health. 2003;62:3–79.
Tosoh method sheet for Hemoglobin A1c.
BioRad variant hemoglobin A1c program. Instruction manual.
Jeppsson JO, Kobold U, Barr J, et al. Approved IFCC reference method for the measurement of HbA1c in human blood. Clin Chem Lab Med. 2002;40:78–89.
Jørgensen T, Borch-Johnsen K, Thomsen TF, Ibsen H, Glumer C, Pisinger C. A randomized non-pharmacological intervention study for prevention of ischaemic heart disease: baseline results Inter99 (1). Eur J Cardiovasc Prev Rehabil. 2003;10:377–86.
Voight BF, Kang HM, Ding J, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4.
Skotte L, Korneliussen TSS, Moltke I, Albrechtsen A. Ancestry specific association mapping in admixed populations. 2015. https://doi.org/10.1101/014001.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Fumagalli M, Moltke I, Grarup N, et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science (80-). 2015;349:1343–7.
Hussain S, Richardson E, Ma Y, et al. Glucokinase activity in the arcuate nucleus regulates glucose intake. J Clin Invest. 2015;125:337–49.
Jørgensen ME, Bjerregaard P, Borch-Johnsen K, Witte D. New diagnostic criteria for diabetes: is the change from glucose to HbA1c possible in all populations? J Clin Endocrinol Metab. 2010;95:E333–6.
Scott LJ, Erdos MR, Huyghe JR, et al. The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat Commun. 2016;7:11764.
Machiela MJ, Chanock SJ. LD link: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2014;31:3555–7.
Caruso M, Ma D, Msallaty Z, et al. Increased interaction with insulin receptor substrate 1, a novel abnormality in insulin resistance and type 2 diabetes. Diabetes. 2014;63:1933–47.
Wang H, Liu L, Zhao J, et al. Large scale meta-analyses of fasting plasma glucose raising variants in GCK, GCKR, MTNR1B and G6PC2 and their impacts on type 2 diabetes mellitus risk. PLoS ONE. 2013;8:e67665.
Grarup N, Moltke I, Albrechtsen A, Hansen T. Diabetes in population isolates: lessons from Greenland. Rev Diabet Stud. 2015;12:320–9.
Acknowledgements
The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk). The B99 and IHIT study was supported by Karen Elise Jensen’s Foundation, NunaFonden, the Medical Research Council of Denmark, the Medical Research Council of Greenland, and the Commission for Scientific Research in Greenland. The study was partially funded by the Novo Nordisk Foundation (grants NNF15OC0017918 and NNF16OC0019986). None of the funding agencies had any role in the study design or in the collection or interpretation of the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Appel, E.V., Moltke, I., Jørgensen, M.E. et al. Genetic determinants of glycated hemoglobin levels in the Greenlandic Inuit population. Eur J Hum Genet 26, 868–875 (2018). https://doi.org/10.1038/s41431-018-0109-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0109-3
