
Abstract
Variants in the SPATA5 gene were recently described in a cohort of patients with global developmental delay, sensorineural hearing loss, seizures, cortical visual impairment and microcephaly. SPATA5 protein localizes predominantly in the mitochondria and is proposed to be involved in mitochondrial function and brain developmental processes. However no functional studies have been performed. This study describes five patients with psychomotor developmental delay, microcephaly, epilepsy and hearing impairment, who were thought clinically to have a mitochondrial disease with subsequent whole-exome sequencing analysis detecting compound heterozygous variants in the SPATA5 gene. A summary of clinical data of all the SPATA5 patients reported in the literature confirms the characteristic phenotype. To assess SPATA5’s role in mitochondrial dynamics, functional studies were performed on rat cortical neurons. SPATA5-deficient neurons had a significant imbalance in the mitochondrial fusion-fission rate, impaired energy production and short axons. In conclusion, SPATA5 protein has an important role in mitochondrial dynamics and axonal growth. Biallelic variants in the SPATA5 gene can affect mitochondria in cortical neurons and should be considered in patients with a neurodegenerative disorder and/or with clinical presentation resembling a mitochondrial disorder.
Similar content being viewed by others
Introduction
Autosomal recessive variants in the SPATA5 (spermatogenesis-associated protein 5, MIM: 613940) gene were recently associated with a specific disease phenotype. Tanaka et al. [1], Kurata et al. [2] and Buchert et al. [3] reported 25 individuals with intellectual disability, microcephaly, hypotonia, spasticity, seizures, sensorineural hearing loss and cortical visual impairment.
The SPATA5 gene and the protein encoded by it were first described by Liu et al. [4] as a spermatogenesis-associated factor (SPAF). The SPAF protein is a member of the AAA (ATPase Associated with diverse Activities) protein subfamily having two highly conserved ATPase modules and a putative mitochondrial matrix-targeting sequence. Liu et al. [4] concluded that SPATA5 gene may be important in spermatogenesis, through remodelling of mitochondria from an orthodox form to a condensed form, as SPAF’s predominant expression was in the spermatogonia and spermatocytes, but not in spermatids, and subcellularly in the inner membrane and matrix of mitochondria. However, no functional studies to link variants in the SPATA5 gene to mitochondrial dysfunction have been done. In addition, the recent association of SPATA5 variants with a neurodegenerative disease [1,2,3], suggests a role of the SPATA5 gene not only in spermatogenesis, but also in neuronal development.
This paper reports five new patients from four families with compound heterozygous variants in the SPATA5 gene and reviews the clinical features and reported mutations of all the patients described so far.
To analyse the pathomechanisms of SPATA5 gene deficiency on a cellular level, functional studies on rat cortical neurons were undertaken. An association with neuronal development and mitochondrial dysfunction is suggested.
Patients
Five patients from four different families were enroled in this study. The first family with two affected individuals (Patients 1 and 2) was identified at the Department of Clinical Genetics, Tartu University Hospital, Tartu, Estonia. Three other families with single affected cases (Patients 3, 4 and 5) were detected at the Institute of Human Genetics, Technical University Munich, Germany. The detailed information about the patients is given in Table 1 and in the Supplementary File. Common features for all the patients were global developmental delay, moderate microcephaly, myoclonic/tonic–clonic epilepsy, abnormal EEG, brain atrophy and/or delayed myelination (Supplementary Fig. S1A–D), sensorineural hearing loss, hyperreflexia, spasticity and dystonia. The facial phenotype of Patient 3 is illustrated on Supplementary Fig. S1E, F. The patients were suspected clinically to have a mitochondrial disorder. However, screening analyses for metabolic disorders including mitochondrial diseases from blood, urine and cerebrospinal fluid were mostly unremarkable. Mitochondrial respiratory chain enzyme analyses from muscle showed combined defects of complex I and IV in Patient 5, but were normal in Patient 1 (from cultured skin fibroblasts) and Patient 3 (from muscle). Whole-exome sequencing (WES) was performed in Patients 1, 3, 4 and 5 in the context of two separate research projects.
Methods
The study of the Estonian family was approved by Research Ethics Committee of the University of Tartu (approval date 17/11/2014 and number 242/M-10). The ethics committee of the Technical University Munich approved the study of Patients 3, 4 and 5.
Whole-exome sequencing
WES analysis of the Patients 1, 3, 4 and 5 was carried out using methods described earlier [5, 6] and detected compound heterozygous variants in the SPATA5 gene. All the variants were submitted to the LOVD (http://databases.lovd.nl/shared/genes/SPATA5) with individual IDs 00103212-00103215.
Functional studies
To analyse the functional consequences of SPATA5 gene deficiency on the central nervous system, the effects of SPATA5 shRNA silencing on rat cortical neurons were investigated using the following methods.
Plasmids
Plasmids expressing scrambled shRNA or shRNA targeted against rat SPATA5 were obtained from SABiosciences and were previously validated by PCR. Plasmids expressing GW1-PercevalHR (49082) and neuron-specific pAAV-hSyn-DsRedExpress (22907) were obtained from Addgene. Mito-KikGR1 was constructed as described earlier [7].
Neuronal cultures
Primary cultures of rat cortical cells were prepared from neonatal Wistar rats as described [8]. Neurons were grown in Neurobasal A medium supplemented with B27 with or without Phenol Red on poly-L-lysine-coated 96-well white plates, 35-mm plastic or glass bottom dishes. All culture media and supplements were obtained from Life Sciences. For transfection of cells growing on glass bottom dishes, the conditioned medium was replaced with 100 μl Opti-MEM I medium containing 2% Lipofectamine 2000 and 1–2 μg total DNA containing an equal amount of each plasmid. The dishes were incubated for 3–4 h, after which fresh medium was added.
Mitochondrial fusion and fission rate analysis
For mitochondrial fusion and fission rate analysis, cortical neuronal cultures were transfected with mito-KikGR1 plasmid and plasmids of interest as described earlier [7] and examined at DIV 7–8 by a laser scanning confocal microscope (LSM 780, Carl Zeiss Microscopy GmbH). For fusion acquisition, mito-KikGR1 was illuminated with a 488-nm argon laser line to visualize the intense green mitochondrial staining. Selected mitochondria were then photoconverted to red using a 405-nm diode laser and illuminated using a 561 nm DPSS laser. The images were taken at 10-s intervals for 10 min, the fate of all activated mitochondria was followed throughout the time-lapse, and the fusion and fission events were recorded.
ATP/ADP ratio measurement
Neurons expressing the ATP/ADP ratio sensor PercevalHR at neuronal endings were excited using a 405 nm diode laser and a 488 nm line of an Argon laser and collected using a 494–553 nm emission window. The ratio of fluorescence intensities, when exciting at 488 nm divided by 405 nm (F488 nm/F405 nm), was calculated from the collected signal from 50 axonal endings from five dishes per group.
Axonal growth
For neuronal maturation experiments, cortical neurons were transfected at day 1 in vitro (DIV1) with a plasmid expressing neuron-specific pAAVhSyn-DsRed1 and plasmids of interest. For the analysis of axonal growth, images of cultured cortical neurons at DIV4 were captured using an Olympus IX70 inverted microscope with a 20x objective and traced manually using Neurolucida software (MBF Bioscience) and Fiji [9].
Immunohistochemistry
Neurons, HeLa, COS7 and SHSY5Y cells were fixed using 4% paraformaldehyde solution in NeurobasalTM A containing 5% sucrose for 10 min at 37 °C. Fixed cells were permeabilised using 0.1% Triton X-100 in PBS for 7 min and then blocked using 10% normal goat serum and 3% BSA for 60 min at room temperature. The cells were then incubated with the primary antibodies rabbit anti-myc (1:300, ab 9106, Abcam, USA) and mouse anti-TOM20 (1:200, ab 56783, Abcam, USA) in the presence of 10% normal goat serum and 3% BSA at 4 °C for 24 h. After washing, the cells were further incubated with respective Alexa-Fluor-488- or Alexa-Fluor-594-conjugated secondary antibodies at room temperature for 1 h and subsequently examined using confocal microscope.
Statistics
Data are presented as the mean ± SEM. T-test or one-way ANOVAs followed by Bonferroni post hoc test were used to compare differences between experimental samples and control groups. P values of <0.05 were considered statistically significant.
Results
Whole-exome sequencing
In Patient 1, after filtering for rare protein altering possibly biallelic variants two heterozygous variants in the SPATA5 gene were identified by WES: c.250C>T, p.(Arg84*) and c.989_991del, p.(Thr330del) (RefSeq NM_145207.2). The c.250C>T is a nonsense variant presumably causing loss-of-function, which appears only in 1 out of 121,318 alleles (i.e. one heterozygous carrier) in the Exome Aggregation Consortium (ExAC) database [10]. The c.989_991del in-frame deletion has been previously reported in multiple patients with SPATA5-related disorder [1,2,3]. Both variants were confirmed by Sanger sequencing in both, Patients 1 and 2. Their mother was confirmed to carry only the c.250C>T variant, DNA from the father was not available for testing.
In Patient 3, after filtering the results of WES with the frequency of online databases (MAF < 0.1% in 1000 genomes, ExAC), pathogenicity prediction programs (PolyPhen-2, Sift) and ClinVar, SPATA5 was the only gene with both variants predicted to be probably damaging (c.554G>A, p.(Gly185Glu)) or having a likely pathogenic rating in ClinVar (c.989_991del, p.(Thr330del)). The compound heterozygous state of the two variants was confirmed by Sanger sequencing, the mother carried the missense variation, the father the deletion.
WES analysis of Patient 4, using the above-described filtering, identified only SPATA5 gene containing a nonsense loss-of-function heterozygous variant, namely c.394C>T, p.(Gln132*). The patient also carried the c.989_991del, p.(Thr330del) in-frame deletion in heterozygous state in the SPATA5 gene. The mother was the carrier of the nonsense variant and the deletion came from the father.
The same filtering method of Patient 5 resulted in only one nonsense loss-of-function variant (c.700C>T, p.(Gln234*)) in the SPATA5 gene. This variant was detected in a heterozygous state predicted to be compound heterozygous with the c.2384C>G, p.(Pro795Arg) missense variant. This assumption was confirmed by Sanger sequencing, where the healthy mother was the carrier of the c.700C>T nonsense variant and the healthy father carried the c.2384C>G missense variant, each in heterozygous state.
Patient 1 was identified in the Estonian research project, which included 21 patients with a clinical suspicion of a mitochondrial disorder, but no genetic diagnosis. These 21 cases were selected from a database of 181 patients whose fibroblast cell cultures have been stored from 2003-2014. Patients 3–5 were identified in the German research project, which included 353 patients with a suspicion of a mitochondrial disorder to whom WES analysis was performed and the three patients with SPATA5 variants were identified. The diagnostic yield was 0.6% and 0.8% in those two cohorts, respectively.
The results of the functional studies on rat cortical neurons
SPATA5 is cytosolic and not co-localizing with the mitochondrial marker
SPATA5 protein has a putative mitochondrial matrix-targeting sequence and has been shown to localize in mitochondria in mouse testis [4]. However, subcellular localization data of SPATA5 in neurons and other cell types has not been reported. To our surprise the localization of overexpressed SPATA5 in cultured primary cortical neurons was dominantly cytosolic and clearly not co-localizing with the mitochondrial marker (Fig. 1). Similar relatively homogenous cytosolic localization was observed in neuronal SH-SY5Y cells and HeLa cells. The signal was less homogenous in COS7 cells, but also clearly not co-localizing with the mitochondrial marker. Unfortunately, we were not able to visualize endogenous SPATA5 as the antibody staining was too weak for co-localization study. It should also be noted that despite having the mitochondrial targeting sequence, several subcellular localization prediction tools like PSORT II [11], Yloc [12] and Cello [13] suggested cytosolic localization of SPATA5.

SPATA5 has a dominantly cytosolic localization and does not co-localize with mitochondria. Overexpressed SPATA5–myc (detected using an anti-myc antibody, green) is showing relatively homogenous localization in cortical neurons, SH-SY5Y and Hela cells. In COS7 cells, part of the SPATA5 was in filamentous network-like structures but as demonstrated by the zoom panels it was nevertheless not co-localizing with mitochondria. Mitochondrial outer membrane was stained using TOM20 antibody
SPATA5 deficiency affects mitochondrial morphology and inhibits mitochondrial dynamics
Previously, it has been suggested that SPATA5 has a role in mitochondrial morphogenesis during spermatogenesis [4]. Therefore, we first tested whether the SPATA5 deficiency affects mitochondrial morphology in neurons. Indeed, there was a significant, 20% decrease in mitochondrial length in SPATA5 shRNA-treated neurons (efficiency of SPATA5 shRNA is demonstrated in Fig. 2a) compared with scrambled shRNA-treated controls (Fig. 2b, c). The mitochondrial length is controlled by mitochondrial fusion–fission balance [7]. Therefore, we then measured mitochondrial fusion and fission using photoconvertible mitochondria targeted Kikume Green-Red, which enables the quantification of fusion events between green- and red-emitting mitochondria (Fig. 2d). There was a slight statistically non-significant decrease in the number of fusion events in SPATA5 shRNA-treated neurons and slight statistically non-significant increase in the number of fission events in SPATA5 shRNA-treated neurons (Fig. 2e, f). However, together these changes led to two fold statistically significant decrease in fusion–fission ratio suggesting strong imbalance between the fusion and fission events in neuronal axons (Fig. 2g).

SPATA5 deficiency impairs mitochondrial dynamics and ATP production. a Relative mRNA expression levels in neuronal PC cells transfected with scrambled or SPATA5 shRNA. The expression level of SPATA5 was normalized to CYC. b, c Neurons were transfected with mito-DsRed, GFP and scrambled shRNA or SPATA5 shRNA. SPATA5 shRNA neurons demonstrate significantly reduced mitochondrial length (n = 16 neurons in each group). d–g Primary cortical neurons were transfected with the photoconvertible mitochondrially targeted construct mito-Kikume—Green and scrambled shRNA or SPATA5 shRNA. Selected mitochondria were irradiated using a 405-nm laser line, thereby converting mito-Kikume—Green into mito-Kikume—Red. Fusion events between mito-Kikume—Green and photoactivated mito-Kikume—Red mitochondria are visible when mitochondria become yellow after mixing of the contents of the red and green mitochondria (white arrows) and fission events when these mitochondria split (yellow arrows) (d). In primary cortical neurons, SPATA5 shRNA tends to decrease the fusion rate (e) and increase the fission rate (f) leading to significant decrease in fusion–fission ratio (g) (four fields per dish were imaged and at least four dishes per condition were used). h–i Neurons were transfected with the ATP sensor Perceval and scrambled SPATA5 shRNA or SPATA5 shRNA plus shRNA insensitive human SPATA5 encoding plasmid. FRET (F488), CFP (F405) and pseudocolour ratiometric (F488/F405) images of an axonal ending of control neuron expressing PercevalHR are depicted (h). SPATA5-deficient neurons show a lower cytosolic ATP level as compared to the control (i). Note that the lower ATP levels in SPATA5 deficient neurons is restored by wt SPATA5 overexpression (n = 50 neurons)
SPATA5 deficiency is associated with decreased cellular ATP
To test whether the changes in mitochondrial morphology and dynamics could affect the energetic status of the neurons a quantitative analysis of cell ATP/ADP ratio was performed using the genetically encoded fluorescent ratiometric probe PercevalHR, which senses ATP/ADP ratio (Fig. 2h) [14]. Neurons were first transfected with SPATA5 shRNA encoding plasmid and compared with scrambled shRNA-treated controls. Further experiments performed at axonal endings of scrambled or SPATA5 shRNA expressing neurons showed a statistically significant 12% decrease in signal in the SPATA5 shRNA group, suggesting a decrease in ATP levels (Fig. 2i). Overexpression of human shRNA-insensitive SPATA5 restored the ATP/ADP ratio in the SPATA5 shRNA-treated group demonstrating the specificity of shRNA.
SPATA5 deficiency delays neuronal development
The results demonstrate that SPATA5 deficiency delayed the development of cortical neurons markedly. The longest axon was significantly shorter in developing DIV4 SPATA5 shRNA-transfected neurons (Fig. 3). Similarly, overexpression of human shRNA-insensitive SPATA5 restored the axonal growth in the SPATA5 shRNA-treated group.

SPATA5 deficiency impairs neuronal growth. Primary cortical neurons were transfected with the neuronal marker pAAV-hSyn- DsRed1 and scrambled shRNA or SPATA5 shRNA at DIV 1, and neuronal morphology was assessed at DIV 4. a Microscopy images (above) and examples of Neurolucida reconstructions (below) are depicted. b SPATA5 deficiency retards growth of the longest axon that is restored by wt SPATA5 overexpression (n = 30 neurons in each group)
Discussion
Including these 5 subjects, 30 patients with homozygous or compound heterozygous variants in the SPATA5 gene have been reported in the literature [1,2,3]. All patients share similar features and thus a characteristic phenotype can be described.
Symptoms start in early infancy with all of the patients developing global developmental delay with severely limited speech or no words, hearing impairment (most frequently sensorineural hearing loss, 77%), and a pathological EEG with symptomatic epilepsy being present in 73% of patients. Other prominent features are microcephaly (90%), gastrointestinal problems (73%), abnormal brain MRI (67%, predominantly brain atrophy and delayed myelination), visual impairment (most frequently cortical visual impairment, 41%) and no or reduced eye contact (48%). The patients tend to have a combination of axial hypotonia and peripheral hypertonia with spasticity. Detailed clinical data of all reported patients is summarized in Table 2.
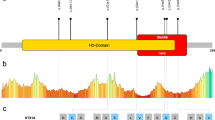
Together, 25 different variants have been found across the gene: 14 missense, 5 nonsense, 3 frameshift variants, 2 in-frame deletions and 1 intronic splice-site variant. These are illustrated in Fig. 4. The most common reported variant is an in-frame deletion (c.989_991del, p.(Thr330del)) that was seen in 6 out of 18 different families (33%) and in 8 of the 30 patients. Three other variants (p.(Arg84Gln); p.(Arg186*); p.(Asp628Gly)) have occurred in two different families each. Interestingly, there is a homozygous individual with the p.(Thr330del) variant in the ExAC database [10], but also one patient described by Tanaka et al. [1] has a homozygous p.(Thr330del) variant. There are no mutation hot spots in the gene and there is no clear association between the location of the variants and severity of the phenotype. The siblings in Tanaka et al. [1] underlie this fact, as the younger sister has a milder phenotype than her elder brother, although they have the same genotype (NM_145207.2: c.2351G>A and c.269G>T). The precise investigation of the 3D structure of the protein could help for better understanding of the effects of the variants.

The schematic figure of the SPATA5 gene with the variants reported so far. a Exons are shown as grey boxes. All six variants from our study are shown above, and all published are shown below the gene. The loss-of-function variants are tagged with bold black lines. b The putative mitochondrial targeting sequence and the two AAA domains are marked with dashed lines
Previous researchers have suggested that SPATA5 is associated with mitochondrial morphology and function [4]. This study shows that SPATA5 deficient neurons have a significant imbalance of mitochondrial fusion and fission events leading to mitochondrial shortening that in turn result in decreased ATP production at axonal endings. This confirms that SPATA5 deficiency alters the morphology and function of mitochondria in neurons and therefore can be indirectly categorized under mitochondrial disorders. This also supports the patients’ phenotype and the clinicians’ initial suspicion of a mitochondriopathy.
In contrast to Liu et al. [4] suggesting that SPATA5 is localized in the inner membrane or matrix of mitochondria in mouse testis, our experiments in cortical neurons, SH-SY5Y, Hela and COS7 cells showed that it was dominantly cytosolic. We observed no co-localization with mitochondria suggesting that the putative mitochondrial targeting sequence might not be sufficient to drive SPATA5 to the mitochondria in these cell types. It should be noted here that Liu et al. [4] did also demonstrate that SPATA5 and the mitochondrial marker signals do not overlap. There are many examples where non-mitochondrial proteins play an active role in mitochondrial function. These proteins control mitochondrial biogenesis, mitochondrial trafficking and anchoring, as well as mitochondrial quality control mechanisms (e.g. mitophagy).
For example, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is caused by variants in the TYMP gene encoding cytosolic thymidine phosphorylase. These variants cause loss of thymidine phosphorylase activity and lead to accumulation of thymidine and deoxyuridine. This impairs mtDNA replication leading to accumulation of point mutations, deletions, and depletion of mtDNA [15,16,17]. Variants in the gene encoding KIF5A (Kinesin Family Member 5A), a protein that interacts with mitochondrial adaptor proteins and is responsible for mitochondrial transport in axons, are leading to hereditary spastic paraplegia [18, 19]. Variants in Parkin, a cytosolic ubiquitin E3 ligase known to be a crucial component of mitochondrial quality control mechanism, are associated with hereditary Parkinson’s disease [20, 21]. Another Parkinson’s disease related protein LRRK2, known to be involved in the regulation of mitochondrial dynamics and function, is also primarily localized to the cytoplasm [22]. Variants in the GBA gene encoding lysosomal glucocerebrosidase cause Gaucher disease and increase susceptibility to Parkinson’s disease. Elegant paper by Osellame et al. [23] shows that this is related to downregulation of mitophagy and the ubiquitin–proteasome system resulting in accumulation of dysfunctional and fragmented mitochondria leading in turn to impaired energy and free radical homoeostasis.
The majority of Wolfram syndrome cases are caused by variants in the WFS1 (Wolfram syndrome 1) gene encoding an endoplasmic reticulum (ER) protein. However, the clinical features of the syndrome resemble mitochondrial disease symptoms [24]. We have recently shown that down-regulation of WFS1 in neurons leads to dramatic changes in mitochondrial dynamics (inhibited mitochondrial fusion, altered mitochondrial trafficking and augmented mitophagy), which results in lower levels of ATP and inhibits neuronal development [25]. Another example of the ER protein variant responsible for mitochondrial damage is RYR1 (ryanodine receptor type 1) variant in central core disease [26]. One of the characteristics of this type of myopathy is the lack of mitochondria within the centres of muscle fibres. Therefore, our results allow to suggest that similarly to these proteins, SPATA5 is indirectly or directly interacting with mitochondria and required for their proper dynamics. Nevertheless, we cannot completely exclude a possibility that a small fraction of SPATA5 is still localized to the mitochondria and controls the mitochondrial function from there.
This study also shows that the SPATA5 gene plays an important role in the development of cortical neurons. Local ATP supply is critical for axonal growth. In neurons, most of the ATP supply is produced in the mitochondria and decline in ATP production could impair the axonal growth. Depletion of mitochondria at or before axogenesis prevents axon formation [27]. Similarly, a lack of synaptic or terminal axonal mitochondria results in aberrant organelle transport and dysfunctional synapses [28, 29]. Vaarmann et al. [30] and Cagalinec et al. [25] have recently shown that increased mitochondrial ATP production in axonal endings supports axonal growth and suggests that mitochondrial ATP production is required for proper neuronal development.
In conclusion, SPATA5 protein is required to sustain mitochondrial morphology, dynamics and ATP production in neurons and its deficiency leads to impaired axogenesis in vitro, in primary cortical neurons. SPATA5 deficiency results in a syndrome with severe global developmental delay, severe speech impairment, hearing loss, abnormal electroencephalogram and microcephaly. Biallelic variants in the SPATA5 gene can affect mitochondria in cortical neurons and should be considered in patients with a neurodegenerative disorder and/or with clinical presentation resembling a mitochondrial disorder.
References
Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet. 2015;97:457–64.
Kurata H, Terashima H, Nakashima M, Okazaki T, Matsumura W, Ohno K, et al. Characterization of SPATA5-related encephalopathy in early childhood. Clin Genet. 2016;90:437–44.
Buchert R, Nesbitt AI, Tawamie H, Krantz ID, Medne L, Helbig I, et al. SPATA5 mutations cause a distinct autosomal recessive phenotype of intellectual disability, hypotonia and hearing loss. Orphanet J Rare Dis. 2016;11:130.
Liu Y, Black J, Kisiel N, Kulesz-Martin MF. SPAF, a new AAA-protein specific to early spermatogenesis and malignant conversion. Oncogene. 2000;19:1579–88.
Pajusalu S, Reimand T, Ounap K. Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am J Med Genet A. 2015;167A:1913–5.
Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–9.
Cagalinec M, Safiulina D, Liiv M, Liiv J, Choubey V, Wareski P, et al. Principles of the mitochondrial fusion and fission cycle in neurons. J Cell Sci. 2013;126:2187–97.
Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, et al. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J Biol Chem. 2009;284:21379–85.
Zhao B, Choi CH, Bhuripanyo K, Villhauer EB, Zhang K, Schindelin H, et al. Inhibiting the protein ubiquitination cascade by ubiquitin-mimicking short peptides. Org Lett. 2012;14:5760–3.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Horton P, Nakai K. Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc Int Conf Intell Syst Mol Biol. 1997;5:147–52.
Briesemeister S, Rahnenfuhrer J, Kohlbacher O. YLoc--an interpretable web server for predicting subcellular localization. Nucleic Acids Res. 2010;38:W497–W502.
Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006;64:643–51.
Tantama M, Martinez-Francois JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013;4:2550.
Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994;44:721–7.
Papadimitriou A, Comi GP, Hadjigeorgiou GM, Bordoni A, Sciacco M, Napoli L, et al. Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology. 1998;51:1086–92.
Nishigaki Y, Marti R, Copeland WC, Hirano M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J Clin Invest. 2003;111:1913–21.
Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet. 2002;71:1189–94.
Ebbing B, Mann K, Starosta A, Jaud J, Schols L, Schule R, et al. Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum Mol Genet. 2008;17:1245–52.
Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803.
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31.
Niu J, Yu M, Wang C, Xu Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J Neurochem. 2012;122:650–8.
Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013;17:941–53.
Barrett TG, Bundey SE, Fielder AR, Good PA. Optic atrophy in Wolfram (DIDMOAD) syndrome. Eye. 1997;11(Pt 6):882–8.
Cagalinec M, Liiv M, Hodurova Z, Hickey MA, Vaarmann A, Mandel M, et al. Role of mitochondrial dynamics in neuronal development: Mechanism for Wolfram syndrome. PLoS Biol. 2016;14:e1002511.
Lynch PJ, Tong J, Lehane M, Mallet A, Giblin L, Heffron JJ, et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci USA. 1999;96:4164–9.
Mattson MP, Partin J. Evidence for mitochondrial control of neuronal polarity. J Neurosci Res. 1999;56:8–20.
Lee CW, Peng HB. The function of mitochondria in presynaptic development at the neuromuscular junction. Mol Biol Cell. 2008;19:150–8.
Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–78.
Vaarmann A, Mandel M, Zeb A, Wareski P, Liiv J, Kuum M, et al. Mitochondrial biogenesis is required for axonal growth. Development. 2016;143:1981–92.
Acknowledgements
We thank all families for their kind cooperation. Furthermore, we thank the contribution of the medical-laboratory assistants of our institutes. This work was supported by the Estonian Research Council grant PUT355 and IUT2-5, and the European Regional Development Fund (Project No. 2014-2020.4.01.15-0012), and by the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (grant #FKZ 01ZX1405C).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Sanna Puusepp and Reka Kovacs-Nagy contributed equally to this work.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Puusepp, S., Kovacs-Nagy, R., Alhaddad, B. et al. Compound heterozygous SPATA5 variants in four families and functional studies of SPATA5 deficiency. Eur J Hum Genet 26, 407–419 (2018). https://doi.org/10.1038/s41431-017-0001-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0001-6
This article is cited by
-
Computational and mitochondrial functional studies of novel compound heterozygous variants in SPATA5 gene support a causal link with epileptogenic encephalopathy
Human Genomics (2023)
-
Whole‐exome sequencing of a Saudi epilepsy cohort reveals association signals in known and potentially novel loci
Human Genomics (2022)
-
Visualizing maturation factor extraction from the nascent ribosome by the AAA-ATPase Drg1
Nature Structural & Molecular Biology (2022)
-
Structural dynamics of AAA + ATPase Drg1 and mechanism of benzo-diazaborine inhibition
Nature Communications (2022)
-
4q27 deletion and 7q36.1 microduplication in a patient with multiple malformations and hearing loss: a case report
BMC Medical Genomics (2020)
