
Abstract
Retinoblastoma is the most common eye cancer in children. Numerous families have been described displaying reduced penetrance and expressivity. An extensive molecular characterization of seven families led us to characterize the two main mechanisms impacting on phenotypic expression, as follows: (i) mosaicism of amorphic pathogenic variants; and (ii) parent-of-origin-effect of hypomorphic pathogenic variants. Somatic mosaicism for RB1 splicing variants (c.1960+5G>C and c.2106+2T>C), leading to a complete loss of function was demonstrated by high-depth NGS in two families. In both cases, the healthy carrier parent (one with retinoma) showed a variant frequency lower than that expected for a heterozygous individual, indicating a 56–60% mosaicism level. Previous evidences of a ~3-fold excess of RB1 maternal canonical transcript led us to hypothesize that this differential allelic expression could influence phenotypic outcome in families at risk for RB onset. Accordingly, in five families, we identified a higher tumor risk associated with paternally inherited hypomorphic pathogenic variants, namely a deletion resulting in the loss of 37 amino acids at the N-terminus (c.608-16_608del), an exonic substitution with a “leaky” splicing effect (c.1331A>G), a partially deleterious substitution (c.1981C>T) and a truncating C-terminal variant (c.2663+2T>C). The identification of these mechanisms changes the genetic/prenatal counseling and the clinical management of families, indicating a higher recurrence risk when the hypomorphic pathogenic variant is inherited from the father, and suggesting the need for second tumor surveillance in unaffected carriers at risk of developing adult-onset cancer such as osteosarcoma or leiomyosarcoma.
Similar content being viewed by others


Introduction
Retinoblastoma (RB; OMIM #180200) is the most common pediatric intraocular tumor, affecting about 1 in 15,000–20,000 live births [1]. It is caused by biallelic inactivation of RB1, the first characterized tumor suppressor gene, located at 13q14.2 [2, 3]. The most frequent form is the non-hereditary one (60%), with both inactivating events occurring in cells of the developing retina. In this form, the tumor is confined to a single eye (unilateral) and develops after the first year of life. The hereditary form (40%) is due to a RB1 germline predisposing variant and subsequent somatic inactivation of the other allele, mostly occurring through mitotic recombination/nondisjunction or large deletions [4]. In this form, the mean age of diagnosis is anticipated (within the first year of life) and cancer develops as multiple foci (multifocal), usually involving both eyes (bilateral).
Hereditary RB is transmitted according to an autosomal dominant pattern; the penetrance is incomplete and generally reported as equal to 90%. However, it is common to observe families in which a significant fraction of pathogenic variant carriers remains unaffected or develops only unilateral tumors or benign lesions called retinomas [5], a phenomenon that has always posed a question about the underlying mechanisms. Pathogenic variants reducing RB1 expression or partially inactivating pRB activity have been associated to milder phenotypic expression [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. However, the description of low-penetrant families bearing classical “null” variants and the observation that penetrance/expressivity can vary significantly between and within families with identical pathogenic variants has led to speculate that other mechanisms may be determinant [17, 21,22,23,24].
As early as the eighties, mosaicism has been established as an important cause of phenotypic variability [25]. Especially in disorders such as RB, in which a high proportion of cases represents new variants, this mechanism is expected to occur and to influence phenotypic expression [26]. In 1998 Sippel and coworkers investigated this phenomenon in a large cohort and documented mosaicism for the initial RB1 pathogenic variant in ~10% of families [27]. Subsequently, the employment of more sensitive techniques has allowed the detection of even low-level mosaicism and the identification of mosaic RB cases has increased up to 30%, but the impact of this phenomenon on phenotypic expression has been poorly investigated [28,29,30].
Clinical penetrance can also be influenced by the gender of the transmitting parent (parent-of-origin effect), as demonstrated in different diseases such as myoclonus dystonia [31] or paraganglioma [32]. The best characterized mechanism underlying this phenomenon is genomic imprinting [33]. An association between parent-of-origin-dependent allelic expression and phenotype was already observed in 2002 for a pathogenic splicing RB1 variant segregating in two families [15]. In 2009, a ~3-fold excess of the RB1 maternal canonical transcript due to an imprinting mechanism was demonstrated [34]. As a consequence, maternally inherited pathogenic variants with hypomorphic effect may retain sufficient suppressor activity to prevent tumor development. In accordance, Eloy and collaborators demonstrated a parent-of-origin effect in low-penetrance pedigrees segregating the common c.1981C>T/p.(Arg661Trp) pathogenic substitution and observed the same phenomenon for other weaker alleles [20].
Herein we investigated mosaicism and parent-of-origin effect as the two main mechanisms capable to explain reduced penetrance/expressivity in familial RB. Somatic mosaicism for splice site pathogenic variants (c.1960+5G>C and c.2106+2T>C) predicted to result in protein truncation and producing only the mutated transcript was identified in two families. A parent-of-origin effect was demonstrated in five families harboring hypomorphic pathogenic RB1 variants: two already reported exonic substitutions (c.1981C>T and c.1331A>G) and two new splice site variants (c.608–16_608del and c.2663+2T>C).
Materials and methods
Families
Diagnosis of RB was established at the Retinoblastoma Referral Center of Siena (Ophtalmology Department, AOUS) and in external Institutes (Istituto Gaslini of Genova and Pio Istituto Oftalmico of Milano). Families underwent genetic counseling and blood samples were collected in EDTA-containing tubes and sent to the Unit of Medical Genetics of Siena for RB1 analysis (Sanger sequencing and MLPA). When a germline predisposing variant was found in the proband, molecular testing was extended to relatives. Identified RB1 pathogenic variants were submitted to LOVD database (http://rb1-lsdb.d-lohmann.de) with the following IDs: RB1_00019 (c.1981C>T), RB1_01265 (c.1960+5G>C), RB1_01248 (c.2106+2T>C), RB1_00280 (c.1331A>G), RB1_02115 (c.608-16_608del), RB1_01664 (c.2663+2T> C). To assess penetrance/expressivity, the diseased-eye-ratio (der) was calculated [8]. The DER is defined as the ratio between the number of affected eyes and the number of variant carriers in a family. A DER <1.5 is consistent with incomplete penetrance. All first-generation carriers were excluded from DER calculation to avoid any bias relating to mosaicism. Additional blood samples from probands and their relatives were collected in PAXgene tubes for RNA isolation. Informed consent was obtained from all adult patients and parents of affected children.
DNA/RNA isolation
Genomic DNA was extracted from EDTA peripheral blood samples using MagCore HF16 (Diatech Lab Line). RNA was isolated from whole blood stabilized in PAXgene Blood RNA tubes (PreAnalytiX®, Qiagen) following manufacturer’s instructions. RNA quality was evaluated by NanoDrop 2000 Spectrophotometer (Thermo Scientific).
High-depth NGS
To investigate mosaicism for RB1 pathogenic variants (GeneBank Accession L11910.1; transcript NM_000321.2), the specific exonic/intronic sequences were analyzed by next-generation sequencing (NGS) at high-depth (at least 1000× allowing to detect a minimum of 5% of mosaicism) using Roche 454 GS Junior platform as previously described [30]. Linearity and limit of detection (LOD) of the NGS assay were previously assessed by drawing calibration curves for different RB1 variants [30]. DNA samples from healthy controls and mutated offspring of familial RB cases were used to verify that the method was capable to distinguish mosaic variants from background errors and heterozygous variants, respectively [30]. Each sample was analyzed three times [30]. Target regions were amplified using fusion primers reported in Supplementary Table 1. Variants in silico analysis was carried out using the Interactive Biosoftware Alamut v2.3 (Interactive Biosoftware, Version 2.7) [35]. Relating to the c.1981C>T pathogenic substitution, the above described protocol was also employed to quantify mutant/wild-type allelic expression. cDNA, retrotranscribed from patient’s RNA, was amplified using primers reported in Supplementary Table 1.
Minigene assays
A portion of RB1, containing the exon close to or harboring the variant of interest and part of the upstream and downstream, was amplified from patient’s DNA (oligonucleotides and PCR conditions are available upon request). Concerning the c.1331A>G allele, exon 13 and part of the flanking introns were amplified from human genomic DNA and the specific variant was introduced by site-directed mutagenesis using the QuikChange II site-directed mutagenesis kit (Stratagene). We employed as backbone the β-globin-pCDNA3.1 vector [36] and obtained the hybrid minigene constructs as previously described [37]. For expression analysis, we selected one clone containing the wild-type allele, one clone with each variant and the empty vector. Minigene expression assay was carried out on Y79 cells (ATCC® HTB18™), a human retinoblastoma cell line. Cells were cultured in suspension in RPMI 1640 medium (Gibco Thermo Fischer Scientific) supplemented with 20% fetal bovine serum. After 24 h of serum-deprivation, cells were transfected with Lipofectamine® 2000 according to the manufacturer’s protocol. After 24 h, total RNA was extracted using the TRIzol kit (Thermo Fischer Scientific) and retrotranscribed. The cDNA was amplified using primers located on β-globin exon 2 and 3 to avoid eventual interference from the endogenous gene and PCR fragments were analyzed as previously reported [37].
RNA analysis
RNA was retrotranscribed into cDNA using Qiagen RT-PCR kit (QuantiTect® Reverse Transcription Kit) and 1 µg of total RNA per reaction. Amplification was conducted on a Thermal Cycler 2720 (Applied Biosystems) using primers reported in Supplementary Table 1. Thermal cycling was the following: 95 °C for 5 min, followed by 35 cycles at 95 °C for 30 s, at the specific annealing temperature for 30 s, at 72 °C for 30 s, followed by a final extension at 72 °C for 5 min. PCR products were separated by electrophoresis on a 3% agarose gel. When multiple bands were obtained, cDNA fragments were separately sequenced after extraction by Zyomoclean Gel DNA Recovery kit (Zymo research). Relating to the c.1981C>T substitution, total sample was purified using AMPure PCR purification system (Agencourt). DNAs were sequenced by Sanger sequencing using the PE Big Dye Terminator Cycle Sequencing Kit on an ABI Prism 3130 analyser (Applied Biosystems). The data were analyzed using the Sequencher version 4.9 software.
RealTime qRT-PCR
Primers were specifically designed to quantify mutant and wild-type transcripts (Supplementary Table 2). GUSB was selected as housekeeping gene. Quantitative PCR was carried out in single-plex reactions in a 96-well optical plate with FastStart SYBR Green Master Mix (Roche) on an ABI Prism 7700 Sequence Detection System (Applied Biosystems). Experiments were performed in triplicates in a final volume of 20 ul with 25 ng of cDNA and 25 nM of each primer, following the SYBR Green protocol (Roche). Standard thermal cycling conditions were: 2 min at 50 °C and 10 min at 95 °C; 40 cycles at 95 °C for 15 s and 60 °C for 1 min; dissociation stage 95 °C for 15 s, 60 °C for 20 s and 95 °C for 15 s. The data were analyzed using the comparative Ct method [38]. Results were normalized respect to the expression of the wild-type transcript in unaffected members and the total transcript expression obtained amplifying a region not involved in splicing aberrations in carrier subjects (Supplementary Table 2).
Results
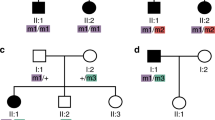
Herein, we investigated the molecular mechanisms underlying phenotypic expression in seven RB families (five novel and two already reported) (Fig. 1) (Table 1) [14, 17]. In these families, traditional sequencing had previously shown the presence of six pathogenic RB1 variants: four intronic (c.608-16_608del, c.1960+5G>C, c.2106+2T>C and c.2663+2T>C) and two exonic variants (c.1331A>G and c.1981C>T) (Fig. 1) [14, 17]. Analysis of RNA directly isolated from patient’s blood samples and/or minigene assays in an RB cell line (Y79) allowed to characterize the consequences of splicing variants and to investigate a possible “leaky” effect. High-depth NGS and wild-type/mutant allelic expression analysis allowed to characterize the molecular mechanisms explaining phenotypic presentations, as follows: (I) somatic mosaicism of amorphic pathogenic variants; and (II) parent-of-origin-effect of hypomorphic pathogenic variants (Table 2).

Pedigrees of RB families. Blackened symbols indicate patients with bilateral retinoblastoma; half-blackened symbols indicate patients with unilateral retinoblastoma; half-gray symbols indicate subjects with retinoma; crosshatched symbols indicate subjects with other RB1-related neoplasms: osteosarcoma in family 3, leiomyosarcoma in family 4 and lipomas in family 7; OC obligated carriers; dashed symbols: deceased subjects. Pedigree of family 7 has been taken from Genuardi et al. 2001 [14]. The RB1 variants shown are described using the NM_000321.2 transcript reference sequence

Minigene assay results. RT-PCR analysis of the RB1 minigene constructs expressed in Y79 cells using vector-specific primers. A schematic representation of the hybrid minigene constructs is depicted on the left; the dotted lines indicate normal splicing. Each construct contains the exon close to, or harboring the variant of interest and part of the upstream and downstream introns of RB1 gene. In a the construct contains the exon 7 of the RB1 gene; c.608–16_608del, construct containing the c.608-16_608del variant. In b, the construct contains exon 19 of the RB1 gene. Legends as above, c.1960+5G>C, construct containing the c.1960+5G>C variant; In c, the construct contains exon 20 of the RB1 gene. c.2106+2T>C, construct containing the c.2106+2T>C variant. In d, the construct contains exon 25 of the RB1 gene. c.2663+2T>C, construct containing the c.2663+2T>C variant. In e, the construct contains exon 13 of the RB1 gene. c.1331A>G, construct containing the c.1331A>G variant. M: 1 kb plus molecular marker (Invitrogen); Empty empty β-globin-pCDNA3.1 vector; WT wild-type construct. Transcript refseq ID: NM_000321.2

High-depth NGS in families 2 and 5. Sequencing results are showed as percentage variant of total number of reads. a In family 2, the c.1960+5G>C pathogenic variant was detected at a percentage of ~50% in the proband with retinoblastoma and ~28% in the unaffected mother. b In family 5, the c.2106+2T>C pathogenic variant was detected at a percentage of ~50% in the proband with retinoblastoma and ~ 34% in the father with retinoma. Transcript refseq ID: NM_000321.2.
Clinical presentations
Family 1
The proband is the first child of healthy non-consanguineous Chinese parents (Fig. 1 and Table 1). From the paternal side, it was reported a 2nd degree cousin affected by bilateral RB (IV2) diagnosed at 11 months of age. Proband, at the age of 2 years, was referred to an ophthalmologist because of onset of anisocoria in his right eye. Brain and spinal MRI revealed an expansive partially calcified lesion suggestive for RB extending through the papilla to the proximal portion of the ipsilateral optic nerve, without signs of dissemination. The child underwent chemotherapy and successive enucleation. Histological examination confirmed RB diagnosis. After 6 months, brain and spinal MRI screening did not show any residual disease or dissemination. An ophthalmological screening with fundus examination in both parents resulted normal.
Family 2
The proband was born from the second pregnancy of healthy non-consanguineous Greek parents (Fig. 1 and Table 1). The first pregnancy was interrupted at 20 weeks of gestation for oligohydramnios. At the age of 2 months, parents noticed strabismus in the left eye of the child and soon afterwards the diagnosis of bilateral RB was established. Family history was unremarkable.
Family 3
The proband is the first child of non-consanguineous parents (Fig. 1 and Table 1). The father (II2) presented osteosarcoma of the right leg that was surgically resected at the age of 14 years. Fundus examination, performed in the father after RB diagnosis in the son, was referred as normal. At the age of 8 months the proband was referred to an ophthalmologist for the observation of leukocoria in the right eye. Brain MRI revealed a mass of 1.6 cm × 1.1 cm × 7 mm, suggestive for intrabulbar RB. Ophthalmoscopic evaluation revealed a focal lesion spanning >50% of the retinal surface and four additional foci in the nasal retina. In the left eye, a paramacular lesion of the retina of one-disc diameter in size was observed. The child underwent focal and systemic chemotherapy.
Family 4
The female proband is the child of a non-consanguineous couple (Fig. 1 and Table 1). She had the enucleation of the right eye for unilateral RB at the age of 4 years. A bilateral RB was diagnosed in the brother (IV5) at the age of 2 years and 4 months. Histological examination reported an undifferentiated RB of 12 × 17 mm rich of eosinophils involving the optic nerve and the meninges. Chemotherapy and radiotherapy followed enucleation. A single tumor was then identified in the right eye, treated by several photocoagulation sessions. The patient died 5 months later for a recurrence of the disease in the left orbital frontal region. The father died for a leiomyosarcoma at the age of 81 years. A paternal second cousin (V1) developed unilateral RB at the age of 5 years. A cousin of the proband’s father (III1) died at the age of 5 years for RB.
Family 5
This family, already reported in one of our previous studies, was initially classified as a sporadic case, in which the proband had a diagnosis of bilateral RB at the age of 4 months (Fig. 1) (Table 1) [17]. RB started in the right eye and required chemotherapy, and successive enucleation. Histological examination showed an endofitic and esofitic mass, well-differentiated, which did neither infiltrate the anterior chamber nor the optic nerve. A following involvement of the left eye required chemotherapy, laser therapy, radiotherapy and enucleation. After RB diagnosis in the proband, fundus examination in the father (I2) showed the presence of a retinoma that underwent spontaneous calcification in the left eye, leading to a reclassification of this case as a familial RB.
Family 6
This family came to our attention to define the recurrence risk for RB in II2 subject’s newborn (Fig. 1 and Table 1). His two siblings both presented with bilateral RB starting from 3 months of age. The disease required enucleation of both eyes in the older brother, enucleation of the right eye and radiotherapy of the controlateral eye in the younger sister. Anamnestic history was silent for RB in other family members. An ophthalmological screening with fundus examination performed in the father revealed the presence of a retinoma in the left eye.
Molecular analysis
Minigene assays
To characterize the splicing effects of RB1 predisposing variants and investigate a possible “leaky” effect, a hybrid minigene approach was employed. The assay was performed on a human retinoblastoma cell line (Y79 cells) in order to mimick splicing factors landscape in the tumor. Our results showed that all the four intronic variants resulted in aberrant splicing, without the formation of the wild-type transcript, proving that they exert a severe effect on mRNA maturation (Fig. 2a-d). The corresponding wild-type constructs generated instead the expected splicing products with the exception of the plasmid containing exon 19: in this case, splicing was less efficient, showing in addition to the correctly spliced transcript, some degree of intronic retention (with a longer isoform retaining 125 nucleotides of intron 19) (Fig. 2b). Nevertheless, no functional transcripts containing exon 19 were expressed from the mutated construct and two bands were obatained: a fainter upper one corresponding to the same “mis-spliced” transcript present in the wild type and a lower most abundant band showing complete skipping of exon 19 with retention of the same portion of intron 19 found in the longer transcript. Concerning the other variants, minigene assay revealed that the c.608-16_608del causes the skipping of exon 7 (Fig. 2a). Expression of the c.2106+2T>C substitution in the β-globin minigene led to the usage of an upstream alternative 5’ splice site, resulting in the expression of a single transcript containing exon 20 lacking the final 34 nucleotides, thus resulting in a frameshift (Fig. 2c and Table 2). As for the c.2663+2T>C variant, expression analysis confirmed an aberrant splicing pattern, consisting of both exon 25 skipping and intronic retention, in the absence of the normally spliced transcript (Fig. 2d).
Minigene assay was also employed to evaluate the splicing effect of a pathogenic exonic substitution (c.1331A>G) previously reported in a low-penetrant RB family showing a clear parent-of-origin effect with a higher probability to manifest the tumor in case of paternal inheritance [14]. As shown in Fig. 2e, expression of the mutant minigene yielded two bands: sequencing of the lower RT-PCR product confirmed skipping of exon 13, whereas the upper fainter band corresponds to the wild-type transcript, demonstrating a “leaky” effect (hypomorphic variant).
High-depth NGS
On the basis of pedigree characteristics, we tested the hypothesis that carriers who have not developed RB in families 2, 3, 5 and 6 can be mosaic for the predisposing variant. We, therefore, employed high-depth NGS to analyze the percentage of mutated molecules in DNAs isolated from probands and carrier parents. In families 3 and 6, we detected about the same percentage of mutated molecules compatible with a heterozygous state (data not shown). In the other two families, we found a ~50% of mutated molecules in the affected proband in comparison with a lower percentage in the unaffected mother (~28%; family 2) and in the father with retinoma (~34%; family 5), supporting the conclusion that somatic mosaicism can explain phenotypic presentations (Fig. 3a, b and Table 2).

mRNA wt/mutant allelic quantification. a Characterization of the effect of the splicing deletion c.608-16_608del identified in family 1. RT-PCR using primers spanning exons 5-8 showed two products in both the affected proband and the unaffected carrier father: the expected normal-sized transcript (263bp) and a shorter one of 151bp (upper panel). The latter product was not observed in the mother not bearing the variant. Direct sequencing of the shorter product showed the complete skipping of exon 7. At protein level, aberrant transcript with exon 7 skipping is predicted to encode a product lacking 37 amino acids at the amino terminus (middle panel). Real time PCR relative quantification, normalized respect to the total transcript (shaded in grey), showed an inverted ratio between wt and mutant isoform in the affected child (87% vs 13%) respect to the father (14% vs 86%) (lower panel). b Characterization of the effect of the c.2663+2T>C variant identified in family 6. RT-PCR was carried out using primers spanning exons 23-27. An additional shorter product respect to the normal-sized one was observed in mutated family members (I1, II1 and II3) (upper panel). Sanger sequencing of the abnormal transcript showed the complete exclusion of exon 25, resulting in a frameshift that insert a stop codon at amino acid 841 (middle panel). By real time qPCR analysis, the two affected individuals showed a higher (70%) wt allelic expression respect to the mutant one (30%), while the father displayed almost exclusively (~98%) the mutant transcript (lower panel). WT, normal-sized wild-type product; Mut, abnormally spliced product; Ctrl1 and Ctrl2, control samples. Transcript refseq ID: NM_000321.2.
Transcript analysis: quantification of wild-type/mutant allelic expression
On the basis of the pedigrees, a parent-of-origin-effect was hypothesized to explain phenotypic presentations in the previously studied families (3 and 6) and in the remaining families (1 and 4) (Table 1). Transcript analysis with quantification of wt/mutant allele ratio was thus performed on mRNA directly isolated from peripheral blood of available individuals.
RT-PCR for the splicing pathogenic variants c.608-16_608del (family 1) and c.2663+2T>C (family 6) confirmed results obtained by minigene assay: in-frame skipping of exon 7 in the first case and frameshift skipping of exon 25 in the second case (Fig. 4a, b, upper panels (Supplementary Fig. 1 and Table 2). For the c.2663+2T>C variant, RT-PCR did not identify the additional aberrant transcript with intronic retention detected by minigene assay probably due to product instability. At protein level, the aberrant transcript with exon 7 skipping is predicted to encode a product lacking 37 amino acids at the N-terminus (Fig. 4a, middle panel). On the other hand, exon 25 skipping leads to a frameshift, and ultimately to a premature stop codon (aa 841), which is predicted to result in a truncated protein lacking the carboxy-terminus (Fig. 4b, middle panel). For both variants, real-time qPCR expression analysis showed a higher expression of the abnormally spliced products in the unaffected carrier fathers along with an inversion of the ratio between the two differentially spliced isoforms with respect to the affected offspring (Fig. 4, lower panels).
The c.1981C>T substitution was identified in two families (3 and 4) (Fig. 1 and Table 2). In family 4, we had the possibility to investigate mRNA isolated from proband blood sample and quantify wt/mutant allelic expression using high-depth NGS. We found that mutant molecules are ~23%, in accordance with an expected lower expression of a paternally inherited variant (Supplementary Fig. 2).
Discussion
The identification of the molecular bases of penetrance and expressivity represents a key point to better understand disease mechanisms and provide optimal genetic counseling. Several studies suggest that penetrance and expressivity of hereditary RB can be influenced by the nature of the predisposing germline variant [8]. However, different evidences have suggested that this factor alone cannot be sufficient and that other mechanisms are involved. Importantly, MED4 and MDM2 genes have been identified as RB modifiers [39, 40]. More recently, a parent-of-origin effect due to an imprinting mechanism has been demonstrated to impact on phenotypic expression associated with the common c.1981C>T deleterious variant [20]. Herein, an extensive molecular characterization of seven families led us to characterize the following two main mechanisms underlying reduced penetrance/expressivity: (i) mosaicism of amorphic pathogenic variants; and (ii) parent-of-origin-effect of hypomorphic pathogenic variants.
On the basis of pedigrees analysis, we hypothesized that carriers could be mosaic for the predisposing RB1 variant in families 2, 3, 5, and 6. According to this model, the reduced tumor susceptibility would be associated to a decreased pool of retinal cells that can be targeted by a second inactivating event. By using an already published high-depth NGS method designed to distinguish high- and low-level mosaicism, in families 2 and 5, we demonstrated that healthy carrier parents showed RB1 variant frequencies in blood that were less than the 50% expected for a heterozygote. Even if variant levels in the retina could not be established, these findings clearly indicate a mosaic condition and support the conclusion that this mechanism is responsible for the observed clinical presentations [30]. The identified pathogenic variants (c.2106+2T>C and c.1960+5G>C) were already reported in RB patients [17, 41, 42]. By hybrid minigene assay, we showed that both variants provoke an aberrant splicing with a severe effect on mRNA maturation since no wild-type transcript could be generated, suggesting a complete loss-of-function effect. In agreement, a previous study reported that the c.1960+5G>C variant gives rise to a truncated protein without detection of normal pRb [41]. The introduction of NGS has greatly improved the detection of mosaicism and has demonstrated that a high fraction of RB1 sequence variants arises by post-zygotic events [29, 30]. However, the phenotypic impact of mosaicism in RB remains to be determined. Only few low-penetrant families with identified or presumed mosaicism have been reported so far [27, 28]. These pedigrees show the more common “null” alleles, which are expected to completely abolish repressor activity. Unaffected carriers can thus be observed also in the context of a predisposing amorphic RB1 variant and, in this case, mosaicism probably represents the mechanism underlying phenotypic expression. On the other side, mosaicism has not been reported in association with weak alleles, suggesting that this combination is less likely to result in RB.
The remaining five pedigrees were compatible with the hypothesis of a parent-of-origin effect combined with predisposing hypomorphic RB1 variants [14]. According to this model, maternal inheritance is associated with a higher probability of being unaffected or developing only benign retinomas. This model is in agreement with the previous demonstration that RB1 gene is preferentially expressed from the maternal allele due to an imprinting mechanism and is consistent with the hypothesis that maternally inherited variants with hypomorphic effect may retain sufficient suppressor activity to prevent tumor development [34]. In this study, pathogenic variants include the well-known c.1981C>T/p.(Arg661Trp) substitution (families 3 and 4), one exonic variant with splicing impact (family 7), and two splice site changes whose effect at the transcript level has never been previously investigated (families 1 and 6). The c.1981C>T variant is known to partially inactivate pRb and a correlation between the gender of the transmitting carrier and penetrance has been recently demonstrated [20, 43]. As to the c.1331A>G exonic substitution, minigene assay confirmed aberrant splicing and showed that a fraction of normal transcript was still produced by the mutant allele, justifying the milder phenotypic impact [14]. Trans-regulatory factors can change the ratio between wild-type and aberrant isoforms over time and in different tissues, resulting in a modulation of the “leaky” effect and contributing to clinical variability. Concerning intronic variants, our results demonstrated that both changes result in aberrant splicing without evidence of leaky effect. In accordance with the hypothesis of hypomorphic alleles, the c.608-16_608del variant results in the in-frame skipping of exon 7, leading to the loss of 37 aa at the N-terminus. Previous studies on humans and mice agreed that loss of this domain generates partially penetrant phenotypes on tumor incidence [11, 16, 44,45,46,47,48]. More recently, Borysov and colleagues provided a molecular explanation for these findings, showing that this protein portion is important for growth-suppressive functions, but its absence only partially impairs the ability of pRB to inhibit DNA replication and block G1-to-S cell cycle transit [49]. The c.2663+2T>C variant generates an abnormal transcript that, lacking exon 25, is predicted to truncate the protein at the carboxy-terminus. A paucity of truncating variants from exon 25 onwards have been reported in RB patients, suggesting that loss of the C-terminal portion affect unessential protein functions [42]. In families 1 and 6, quantitative analysis showed an inversion of the expression ratio between the two differentially spliced isoforms in carrier fathers (one unaffected and one with retinoma) compared to the affected offspring: the higher expression of the mutated transcript in fathers who had not developed the tumor indicates that the variant is on the maternal allele. The data normalization respect to total expression levels showed that aberrant isoforms represent the main transcripts in both carrier fathers despite different degrees of RNA degradation.
The identification of these molecular mechanisms changes the genetic/prenatal counseling and the clinical management of RB families. In sporadic cases such as families 2 and 5, parents are at increased risk to have other affected children due to a varying degree of mosaicism. Already in 2000, the risk for siblings of RB patients was suspected to be underestimated due to parental mosaicism [50]. NGS now allows to detect even low-level mosaicism and to provide a better estimate of the impact of this phenomenon. A very recent study provided a recurrence risk estimation due to parental mosaicism 266- to 533-fold higher, as compared to the general population [51]. Because of a parent-of-origin effect, unaffected fathers, who are born from mothers with RB, can harbor the predisposing variant with a 50% risk of passing it down to their children who can thus be affected. Interestingly, in families segregating the c.1981C>T variant, we observed extraocular RB1-related cancers such as osteosarcoma or leiomyosarcoma in adult carriers who had not developed RB.
In conclusion, this study indicates that mosaicism of amorphic pathogenic variants and parent-of-origin effect of hypomorphic pathogenic variants significantly impact on phenotypic presentation in RB. Thus, the identification of these mechanisms has important implications for genetic counseling, allowing to provide a better estimation of the recurrence risk, a more accurate prognostic assessment, prenatal diagnosis options and optimal medical surveillance in all at-risk individuals.
References
Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975-2004. Br J Ophthalmol. 2009;93:21–3.
Knudson AG Jr.. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–3.
Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–6.
Cavenee WK, Dryja TP, Phillips RA, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305:779–84.
Harbour JW. Molecular basis of low-penetrance retinoblastoma. Arch Ophthalmol. 2001;119:1699–704.
Sakai T, Ohtani N, McGee TL, Robbins PD, Dryja TP. Oncogenic germ-line mutations in Sp1 and ATF sites in the human retinoblastoma gene. Nature. 1991;353:83–6.
Kratzke RA, Otterson GA, Hogg A, et al. Partial inactivation of the RB product in a family with incomplete penetrance of familial retinoblastoma and benign retinal tumors. Oncogene. 1994;9:1321–6.
Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349–54.
Cowell JK, Bia B, Akoulitchev A. A novel mutation in the promotor region in a family with a mild form of retinoblastoma indicates the location of a new regulatory domain for the RB1 gene. Oncogene. 1996;12:431–6.
Bremner R, Du DC, Connolly-Wilson MJ, et al. Deletion of RB exons 24 and 25 causes low-penetrance retinoblastoma. Am J Hum Genet. 1997;61:556–70.
Otterson GA, Chen W, Coxon AB, Khleif SN, Kaye FJ. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc Natl Acad Sci USA. 1997;94:12036–40.
Schubert EL, Strong LC, Hansen MF. A splicing mutation in RB1 in low penetrance retinoblastoma. Hum Genet. 1997;100:557–63.
Scheffer H, Van Der Vlies P, Burton M, et al. Two novel germline mutations of the retinoblastoma gene (RB1) that show incomplete penetrance, one splice site and one missense. J Med Genet. 2000;37:E6.
Genuardi M, Klutz M, Devriendt K, Caruso D, Stirpe M, Lohmann DR. Multiple lipomas linked to an RB1 gene mutation in a large pedigree with low penetrance retinoblastoma. Eur J Hum Genet. 2001;9:690–4.
Klutz M, Brockmann D, Lohmann DR. A parent-of-origin effect in two families with retinoblastoma is associated with a distinct splice mutation in the RB1 gene. Am J Hum Genet. 2002;71:174–9.
Lefevre SH, Chauveinc L, Stoppa-Lyonnet D, et al. A T to C mutation in the polypyrimidine tract of the exon 9 splicing site of the RB1 gene responsible for low penetrance hereditary retinoblastoma. J Med Genet. 2002;39:E21.
Sampieri K, Hadjistilianou T, Mari F, et al. Mutational screening of the RB1 gene in Italian patients with retinoblastoma reveals 11 novel mutations. J Hum Genet. 2006;51:209–16.
Gamez-Pozo A, Palacios I, Kontic M, et al. Pathogenic validation of unique germline intronic variants of RB1 in retinoblastoma patients using minigenes. Hum Mutat. 2007;28:1245.
Hung CC, Lin SY, Lee CN, et al. Low penetrance of retinoblastoma for p.V654L mutation of the RB1 gene. BMC Med Genet. 2011;12:76.
Eloy P, Dehainault C, Sefta M, et al. A parent-of-origin effect impacts the phenotype in low penetrance retinoblastoma families segregating the c.1981C>T/p.Arg661Trp mutation of RB1. PLoS Genet. 2016;12:e1005888.
Onadim Z, Hogg A, Baird PN, Cowell JK. Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc Natl Acad Sci USA. 1992;89:6177–81.
Dalamon V, Surace E, Giliberto F, Ferreiro V, Fernandez C, Szijan I. Detection of germline mutations in argentine retinoblastoma patients: low and full penetrance retinoblastoma caused by the same germline truncating mutation. J Biochem Mol Biol. 2004;37:246–53.
Sanchez-Sanchez F, Ramirez-Castillejo C, Weekes DB, et al. Attenuation of disease phenotype through alternative translation initiation in low-penetrance retinoblastoma. Hum Mutat. 2007;28:159–67.
Abouzeid H, Schorderet DF, Balmer A, Munier FL. Germline mutations in retinoma patients: relevance to low-penetrance and low-expressivity molecular basis. Mol Vis. 2009;15:771–7.
Hall JG. Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am J Hum Genet. 1988;43:355–63.
Carlson EA, Desnick RJ. Mutational mosaicism and genetic counseling in retinoblastoma. Am J Med Genet. 1979;4:365–81.
Sippel KC, Fraioli RE, Smith GD, et al. Frequency of somatic and germ-line mosaicism in retinoblastoma: implications for genetic counseling. Am J Hum Genet. 1998;62:610–9.
Rushlow D, Piovesan B, Zhang K, et al. Detection of mosaic RB1 mutations in families with retinoblastoma. Hum Mutat. 2009;30:842–51.
Chen Z, Moran K, Richards-Yutz J, et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35:384–91.
Amitrano S, Marozza A, Somma S, et al. Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet. 2015;23:1523–30.
Muller B, Hedrich K, Kock N, et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet. 2002;71:1303–11.
Badenhop RF, Cherian S, Lord RS, Baysal BE, Taschner PE, Schofield PR. Novel mutations in the SDHD gene in pedigrees with familial carotid body paraganglioma and sensorineural hearing loss. Genes Chromosomes Cancer. 2001;31:255–63.
Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14:609–17.
Buiting K, Kanber D, Horsthemke B, Lohmann D. Imprinting of RB1 (the new kid on the block). Brief Funct Genom. 2010;9:347–53.
Houdayer C. In silico prediction of splice-affecting nucleotide variants. Methods Mol Biol. 2011;760:269–81.
Forzan M, Salviati L, Pertegato V, et al. Is CFTR 621+3 A>G a cystic fibrosis causing mutation? J Hum Genet. 2010;55:23–6.
Fallerini C, Baldassarri M, Trevisson E et al. Alport syndrome: impact of digenic inheritance in patients management. Clin Genet. 2016;92:34–44.
Livak KJ. ST: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 1997;25:402–8.
Castera L, Sabbagh A, Dehainault C, et al. MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst. 2010;102:1805–8.
Dehainault C, Garancher A, Castéra L, et al. The survival gene MED4 explains low penetrance retinoblastoma in patients with large RB1 deletion. Hum Mol Genet. 2014;23:5243–50.
Tsai T, Fulton L, Smith BJ, et al. Rapid identification of germline mutations in retinoblastoma by protein truncation testing. Arch Ophthalmol. 2004;122:239–48.
Price EA, Price K, Kolkiewicz K, et al. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J Med Genet. 2014;51:208–14.
Fang W, Mori T, Cobrinik D. Regulation of PML-dependent transcriptional repression by pRB and low penetrance pRB mutants. Oncogene. 2002;21:5557–65.
Goodrich DW. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J Cell Physiol. 2003;197:169–80.
Dryja TP, Rapaport J, McGee TL, Nork TM, Schwartz TL. Molecular etiology of low-penetrance retinoblastoma in two pedigrees. Am J Hum Genet. 1993;52:1122–8.
Sanchez F, Mateu E, Beneyto M, Najera C, Prieto F. A constitutional homozygous mutation in the RB1 gene in a patient with unilateral retinoblastoma. J Med Genet. 2000;37:615–20.
Jakubowska A, Zajaczek S, Haus O, et al. Novel RB1 gene constitutional mutations found in Polish patients with familial and/or bilateral retinoblastoma. Hum Mutat. 2001;18:459.
Riley DJ, Liu CY, Lee WH. Mutations of N-terminal regions render the retinoblastoma protein insufficient for functions in development and tumor suppression. Mol Cell Biol. 1997;17:7342–52.
Borysov SI, Nepon-Sixt BS, Alexandrow MG, Then terminus of the retinoblastoma protein inhibits DNA replication via a bipartite mechanism disrupted in partially penetrant retinoblastomas. Mol Cell Biol. 2015;36:832–45.
Smith JH, Murray TG, Fulton L, O’Brien JM. Siblings of retinoblastoma patients: are we underestimating their risk? Am J Ophthalmol. 2000;129:396–8.
Dehainault C, Golmard L, Millot GA, et al. Mosaicism and prenatal diagnosis options: insights from retinoblastoma. Eur J Hum Genet. 2017;25:381–3.
Acknowledgements
We thank retinoblastoma patients and their families for the participation to the study. We also thank professor Genuardi Maurizio for a critical review of the manuscript. This work was supported by the Italian Ministry of Health Grant GR-2009-1578914 and University of Padova Grant CPDA140508/14 to ET.
Author’s Contributions
VI has performed the experiments and has drafted the manuscript. AR, and FA have made substantial contributions to conception and design of the study, and reviewed the manuscript. AMP has made important contributions in interpretation of molecular results and clinical data collection. EG, ET, VM, EF have contributed to the experiments and data acquisition. TH, SDeF and EG are ophthalmologists that have made RB diagnosis. PT has conducted histopathological studies in available eye samples. VC and AA are neurosurgeons who have dealt with patients treatment. GR and MAM have conducted the genetic counseling to RB families. All authors have given the approval of the final version of the manuscript to be published. They ensure that all aspects of the work are investigated and resolved with accuracy and integrity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Imperatore, V., Pinto, A.M., Gelli, E. et al. Parent-of-origin effect of hypomorphic pathogenic variants and somatic mosaicism impact on phenotypic expression of retinoblastoma. Eur J Hum Genet 26, 1026–1037 (2018). https://doi.org/10.1038/s41431-017-0054-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0054-6

{kind=link}
{kind=link}