
Abstract
Constitutional mismatch repair deficiency (CMMRD) is an autosomal recessively inherited childhood cancer susceptibility syndrome caused by biallelic germline mutations in one of the mismatch repair (MMR) genes. The spectrum of CMMRD-associated tumours is very broad and many CMMRD patients additionally display signposting non-neoplastic features, most frequently café-au-lait macules and other pigmentation alterations. We report on a 13-month-old girl suspected of having CMMRD due to a desmoplastic medulloblastoma and a striking skin pigmentation that included multiple café-au-lait macules, hypopigmented areas and Mongolian spots. Whole-exome sequencing revealed homozygosity for MSH2 variant p.(Leu92Val) and MSH6 variant p.(Val809del), both variants of uncertain significance (VUS). Immunohistochemical analysis of the tumour tissue showed expression of all four MMR proteins and gMSI testing was negative. However, functional assays demonstrated that the cells of the patient displayed methylation tolerance and ex vivo microsatellite instability, which unequivocally confirmed the diagnosis of CMMRD. Taken together, the results render the MSH2 variant unlikely to be responsible for the phenotype, while they are compatible with MSH6-associated CMMRD. This case illustrates the diagnostic strategy of confirming CMMRD syndrome in patients with VUS.
Similar content being viewed by others

Introduction
Constitutional mismatch repair deficiency (CMMRD) caused by biallelic germline mutations in one of the four mismatch repair (MMR) genes confers a high risk for a broad spectrum of malignancies primarily including haematological malignancies, brain tumours and tumours of the Lynch syndrome (LS) spectrum. Typically, patients develop tumours in the first two decades of life and survivors of the primary malignancy are at high risk to develop a second cancer [1]. Therefore, timely diagnosis of CMMRD is important for appropriate anticancer treatment, surveillance and genetic counselling of parents regarding the recurrence risk in siblings as well as their own risk for LS-associated tumours. About two-thirds of CMMRD patients present with pigmentary alterations, most frequently café-au-lait macules (CALM) reminiscent of neurofibromatosis type 1 (NF1) [2], but also a variety of other unspecific pigmentation anomalies. Here we report on a child with desmoplastic medulloblastoma (DMB) and peculiar skin pigmentation who meets the criteria for CMMRD testing [1]. Whole-exome sequencing (WES) revealed homozygous variants of uncertain significance (VUS) in MSH2 and MSH6. The diagnostic strategy allowing us to assign the clinical significance of these variants and finally to confirm CMMRD in the patient is presented.
Results
Case report
The 13-month-old girl suffering from vomiting for several weeks presented with ataxia, convergent strabism and developmental regression at hospitalisation. Magnet resonance imaging of the brain showed massive hydrocephalus caused by a large infratentorial tumour (49 × 41 × 85 mm) with both solid and cystic parts (Fig. 1d). Pathologic analysis of the resected tumour revealed desmoplastic medulloblastoma with a balanced c-Myc and n-Myc status and ß-Catenin negativity. Metastases to the cerebral fluid or spinal cord were excluded. According to the HIT (Hirn-Tumor) guidance protocol, treatment with vincristine, cyclophosphamide, high-dose methotrexate, carboplatin and etoposide and intraventricular methotrexate was initiated and fairly well tolerated under supportive care. Radiotherapy was omitted due to the patient’s age. One year after diagnosis, the child is well and in complete remission.

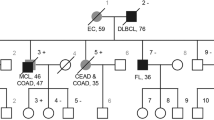
a, b Variegated skin pattern with CAL spots, hypo-pigmented areas and Mongolian and naevus-like spots. c Pedigree of the family; circles, females; squares, males; quarter symbol, heterozygous mutation; half symbol, homozygous mutation; black symbol, MSH2 mutation; red symbol, MSH6 mutation. d Cerebellar medulloblastoma with solid and cystic parts, T2 weighed axial
The patient had a very peculiar skin phenotype with a heterogeneous spectrum of pigmentary alterations including CALM and inguinal freckling reminiscent of NF1, as well as other hyper- and hypo-pigmented skin patches, atypical dermal melanocytosis/Mongolian spots and naevus-like spots (Fig. 1a, b). Neither the healthy 3-year-old sister nor the parents showed these alterations of skin pigmentation. The parents having both Roma background reported non-consanguinity, a pregnancy with a third child who died in utero for unknown reasons and a family history without cancer diagnosis in the three preceding generations (Fig. 1c).
Identification of homozygous variants in MSH2 and MSH6
The family was included in our prospective study ‘Germline mutations in children with cancer’. According to the study protocol, a trio-based WES analysis, including patient and both parents, was performed. Analysis of the WES data for causative variants in the MMR genes revealed a MSH2 missense variant (c.274C>G, p.(Leu92Val)) and a MSH6 3-bp deletion (del) (c.2426_2428delTAG, p.(Val809del)) both present in a homozygous state in the patient and heterozygous in both parents (Fig. 2a).

a WES reveals a homozygous missense variant in MSH2 (c.274C>G, p.(Leu92Val)) and a 3-bp deletion in MSH6 (c.2426_2428delTAG, p.Val809del) in DNA extracted from peripheral blood of the patient. Both variants are heterozygous in the parents. b The MSH2 variant is located in the MutS_I protein domain and the MSH6 variant is located in the MutS_III protein domain. c Sanger sequencing confirmed homozygosity of the two variants in the patient. Both variants are present in a heterozygous state in the parents.
The genome Aggregation Database (gnomeAD) lists the MSH2 variant with an overall minor allele frequency of G = 0.00006. This variant located in the functionally relevant N-terminal MutS-like protein domain (Fig. 2b) affects a highly conserved amino acid and is predicted to be deleterious by several programs (Polyphen, SIFT, MutationTaster). The InSiGHT group database lists this MSH2 variant once with unknown pathogenicity (InSiGHT class 3).
No data on the frequency in unselected individuals is available for the 3-bp del in MSH6. It is predicted to lead to loss of a valine (Val) in a moderately conserved region of the core domain of MSH6. In the InSiGHT database, this variant (2 entries) was classified as variant with unknown pathogenicity (InSiGHT class 3).
Since, both variants were classified as VUS, neither of them could unequivocally confirm the diagnosis of CMMRD. Therefore, we performed additional tests in order to confirm that the clinical phenotype of the child is caused by CMMRD and to render information whether homozygosity of one or both of the VUS is disease causing.
Diagnostic workup
Many inactivating MSH2 missense variants lead at least to expression loss of the heterodimerisation partner MSH6. Therefore, we performed immunohistochemical (IHC) staining on the tumour tissue, which showed nuclear expression of all MMR proteins in neoplastic and non-neoplastic cells (Supplement Fig. 1). Also, gMSI testing performed according to the protocol developed by Ingham et al. [3] did not reveal increased gMSI ratios in the patient’s lymphocytes and, hence, could not confirm CMMRD in the child. Therefore, we next performed methylation tolerance and ex vivo microsatellite instability (evMSI) assays using a lymphoblastoid cell line (LCL) of the patient (Supplementary Material and Methods section). These cells displayed a methylation-tolerance phenotype characteristic of MMR-deficient cells (Fig. 3a). Furthermore, the cell line from the patient achieved the evMSI phenotype as demonstrated by a 1 bp-del in the NR27 microsatellite in the LCL in comparison to peripheral blood lymphocytes (Fig. 3b). Taken together, both assays clearly confirm CMMRD in the patient.

a The lymphoblastoid cell line (LCL) from the patient displayed methylation tolerance. Survival (%) of immortalised lymphoid cells derived from the patient (dark circles) and from an MMR wild-type control (open squares) after two (full lines) or three (dotted lines) rounds of MNNG treatment. b Immortalised lymphoid cells from the patient displayed ex vivo microsatellite instability. Electrophoretograms of fluorescent amplification products for NR21, NR27 and BAT26 microsatellites, respectively. The length of the predominant allele (in base pairs) and the fluorescence intensity are indicated in the box below each profile. A 1 bp deletion (red arrow) occurred at the NR27 locus in the LCL derived from the patient compared with the respective peripheral blood lymphocytes (PBLs)
Discussion
Diagnosing CMMRD has important implications for the management of paediatric cancer patients and their families. Therefore, coming to a definite diagnosis in a suspected patient is of utmost importance. This case illustrates an effective diagnostic strategy to definitely confirm (or refute) CMMRD in patients in whom only MMR gene VUS are identified.
IHC staining of the MMR proteins and analysis of MSI are the preferred diagnostic methods to substantiate suspected CMMRD. However, due to the non-truncating nature of the VUS identified in MSH2 and MSH6, which is compatible with expression of a non-functional protein, IHC was not informative in our patient. This limitation of IHC has already been shown in other CMMRD patients [4,5,6]. It is known that standard MSI testing developed for LS-associated tumours often fails to show MSI in brain tumours of CMMRD patients [7]. Therefore, we performed gMSI testing. This simple and fast assay detects altered dinucleotide-microsatellite alleles present at low level in non-neoplastic cells of individuals with CMMRD due to biallelic PMS2 or MLH1 mutations. CMMRD due to biallelic MSH2 mutations is rare and data are therefore limited, but all cases of MSH2-associated CMMRD analyzed so far also showed increased gMSI ratios [3, 4]. However, since the assay relies on the analysis of dinucleotide-microsatellite markers, it is insensitive towards MSH6-associated CMMRD [3]. Hence, a negative gMSI result, as observed in our patient, is compatible with a constitutive MSH6 defect, but strongly speaks against a defect in PMS2, MLH1 or MSH2. Another approach is based on the evaluation of MSI using mononucleotide markers and methylation tolerance—another functional characteristic of MMR-deficient cells -, in immortalised lymphoblastoid cells (i.e., ex vivo) of suspected patients [4]. This method demonstrated unequivocal results in a case-control study, i.e., abnormal results for both assays were found in all CMMRD patients, whereas normal results for both assays were found in the control cohort [4]. This assay could finally confirm CMMRD also in our patient.
The preserved expression of MSH2 and MSH6 and, in particular, the negative gMSI result speaks against an inactivation of MSH2 in the patient rendering the MSH2 variant p.(Leu92Val) unlikely the disease causing variant. However, these results are compatible with MSH6-associated CMMRD and, consequently, a deleterious effect of the MSH6 variant p.(Val809del). Hence, it is possible that the MSH2 variant is a rare benign variant that happens to be genetically linked to the disease causing mutation in MSH6 (distance between the two variants is ~392 kb). This notion is corroborated by data from the UMD database, which also reports this MSH2 variant as ‘likely neutral’ and together with a different clearly disease-causing MSH6 mutation in a patient with LS. Ultimately, however, functional studies would be needed to come to a final conclusion which of the two variants contributed to which extend to the clinical phenotype of the patient.
Upon confirming CMMRD, our patient was integrated into a cancer surveillance program as proposed by the European consortium ‘C4CMMRD’, the international biallelic MMR deficiency consortium and the consensus statement of the American Association for Cancer Research [8, 9, 12]. According to the InSiGHT database the MSH6 variant was also found in patients tested for LS. We therefore assume that this variant increases the risk for LS-associated tumours in adult heterozygous carriers and recommended that the parents are enroled in a LS surveillance protocol even in absence of LS-associated tumours in their family history. The sister who did not show pigmentation alterations or any other signs of CMMRD was not tested for the MSH6 variant.
A very particular skin pigmentation phenotype was crucial for suspecting CMMRD in our patient. The girl does not only have a large number of hyper- and hypopigmented skin areas, which are frequently reported in CMMRD patients, but also several Mongolian spots. With this very heterogeneous skin pigmentation phenotype she resembles a recently published CMMRD patient with atypical dermal melanocytosis [10] and a patient published by Scott et al. [11]. These patients indicate that also multiple Mongolian spots may belong to the CMMRD-associated skin features.
In conclusion, our case illustrates that WES may be a valid and rapid method to confirm the diagnosis. However, paediatric oncologists and molecular geneticists should be prepared that the interpretation of sequencing data may be challenging and require thorough functional analysis to come to a valid conclusion.
References
Wimmer K, Kratz CP, Vasen HF, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51:355–65.
Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet. 2017;91:507–19.
Ingham D, Diggle CP, Berry I, et al. Simple detection of germline microsatellite instability for diagnosis of constitutional mismatch repair cancer syndrome. Hum Mutat. 2013;34:847–52.
Bodo S, Colas C, Buhard O, et al. Diagnosis of constitutional mismatch repair-deficiency syndrome based on microsatellite instability and lymphocyte tolerance to methylating agents. Gastroenterology. 2015;149:1017–29. e1013
Mork ME, Borras E, Taggart MW, et al. Identification of a novel PMS2 alteration c.505C>G (R169G) in trans with a PMS2 pathogenic mutation in a patient with constitutional mismatch repair deficiency. Fam Cancer. 2016;15:587–91.
Sjursen W, Bjornevoll I, Engebretsen LF, Fjelland K, Halvorsen T, Myrvold HE. A homozygote splice site PMS2 mutation as cause of Turcot syndrome gives rise to two different abnormal transcripts. Fam Cancer. 2009;8:179–86.
Bougeard G, Charbonnier F, Moerman A, et al. Early onset brain tumor and lymphoma in MSH2-deficient children. Am J Hum Genet. 2003;72:213–6.
Tabori U, Hansford JR, Achatz MI, et al. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res. 2017;23:e32–e37.
Vasen HF, Ghorbanoghli Z, Bourdeaut F, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J Med Genet. 2014;51:283–93.
Polubothu S, Scott RH, Vabres P, Kinsler VA, Atypical dermal melanocytosis: diagnostic clue in constitutional mismatch repair deficiency syndrome. Br J Dermatol. 2017;177:e185–6.
Scott RH, Mansour S, Pritchard-Jones K, Kumar D, MacSweeney F, Rahman N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol. 2007;4:130–4.
Carol Durno, C. Richard Boland, Shlomi Cohen, Jason A. Dominitz, Frank M. Giardiello, David A. Johnson, Tonya Kaltenbach, TR. Levin, David Lieberman, Douglas J. Robertson, Douglas K. Rex, Recommendations on Surveillance and Management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2017:152 (6):1605–14.
Acknowledgements
This work was supported by the Elterninitiative Kinderkrebsklinik e.V. Duesseldorf. The authors wish to thank Dr. Claudia Potthoff and Dr. Stefan Balzer for their contribution in sample acquisition, Mrs. Silke Furlan and Mrs. Katayoun Alemazkour for technical assistance, Mrs. Fanny Bouchlis and Mrs. Brigitte Litra for functional assays and Dr. Joerg Schaper for providing the radiological image. We thank the Biological Resource Center, Hôpital Cochin, APHP-Paris France, for the lymphoid cell line establishment.
Author contributions
J.T. and M.K. performed WES data analysis and first drafted the manuscript. K.W. helped writing the manuscript. K.W., M.M., and C.F. critically revised the manuscript for important intellectual content. J.T. performed Sanger sequencing. K.W. and C.F. performed gMSI testing. M.M., O.L., and C.C. performed methylation tolerance assay and evMSI. T.B. obtained informed consent and asked for the family history. T.B., J.I.H., A.B., and M.K. cared for the child. J.F. and J.R. performed the pathological analysis. M.G. was responsible for library preparation, S.G. for the internal SQL database. A.B. and M.K. designed and supervised the project. All authors approved the final manuscript as submitted.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
The study was approved by the local ethics committee and written informed consent was obtained from the parents.
Additional information
Julia Taeubner and Katharina Wimmer authors contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Taeubner, J., Wimmer, K., Muleris, M. et al. Diagnostic challenges in a child with early onset desmoplastic medulloblastoma and homozygous variants in MSH2 and MSH6 . Eur J Hum Genet 26, 440–444 (2018). https://doi.org/10.1038/s41431-017-0071-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0071-5
This article is cited by
-
Using comprehensive genomic and functional analyses for resolving genotype–phenotype mismatches in children with suspected CMMRD in Lebanon: an IRRDC study
Human Genetics (2023)
-
Comprehensive germline-genomic and clinical profiling in 160 unselected children and adolescents with cancer
European Journal of Human Genetics (2021)
-
Medulloblastoma
Nature Reviews Disease Primers (2019)
