
Abstract
Over the course of more than two decades, natural products isolated from various microorganisms and plants have built the foundation for chemical biology research into the mechanism of pre-mRNA splicing. Hand in hand with advances in scientific methodology small molecule splicing modulators have become powerful tools for investigating, not just the splicing mechanism, but also the cellular effect of altered mRNA processing. Based on thorough structure-activity studies, synthetic analogues have moved on from scientific tool compounds to experimental drugs. With current advances in drug discovery methodology and new means of attacking targets previously thought undruggable, we can expect further advances in both research and therapeutics based on small molecule splicing modulators.
Similar content being viewed by others

Introduction
Even with advances in scaffold-based synthesis and natural product-like molecule production, genuine natural products still form a corner stone of chemical biology. However, the path from identification of a new and interesting molecule to a detailed understanding of its mechanism of action, cellular effect and utility in research and in medicine is a long, winding and arduous one. Many compounds prove to act upon different targets than initially thought. Discovery of practical uses as bioprobes or, with a lot of good luck, as therapeutics may take many years. Splicing inhibitors are no exception. While the first molecules were described in 1992, it took till 2007 before their mechanism came to light and only now are we understanding their profound effect on cellular mRNA metabolism, signaling and physiology. Recent studies are testament to the utility of natural products, the tenacity of researchers working on them, as well as significant advances in research technology and methodology.
Initial discoveries
In 1992, herboxidiene, a Streptomyces chromofuscus natural product with strong phytotoxic activity was first described [1, 2] (Fig. 1a, b). It was re-discovered in a different strain of Streptomyces in 2002, this time as one of a group of molecules named GEX1 compounds [3, 4], displaying cytotoxicity in the low to mid-nanomolar range against human tumor cell lines. At the time closer mechanistic studies were not undertaken and the molecular target remained unknown.
Four years later, evaluating whether transcriptional modulators would have antitumor activity, three molecules isolated from a broth of Pseudomonas sp. No.2663. were tested for their effect on the viral SV-40 promoter [5, 6]. The compounds, labeled FR901463, FR901464, and FR901465 led to significant promoter activation. The molecules were cytotoxic at low nanomolar concentration and inhibited the cell cycle in G1 and G2/M phase. FR901464 displayed strong antitumor activity, extending the lifespan of mice carrying the P388 lymphoma cell line and also inhibiting the growth A549 human lung adenoma xenografts. Observing internucleosomal DNA fragmentation at higher concentration, the authors assumed the FR compounds to act directly on chromatin, thereby changing transcriptional activity. Spliceostatin A, a methyl ketal derivative of FR901464 proved more stable in solution at equal potency and became pivotal in identifying the FR compounds’ mechanism of action [7]. Consequently, this family of molecules became known as the spliceostatins.

a The three families of SF3B1-targeting splicing modulators; spliceostatins, pladienolides and herboxidienes. Synthetic and semisynthetic derivates of the original natural products are labeled in orange. b Timeline of discovery or synthesis of the molecules shown in (a). Herboxidienes are labeled in black, spliceostatins in green and pladienolides in blue. c Splicing modulators not targeting SF3B1
In 1994, another report appeared in this journal, presenting a cytotoxic polyketide from a culture of Streptomyces hygroscopicus strain A-9561, isolated in Okinawa [8]. The molecule, named FD-895 proved toxic to doxorubicin-resistant HL-60 leukemia cells, but again mechanistic studies did not commence. Interest in FD-895 reemerged when the structurally related pladienolide family of natural products attracted attention a decade later. Pladienolides were first described in 2004 when a group of 7 related molecules was isolated from a Streptomyces platensis broth, taken from a soil sample from Kanagawa prefecture, Japan [9,10,11]. The initial study aimed at finding new antitumor agents, specifically compounds that would prevent angiogenesis in the hypoxic environment of solid tumors. The screen utilized an alkaline phosphatase reporter driven by the hypoxia inducible factor 1α (HIF1α)-dependent promoter of vascular endothelial growth factor (VEGF) [9, 11].
While it appeared that the active pladienolides did inhibit VEGF expression more strongly than they proved cytotoxic, the IC50 values for both activities only differed around 2–4 fold [9, 11]. Therefore, a specific mechanism targeting HIF1α signaling appeared unlikely. The paper offered a limited amount of structure activity relationship data and noted that pladienolide B (PlaB) inhibited growth of several tumor cell cultures from a panel of 39 cancer cell lines at low nanomolar IC50 values, even cell lines resistant to clinical anticancer agents, such as etoposide, cisplatin, camptothecin, vincristine, taxol and 5-flurouracil. PlaB did not appear to kill cells indiscriminately but was only specifically cytotoxic to some cell lines in the panel, though it remained unclear what drove its selectivity. In mouse xenograft models, PlaB proved effective against tumors from the BSY-1 breast cancer cell line [10]. These initial experiments indicated that pladienolides likely acted by a novel mechanism of action but gave little indication of the molecules’ actual target.
In addition, FD-895 and pladienolides share some superficial structural similarity with the lactimidomycin and isomigrastatin family of translation inhibitors, showing once more that 2D molecular structures are not predictive of activity [12, 13].
Since the herboxidienes, spliceostatins and pladienolides were each discovered assessing completely unrelated activities, nobody would have suspected that, despite representing vastly different structures, the three compounds share virtually identical mechanisms of action. In light of their activity and potency against tumor cells, synthetic routes to producing FR901464 and pladienolide were developed, though owing to an inordinate number of necessary steps, total synthesis for clinical use appeared impractical [14,15,16,17]. The spectrum of available active compounds keeps also increasing by natural means and for a change not from another strain of Streptomyces. The proteobacterium Burkholderia thailandensis MSMB43 yielded thailanstatin A [18, 19]. Not as potent as FR901464 itself, thailanstatin A still inhibits tumor cell growth in the mid to high nanomolar range [20]. In a similar manner, a herboxidiene derivative, RQN-18690A or 18-deoxyherboxidiene, was re-discovered in a screen for angiogenesis inhibitors [21]. It’s activity against human umbilical vein endothelial cells could also be traced back to SF3B1 inhibition.
Despite structural dissimilarity, computational studies indicated that the solution structures of both FR901464 and PlaB adopt similar conformations, centered on their central diene moiety, suggesting that they could bind the same physiological target [22]. Based on this hypothesis fully synthetic molecules centered on a common pharmacophore between FR901464 and PlaB were produced. The analogue displayed cytotoxic activity and induced G2/M cell cycle arrest, but not as potently as the original natural products. Since FR901464 was difficult to synthesize and somewhat unstable in solution, strategies were developed to generate simplified and stable analogues, culminating in the sudemycin family of molecules [23], which also inhibited tumor cell growth, albeit at IC50 values in the micromolar range [24,25,26].
While sudemycins suffered from significantly decreased potency, another synthetic attempt at producing FR901464 analogues yielded meayamycin, which, if anything, proved even more potent than the original natural product [17]. As can be seen in Fig. 1, the chemical space occupied by splicing modulators appears rather accommodating. It took till 2007 before the converging mechanisms of all molecules describes started to become apparent, with PlaB and SSA becoming the first known specific inhibitors of pre-mRNA splicing [27, 28].
Target identification
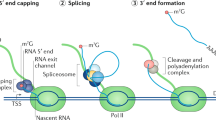
Splicing, the process of removing intronic sequences from a primary transcript, constitutes a key step in gene regulation of eukaryotic cells (reviewed in [29]). The coordinated interplay of several large ribonucleoprotein complexes not only allows producing a readable template for protein synthesis, likely it presents the very key to the complexity of large, multicellular organisms [30]. Every splicing reaction involves a coordinated intramolecular transesterification of an RNA polynucleotide, excising an internal sequence stretch. Via splicing the same primary RNA transcript can yield several different protein isoforms specific to a particular cell type, tissue or developmental stage. This goes so far, that splice isoforms of the same transcript may produce proteins with antagonistic properties (reviewed in [31]). Besides simply cutting out an intronic sequence, splicing of one pre-mRNA may lead to different outcomes. These include the choosing of alternative 3ʹ and 5ʹ splice sites, exon skipping (ES), inclusion of a cassette exon or of mutually exclusive exons (Fig. 2a). In general, an intron is defined by its 5ʹ and 3ʹ splice sites. Toward the 3ʹ end lies the branchpoint sequence containing the adenosine residue whose 2’OH group will conduct the nucleophilic attack on the 5ʹ splice site’s phosphodiester bond, creating a free 3ʹ hydroxyl group, which can then perform an attack on the 3ʹ splice site, connecting two exons and excising a lariat shaped intron (Fig. 2b). This branchpoint A (BPA) is surrounded by the branch-point sequence (BPS) and followed by a poly-pyrimidine tract. Naturally, choosing the right splice site and branch point requires a high degree of regulation and control [29]. Besides the core spliceosome, hundreds of accessory factors, including SR (serine and arginine-rich) proteins, may act as splicing enhancers or repressors, influencing the utilized splice site.

a Types of alternative splicing events with illustrative examples. Green denotes canonical exons, while included alternative splice sites or exons are indicated in blue, mutually exclusive alternative exons in gray [31]. b Simplified overview of spliceosome formation and pre-mRNA splicing
The process begins with E complex formation, where the U1 snRNP marks the 5ʹ splice site, while accessory factors SF1 and the dimeric U2AF bind toward the 3ʹ end, preparing arrival of the U2 snRNP. The U2 snRNP binds to the branch point sequence, displacing SF1, thereby forming A complex. During A complex formation the BPS will form a stable helix with the branchpoint interacting stem loop (BSL) of the U2 snRNA. The BPS is moderately conserved and the degree of complementarity to the U2 snRNA, especially within the BSL, determines the strength of the splice site, with better matches accounting for higher strength. At this point, the pre-mRNA is still intact. The actual cleavage reaction requires binding of the U4/U5/U6 tri-snRNP complex and displacement of U1 and U4. This review will focus on A complex formation, as SSA, PlaB, the herboxidienes and all their derivatives act on this step, specifically on the SF3b subcomplex of the U2 snRNP [32].
The U2 snRNP consists of the U2 snRNA polynucleotide, whose base-pairing to the branch point sequence maintains specificity, and a set of two protein subcomplexes SF3a and SF3b, as well as a set of Sm proteins which form a ring structure around the nascent transcript and enhance splicing fidelity [33]. The SF3b subcomplex, at its core consists of the proteins SF3B1 through SF3B6, PHF5A, TAT-SF1 and the ATPase PRP5. The largest SF3B subunit, SF3B1, contains two N-terminal domains for interaction with U2AF and PHF5A, followed by 20 tandem HEAT (Huntingtin, elongation factor 3, protein phosphatase 2A, target of rapamycin) repeats [34]. SF3B1 forms a long, superhelical structure, curving around PHF5A and contacting part of the U2 snRNA and many key protein factors, including PRP5 and TAT-SF1. Currently, it is thought that U2 finds the right position on the intron by interaction with U1, SF1, and U2AF. At that point the U2 BSL seems protected by TAT-SF1 and part of PRP5. RNA binding leads to PRP5 activation and the ATP-dependent displacement of TAT-SF1, such that BPS and U2 BSL can begin base-pairing, eventually disrupting the stem loop and forming an RNA-RNA double helix between U2 and pre-mRNA. In the early stages of A complex formation SF3B1 is present in an open conformation but will clamp down onto the U2-mRNA helix adopting a closed conformation, thereby also displacing PRP5. In addition, SF3B1 and PHF5A together form a pocket to protect the reactive BPA [35]. Mutations in SF3B1 have been implicated in myelodysplastic syndromes (MDS), chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML) and a number of late-stage cancers. Mutations appear to primarily cluster around the more N-terminal HEAT repeats with changes in R625, K666 and K700 leading to alternative BPS usage and cryptic 3′ splice site selection [36,37,38,39]. Especially HEAT repeat 6 and surroundings appear as mutational hotspots, likely affecting PRP5 binding and function, thereby reducing the fidelity of branch point selection.
Since 2007 further characterization of SF3B1’s prominent role in splicing has become possible thanks to specific chemical probes. Derivatives of FR901464 and pladienolide finally allowed target identification [27, 28]. A biotinylated version of SSA, as well as derivates of PlaB incorporating radiolabels, fluorescent markers and photo-crosslinkers enabled identifying the SF3B complex as the true molecular target. Further experimentation confirmed that SSA, PlaB, herboxidienes and related compounds did interrupt pre-mRNA splicing [40, 41]. In in vitro studies the U2 snRNP interacted more weakly with the pre-mRNA in presence of inhibitor and proved prone to bind at cryptic splice sites. Furthermore, inhibition appeared to occur before ATP hydrolysis, which means that A complex formation was not completed.
It was soon appreciated that in vivo these molecules do not necessarily interrupt splicing in total but modulate cellular splicing behavior. On a global scale, rather than exclusively causing intron retention, a large percentage of aberrant splicing events leads to ES with a smaller fraction displaying altered 3ʹ and 5ʹ splice sites or alternative incorporation of otherwise excluded exons. Percentages differ, depending on experimental conditions and small molecule used, but it appears that intronic GC content affects the choice between ES and intron retention [42, 43]. Resistance mutations to the modulators cluster to HEAT repeats 15 and 16 on SF3B1 and also Y36 on PHF5A. The resistance mutations against the splice modulators on SF3B1 fell much further toward the C-terminus than the mutational hotspots associated with disease phenotypes. Structural studies confirmed the resistance mutations to lie around the actual drug binding site (Fig. 3) [42]. While structural data is currently limited to PlaB, the observed hourglass-shaped binding site with the diene moiety in its “neck” agrees with the idea of a common pharmacophore between the different molecular families (Fig. 3d). Drug binding to SF3B1 arrests the complex in its open conformation, thereby preventing completion of A complex formation [44] (Fig. 3a–c). While initial studies, based on the location of resistance mutations, suggested that the splicing modulators competed with the BPA, in light of structural evidence, it appears more likely that the molecules compete with the intron for the open conformation of the complex. This agrees with in vitro observations that once A complex formation is complete, PlaB loses its effect [45, 46].
While for all practical purposes, SSA, PlaB, herboxidienes and synthetic colleagues employ the same molecular mechanism, their cellular effects are not necessarily identical. Weaker binders, such as sudemycin D6 or herboxidiene tend to mainly induce ES but less intron retention compared to the more potent SSA or PlaB [42]. This again, appears to relate to intronic GC content with a lower GC percentage making the splice site more resistant to modulator activity compared to introns with high GC content. These observations strengthen the idea that not only the mechanism of action, but also relative potency of the binding molecule determine the efficacy of a particular modulator.

a Structure of SF3B1 in open conformation. b PlaB arrests SF3B1 in its open conformation. c SF3B1 in closed conformation. The closed conformation in (c) is based on structural data from B complex SF3B1, but is thought to be highly similar to its shape in A complex. SF3B1 is represented in green, PHF5A in yellow, PlaB in red, pre-mRNA in blue and U2 snRNA in orange. d Depiction of the splicing modulator binding site. Pladienolide (red) binds at the interface of SF3B1 (green) and PHF5A (yellow). Central element to the binding site is the diene moiety in the middle of PlaB (and also of spliceostatin and herboxidiene), fitting through a narrow passage between the two binding proteins. Structural information taken from PDB 5IFE (open), 6EN4 (PlaB-bound) and 6FF4 (closed conformation) [35, 44, 120]
Honorable mention
While pladienolide and SSA’s effect on splicing were serendipitous discoveries, isoginkgetin not only stands out from the molecules discussed thus far as a plant natural product, isolated from leaf extracts of Ginkgo biloba [47], it is also the first actively sought splicing inhibitor (Fig. 1c). Using a reporter with the luciferase coding sequence sitting downstream of a canonical splice site, isoginkgetin emerged from screening a chemical library. While thought to prevent binding of the U4/U5/U6 tri-snRNP to A complex, its mechanism has not been described in as much detail as the SF3B1-targeting compounds, likely also owing to much weaker activity. To this day its binding site remains unknown [48].
Recent studies suggest that isoginkgetin not only inhibits the proteasome [49] but also transcriptional elongation, which may indicate that its cellular effect is much messier than originally anticipated [50]. Especially the latter issue may be difficult to resolve. The original study showed stalled B complex formation in vitro requiring no de novo pre-mRNA synthesis, while the work on transcriptional elongation relied on directional transcriptome analysis. It remains possible that the different behavior on RNA polymerase observed between SF3B targeting molecules and isoginkgetin reflects differences in mechanism with isoginkgetin blocking splicing at a later step, producing a different cellular phenotype. A more recent study on hinokiflavone, a different plant flavonoid, reported inhibition of the SENP de-SUMOylation enzyme, albeit at fairly high concentrations. While detailed mechanistic data is still lacking, the report suggested that hyper-SUMOylation of splicing factors may interfere with proper B complex assembly [51].
Cellular consequences
Since splicing constitutes a fundamental regulatory step at the upstream end of gene expression the downstream cellular consequences of splicing manipulation are numerous. Many different aspects of cellular dysregulation brought about by splicing modulation may each contribute to the observed anticancer activity of these molecules (Table 1).
Besides changing splicing patterns and causing intron retention, changed splicing behavior also leads to alterations in RNA production itself [52, 53]. Genome wide transcription decreased ~20–30% upon SSA treatment [54]. Owing to altered splicing of some effector proteins such as IκBα, ERK and NF-κB signaling pathways displayed increased activity [27, 55]. Furthermore, splicing perturbation leads to slowed RNA polymerase II (RNAPII) progression with the enzyme increasingly failing to reach the end of its transcripts and concomitant decrease in Ser-2 phosphorylation on the RNAPII C-terminal domain [56, 57].
It is important to keep in mind that the snRNPs are not present in stoichiometric proportions, but that U1 concentration far exceeds the level of U2 or U4/U5/U6. Besides marking the 5ʹ splice site, U1 protects mRNA from premature cleavage and poly-adenylation [58, 59]. A recent study observed that SSA treatment led to aberrant poly-adenylation in the MALAT1 (Metastasis Associated Lung Adenoma Transcript 1), a long non-coding RNA (lncRNA), as well as a few canonical mRNA transcripts [60]. MALAT1 is the most abundantly expressed lncRNA in nuclear speckles [61]. Its elevated expression is commonly observed in metastasis-prone tumors [62]. MALAT1 plays an important role in regulating transcription, alternative splicing and posttranscriptional processing [63]. It is known to bind a number of splicing factors, including SRSF1 and hnRNPC, modulating their activities [64, 65] Under normal circumstances MALAT1 is strictly located in the nucleus, but under SSA treatment its polyadenylated form is exported to the cytoplasm. While the currently known splicing modulators do not directly alter U1 activity, impeded U2 function and the subsequent buildup of improperly processed transcripts seem to eventually exhaust the pool of available U1 snRNP and cause further cellular effects from an unexpected source. Importantly, aberrant poly-adenylation was only observed in presence of nonfunctional U2 snRNP, but not when U2 itself was depleted. The acronym MALAT1’s meaning indicates the transcript’s association with cancer [66,67,68]. Whether its aberrant poly-adenylation and export to the cytosol has any bearing on the antitumor activity of splicing modulators or whether it presents a phenomenon of limited scope and relevance remains to be seen, especially when considering that only a fraction of MALAT1 undergoes poly-adenylation.
The sheer amount of misspliced RNAs appears to overwhelm quality control mechanisms, including nonsense-mediated decay. Unspliced pre-mRNA accumulates in the nucleus, with nuclear speckles also increasing in size [27, 69]. Some improperly processed transcripts escape to the cytoplasm and are translated into protein. Initial studies observed production of truncated polypeptides, including a shortened, constitutively active version of the cell cycle regulator p27, labeled p27* [70,71,72]. p27* production and subsequent G1 cell cycle arrest likely account for part of the observed antitumor effect, though it does not explain the full extent of specific toxicity toward some cell lines [73].
Specificity likely derives from a particular tumor’s transcriptome and splicing state. Beyond SF3B1 itself many other splicing factor mutations have been implicated in hematopoietic malignancies. U2AF, SRSF1 and 2 or RBM39 may contain mutually exclusive point mutations on specific residues affecting splice site selection [74, 75]. Cancer cells bearing splicing factor mutations appear particularly sensitive to splicing modulators, with two perturbations in the same cellular system proving synthetic lethal [75,76,77,78,79].
Splice isoforms of VEGF may stimulate or inhibit angiogenesis, depending on whether exon 8 is spliced at a proximal (pro-angiogenic) or a distal (anti-angiogenic) splice site [80,81,82]. SSA treatment leads to a decrease in VEGF expression in malignant tumors, another aspect that may explain part of the antitumor activity of splicing modulators [54].
In a similar vein, CLL relies on high levels of the Bcl2 family apoptosis regulator Mcl-1 for survival. Mcl-1’s long splice isoform (Mcl-1L) has antiapoptotic activity, while the short variant (Mcl-1S) is pro-apoptotic. Splicing modulators appear to favor production of the short isoform [83, 84].
Yet another aspect in the relation between splicing and cancer has garnered attention in recent years, as myc-dependent tumors appear particularly sensitive to splicing modulators. One link connects the arginine methyl transferase PRMT5, which acts on the Sm proteins [85]. It appears that Sm protein assembly aids in the splicing of introns with weak 5ʹ splice sites, leading to increased ES. In addition, myc also drives expression of splicing factor SRSF1, an oncogenic protein in its own right [86, 87].
Some myc-driven tumors seem particularly sensitive to small molecule splicing inhibitors, possibly because the small molecule shifts an already precarious balance in splicing toward acute cytotoxicity [70, 85, 88, 89].
Perhaps focusing on isolated cells in tissue culture alone is not sufficient to appreciate the physiological role and clinical potential of splicing modulators. A recent study observed formation of double-stranded RNA species from intronic sequences upon treatment with sudemycin D6 or the more recently described synthetic PlaB derivative H3B-8800 in myc-driven triple-negative breast cancer [70]. These double-stranded RNAs appear to trigger cellular antiviral immune responses and subsequent cell death.
With mutated splicing factors, leading to altered 5ʹ and 3ʹ splice sites, proteome composition significantly changes, potentially providing a generous source of neoantigens [90,91,92]. A study of 32 TCGA cancer types from 8705 patients investigated the effect of alternative splicing on cancer-specific markers presented on cellular MHC-I antigen presenting complexes [91]. It found a number of tumor-specific markers absent in normal cells, which may act as neoantigens and allow development of mRNA vaccines or other immunotherapies [93]. Splicing modulation very likely alters proteome content and therefore also presented antigens, providing a potential source for immunotherapy. Data generated with isoginkgetin indicates that splicing modulation does lead to presentation of neoantigens on MHC-I complexes [94].
It was appreciated early on that splicing modulators might not only prove potent bioprobes in the laboratory but also provide clinical benefits, especially in cancer treatment. The use of splicing modulators in treating malignancies might be a case of needing to find a suitable disease for the right medication.
Clinical trials
The first splicing modulator to enter the clinic was pladienolide’s semisynthetic derivative E7107. The molecule’s development preceded the identification of its molecular target [95] and E7107 initially showed promise in xenograft models, displaying a wide therapeutic window. Human trials against solid tumors were halted owing to dose-limiting toxicity but showed little or no clinical benefit [96, 97], though splicing modulation was observed.
Given that splicing modulators appear most efficacious against hematopoietic cancers, one could argue that the initial E7107 trials were using the right medication on the wrong disease. While research on E7107 seems to continue, it appears that focus is shifting to a different compound [98].
A more recently published synthetic pladienolide derivative may hold more promise. H3B-8800 is orally bioavailable and appears efficacious against tumors with splicing factor mutations, not limited to SF3B1 but also encompassing U2AF and SRSF2 [76]. The study reiterated differences in physiological behavior even between two related molecules with identical mechanisms of action. H3B-8800 and E7107 displayed different behavior in cell killing and splicing modulation with H3B-8800 preferentially retaining short (<300 bp) GC-rich introns with a weak BPS. These preferences did not appear as pronounced in the experimental system when using E7107. The authors argued that H3B-8800’s selectivity for short GC-rich introns especially impacted expression of other splicing regulators, including U2AF, thereby further disrupting proper mRNA processing.
Initial results from phase I clinical trials against MDS, AML and CML indicated that H3B-8800 acts on its target at similar doses as in preclinical xenograft studies [99] and leads to altered mRNA splicing. However, while the drug appeared to act as expected, clinical responses to splicing modulator treatment were not observed. Trials are still in progress with completion expected by 2022.
With the varied effect on gene expression, depending on the chosen inhibitor and its cellular context, predicting the benefit of splicing modulation for a particular disease proves exceedingly difficult and necessitates the search for suitable prognostic markers. Perhaps, future promise lies in more specific, targeted inhibitors working on individual splicing factors, rather than throwing the entire pre-mRNA processing out of whack.
New tools
The identification of the sulfonamide anticancer drug indisulam points in a promising direction (Fig. 1c). Originally identified in a screen for molecules with antitumor activity [100, 101], indisulam appeared efficacious in xenograft models [102]. In clinical trials against solid tumors, indisulam elicited a response in some 10% of patients with no means of predicting which group of people treated would benefit from taking sulfonamides. Two independent research efforts, using a forward genetic [103] or proteomic approach [104] respectively, realized that indisulam and related sulfonamides E7820, tasisulam and CQS act as a molecular glue, tying the U2AF-related splicing factor RBM39 to DCAF15, an adaptor protein of the CUL4-DDB1-DDA1-DCAF15 E3 ubiquitin ligase complex (Fig. 4a). This association leads to RBM39 polyubiquitination and proteasome-dependent degradation. RBM39 has been identified as a coactivator for several transcription factors and a regulator of receptor-dependent alternative splicing, including splicing of VEGF [105, 106].

a Depiction of indisulam acting as a molecular glue, targeting RBM39 for proteasomal degradation. b Simplified depiction of the mechanism of branaplam (NVS-SM1) in increasing SMN2 exon 7 splicing efficiency. Risdiplam is thought to act in the same manner.
In absence of the sulfonamide DCAF15 and RBM39 have no affinity toward each other. Under indisulam treatment intron retention and ES were observed with only a few cases of changed 3ʹ and 5ʹ splice sites [103], as well as changes in the splice pattern of VEGF [104]. Mutational and structural studies identified a degron motif present in RBM39 and also RBM23, another U2AF-related splicing factor, which destined RBM23 and RBM39 to sulfonamide-dependent proteasomal degradation. In clinical phase II trials, about 1/3 of patients with chronic or relapsed acute myeloid leukemia (AML) responded to indisulam in combination with the nucleotide analogue cytarabine and DNA topoisomerase inhibitor idarubicin [107]. Perhaps, using the emerging targeted protein degradation technology to individually destroy a particular splicing factor holds promise in providing specific therapies with fewer side effects and more predictable clinical behavior.
RNA-binding modulators
In addition, a further field of small molecule modulators in splicing has emerged, relying on compounds that primarily interact with RNA, not protein [108]. It was this area of investigation that led to the first clinically approved therapeutics altering splicing behavior to treat spinal muscular atrophy (SMA) [109].
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disorder, occurring in about 1 out of 11,000 life births. A loss-of-function mutation in the survival of motor neurons 1 (SMN1) gene prevents cells from producing sufficient levels of SMN protein, essential for normal development and cellular homeostasis, not limited to motor neurons. A second gene, SMN2, is present in human cells, but a translationally silent C to T mutation at the 5ʹ end of exon 6 produces a weak splice site, leading to exon 7 exclusion. Consequently, only ~10% of functional SMN protein is produced, as resultant SMNΔ7 protein without exon 7 is unstable and inactive. Chemical screening for molecules enhancing splicing from a reporter gene containing the SMN2 exon6-intron7 splice site followed by optimization through medicinal chemistry led to NVS-SM1, the first molecule to specifically alter splicing behavior at a particular splice site (Fig. 1c). NVS-SM1, also labeled branaplam, enhances the interaction between the SMN2 pre-mRNA and the U1 snRNA, thereby turning a weak splice site into a strong one (Fig. 4b). While branaplam is still in clinical trials, three further therapies against SMA have gained FDA approval in recent years.
The first approved medication, nusinersen, relies on anti-sense oligonucleotides (ASO), and while efficacious, requires repeated intrathecal injections, making prolonged treatment expensive, risky and somewhat impractical. The nusinersen ASO acts by displacing the hnRNP A1/A2 splicing repressor from intron 7 of the SMN2 gene, enhancing exon 7 inclusion [110]. The second approved treatment, marketed as zolgensma, is a gene therapy, delivering functional SMN1 protein through an adeno-associated virus vector, aimed at infants under 2 years of age [111]. Branaplam phase 1 and 2 clinical trials were halted over toxicity concerns from an animal study but seem to have meanwhile resumed again (clinicaltrials.gov NCT02268552). At the beginning of 2021, branaplam did receive orphan drug status from the FDA, though as a treatment for Huntington’s disease. With branaplam trials halted over safety concerns, risdiplam, a competing compound became the first FDA approved oral medication for SMA. Likely sharing the same general mechanism with branaplam, risdiplam constitutes the result of extensive structural optimization, which seems to have yielded a molecule with higher specificity, less off-target toxicity and a more favorable pharmacokinetic profile [112].
While branaplam and risdiplam represent the best studied examples of compounds specifically changing splicing behavior at a particular exon, a recent report identified a small molecule, dubbed RECTAS, changing splicing behavior of the IKAP gene involved in familial dysautonomia, a hereditary sensory neuropathy [113]. Judging by its activity in vitro RECTAS may very well interact directly with the splicing or RNA-processing machinery. Recent findings indicated RECTAS’s potential in the treatment of Parkinson’s disease [114], however, its target still awaits discovery [115].
Outlook
The discussed molecules constitute by no means the only way of pharmacologically changing splicing behavior. A whole body of research has evaluated the effects of CLK and DYRK kinase inhibitors on changing SR protein behavior and thereby alternative splicing. These signaling modulator show some promise in treating several diseases caused by underlying splicing defects, including Duchenne muscular dystrophy, familial dysautonomia and certain types of cystic fibrosis [116, 117]. A discussion of the role of signal transduction in splicing deserves its own review [118].
From a medicinal point of view, RNA specific medications and molecules exploiting the protein degradation machinery point in the future direction of chemical splicing modulation. This activity is not limited to mammalian mRNA processing, but promising results have also been obtained with molecules targeting group II self-splicing introns in pathogenic yeast [119], potentially providing a pathogen specific target to fight opportunistic Candida parapsilosis infections. While drug resistance should not become an issue in treating muscular atrophy, one needs to keep in mind that against cancer increasing specificity in mechanism also increases the chances of drug-specific resistance. Therefore, the potential of relatively broad inhibitors acting on novel targets should not be underestimated.
Splicing modulators provide great study objects for the promises and challenges in chemical biology. They display both the power of natural products as well as the necessity for medicinal chemistry in providing sufficient quantities of stable and potent molecular effectors. Furthermore, they demonstrate how structurally disparate molecules may share a binding site and mechanism of action, yet how small differences in binding may bring about very different cellular outcomes. With splicing modulators perturbing a fundamental process in gene expression control, the need to understand the cellular effects beyond the interaction between drug and target molecule becomes ever more apparent. Even that may not suffice when trying to understand the workings of a small molecule in an organismal context. The next years will show whether splicing modulators targeting SF3B will mature into efficacious therapeutics. In laboratory research, they have already proven themselves powerful bioprobes into the basis of gene expression and we can expect them to provide powerful tools for future discoveries.
References
Isaac BG, Ayer SW, Elliott RC, Stonard RJ. Herboxidiene - a Potent Phytotoxic Polyketide from Streptomyces Sp A7847. J Org Chem. 1992;57:7220–6.
Miller-Wideman M, Makkar N, Tran M, Isaac B, Biest N, Stonard R. Herboxidiene, a new herbicidal substance from Streptomyces chromofuscus A7847. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J Antibiot (Tokyo). 1992;45:914–21.
Sakai Y, Tsujita T, Akiyama T, Yoshida T, Mizukami T, Akinaga S, et al. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J Antibiot (Tokyo). 2002;55:863–72.
Sakai Y, Yoshida T, Ochiai K, Uosaki Y, Saitoh Y, Tanaka F, et al. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. I. Taxonomy, production, isolation, physicochemical properties and biological activities. J Antibiot (Tokyo). 2002;55:855–62.
Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, et al. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo). 1996;49:1204–11.
Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo). 1996;49:1196–203.
Motoyoshi H, Horigome M, Ishigami K, Yoshida T, Horinouchi S, Yoshida M, et al. Structure-activity relationship for FR901464: a versatile method for the conversion and preparation of biologically active biotinylated probes. Biosci Biotechnol Biochem. 2004;68:2178–82.
Seki-Asano M, Okazaki T, Yamagishi M, Sakai N, Takayama Y, Hanada K, et al. Isolation and characterization of a new 12-membered macrolide FD-895. J Antibiot (Tokyo). 1994;47:1395–401.
Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo). 2004;57:188–96.
Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation. J Antibiot (Tokyo). 2004;57:180–7.
Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. I. Taxonomy, fermentation, isolation and screening. J Antibiot (Tokyo). 2004;57:173–9.
Ju J, Rajski SR, Lim SK, Seo JW, Peters NR, Hoffmann FM, et al. Lactimidomycin, Iso-migrastatin and Related Glutarimide-Containing 12-Membered Macrolides Are Extremely Potent Inhibitors of Cell Migration. J Am Chem Soc. 2009;131:1370–71.
Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–17.
Albert BJ, Sivaramakrishnan A, Naka T, Koide K. Total synthesis of FR901464, an antitumor agent that regulates the transcription of oncogenes and tumor suppressor genes. J Am Chem Soc. 2006;128:2792–3.
Thompson CF, Jamison TF, Jacobsen EN. FR901464: Total synthesis, proof of structure, and evaluation of synthetic analogues. J Am Chem Soc. 2001;123:9974–83.
Horigome M, Motoyoshi H, Watanabe H, Kitahara T. A synthesis of FR901464. Tetrahedron Lett. 2001;42:8207–10.
Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, Koide K. Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J Am Chem Soc. 2007;129:2648–59.
He H, Ratnayake AS, Janso JE, He M, Yang HY, Loganzo F, et al. Cytotoxic Spliceostatins from Burkholderia sp. and Their Semisynthetic Analogues. J Nat Prod. 2014;77:1864–70.
Liu X, Biswas S, Berg MG, Antapli CM, Xie F, Wang Q, et al. Genomics-guided discovery of thailanstatins A, B, and C As pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J Nat Prod. 2013;76:685–93.
Donaldson WA. Syntheses of spliceostatins and thailanstatins: a review. Beilstein J Org Chem 2020;16:1991–2006.
Kakeya H, Kaida D, Sekiya H, Nagai K, Yoshida M, Osada H. RQN-18690A (18-deoxyherboxidiene) targets SF3b, a spliceosome component, and inhibits angiogenesis. J Antibiot (Tokyo). 2016;69:121–3.
Lagisetti C, Pourpak A, Jiang Q, Cui X, Goronga T, Morris SW, et al. Antitumor compounds based on a natural product consensus pharmacophore. J Med Chem. 2008;51:6220–4.
Lagisetti C, Pourpak A, Goronga T, Jiang Q, Cui X, Hyle J, et al. Synthetic mRNA splicing modulator compounds with in vivo antitumor activity. J Med Chem. 2009;52:6979–90.
Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011;278:4870–80.
Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol. 2011;6:582–9.
Lagisetti C, Palacios G, Goronga T, Freeman B, Caufield W, Webb TR. Optimization of antitumor modulators of pre-mRNA splicing. J Med Chem. 2013;56:10033–44.
Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3:576–83.
Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570–5.
Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 2008;14:802–13.
Liu G, Mattick JS, Taft RJ. A meta-analysis of the genomic and transcriptomic composition of complex life. Cell Cycle. 2013;12:2061–72.
Kaida D, Schneider-Poetsch T, Yoshida M. Splicing in oncogenesis and tumor suppression. Cancer Sci. 2012;103:1611–6.
Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 2016;22:350–9.
Urlaub H, Raker VA, Kostka S, Luhrmann R. Sm protein-Sm site RNA interactions within the inner ring of the spliceosomal snRNP core structure. EMBO J. 2001;20:187–96.
Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Disco. 2012;11:847–59.
Zhang Z, Will CL, Bertram K, Dybkov O, Hartmuth K, Agafonov DE, et al. Molecular architecture of the human 17S U2 snRNP. Nature. 2020;583:310–3.
Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13:1033–45.
Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615.
Yin S, Gambe RG, Sun J, Martinez AZ, Cartun ZJ, Regis FFD, et al. A Murine Model of Chronic Lymphocytic Leukemia Based on B Cell-Restricted Expression of Sf3b1 Mutation and Atm Deletion. Cancer Cell. 2019;35:283–96.e5.
Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, et al. Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol Cell. 2019;76:82–95.e7.
Effenberger KA, Anderson DD, Bray WM, Prichard BE, Ma N, Adams MS, et al. Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J Biol Chem. 2014;289:1938–47.
Hasegawa M, Miura T, Kuzuya K, Inoue A, Won KiS, Horinouchi S, et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol. 2011;6:229–33.
Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun. 2017;8:15522.
Amit M, Donyo M, Hollander D, Goren A, Kim E, Gelfman S, et al. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012;1:543–56.
Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, et al. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol Cell. 2018;70:265–73.e8.
Bertram K, Agafonov DE, Dybkov O, Haselbach D, Leelaram MN, Will CL, et al. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell. 2017;170:701–13. e11
Plaschka C, Lin PC, Nagai K. Structure of a pre-catalytic spliceosome. Nature. 2017;546:617–21.
Briancon-Scheid F, Lobstein-Guth A, Anton R. HPLC Separation and Quantitative Determination of Biflavones in Leaves from Ginkgo biloba. Planta Med. 1983;49:204–7.
O’Brien K, Matlin AJ, Lowell AM, Moore MJ. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J Biol Chem. 2008;283:33147–54.
Tsalikis J, Abdel-Nour M, Farahvash A, Sorbara MT, Poon S, Philpott DJ, et al. Isoginkgetin, a Natural Biflavonoid Proteasome Inhibitor, Sensitizes Cancer Cells to Apoptosis via Disruption of Lysosomal Homeostasis and Impaired Protein Clearance. Mol Cell Biol. 2019;39:e00489–18.
Boswell SA, Snavely A, Landry HM, Churchman LS, Gray JM, Springer M. Total RNA-seq to identify pharmacological effects on specific stages of mRNA synthesis. Nat Chem Biol. 2017;13:501–7.
Pawellek A, Ryder U, Tammsalu T, King LJ, Kreinin H, Ly T, et al. Characterisation of the biflavonoid hinokiflavone as a pre-mRNA splicing modulator that inhibits SENP. Elife. 2017;6:e27402.
Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012;22:1616–25.
Wu G, Fan L, Edmonson MN, Shaw T, Boggs K, Easton J, et al. Inhibition of SF3B1 by molecules targeting the spliceosome results in massive aberrant exon skipping. RNA. 2018;24:1056–66.
Furumai R, Uchida K, Komi Y, Yoneyama M, Ishigami K, Watanabe H, et al. Spliceostatin A blocks angiogenesis by inhibiting global gene expression including VEGF. Cancer Sci. 2010;101:2483–9.
Khan K, Schneider-Poetsch T, Ishfaq M, Ito A, Yoshimoto R, Mukaida N, et al. Splicing inhibition induces gene expression through canonical NF-kappaB pathway and extracellular signal-related kinase activation. FEBS Lett. 2014;588:1053–7.
Koga M, Satoh T, Takasaki I, Kawamura Y, Yoshida M, Kaida D. U2 snRNP is required for expression of the 3’ end of genes. PLoS ONE. 2014;9:e98015.
Sousa-Luis R, Dujardin G, Zukher I, Kimura H, Weldon C, Carmo-Fonseca M, et al. POINT technology illuminates the processing of polymerase-associated intact nascent transcripts. Mol Cell. 2021;81:1935–50.e6.
Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–8.
Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64.
Yoshimoto R, Chhipi-Shrestha JK, Schneider-Poetsch T, Furuno M, Burroughs AM, Noma S, et al. Spliceostatin A interaction with SF3B limits U1 snRNP availability and causes premature cleavage and polyadenylation. Cell Chem Biol. 2021;S2451-9456:00111-2.
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38.
Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–41.
Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159:188–99.
Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29:3082–93.
Yang F, Yi F, Han X, Du Q, Liang Z. MALAT-1 interacts with hnRNP C in cell cycle regulation. FEBS Lett. 2013;587:3175–81.
Arun G, Diermeier S, Akerman M, Chang KC, Wilkinson JE, Hearn S, et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016;30:34–51.
Amodio N, Stamato MA, Juli G, Morelli E, Fulciniti M, Manzoni M, et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia. 2018;32:1948–57.
Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–9.
Yoshimoto R, Kaida D, Furuno M, Burroughs AM, Noma S, Suzuki H, et al. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA. 2017;23:47–57.
Bowling EA, Wang JH, Gong F, Wu W, Neill NJ, Kim IS, et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021;184:384–403.e21.
Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015;29:63–80.
Carvalho T, Martins S, Rino J, Marinho S, Carmo-Fonseca M. Pharmacological inhibition of the spliceosome subunit SF3b triggers exon junction complex-independent nonsense-mediated decay. J Cell Sci. 2017;130:1519–31.
Satoh T, Kaida D. Upregulation of p27 cyclin-dependent kinase inhibitor and a C-terminus truncated form of p27 contributes to G1 phase arrest. Sci Rep. 2016;6:27829.
Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22:672–8.
Lee SC, North K, Kim E, Jang E, Obeng E, Lu SX, et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell. 2018;34:225–41.e8.
Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24:497–504.
Shirai CL, White BS, Tripathi M, Tapia R, Ley JN, Ndonwi M, et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat Commun. 2017;8:14060.
Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell. 2016;30:404–17.
Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell. 2019;35:369–84.e7.
Nowak DG, Woolard J, Amin EM, Konopatskaya O, Saleem MA, Churchill AJ, et al. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci. 2008;121:3487–95. (Pt 20).
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9.
Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62:4123–31.
Larrayoz M, Blakemore SJ, Dobson RC, Blunt MD, Rose-Zerilli MJ, Walewska R, et al. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia. 2016;30:351–60.
Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010;142:625–36.
Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96–100.
Das S, Anczukow O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012;1:110–7.
Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220–8.
Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–8.
Hepburn LA, McHugh A, Fernandes K, Boag G, Proby CM, Leigh IM, et al. Targeting the spliceosome for cutaneous squamous cell carcinoma therapy: a role for c-MYC and wild-type p53 in determining the degree of tumour selectivity. Oncotarget. 2018;9:23029–46.
Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, et al. Intron retention is a source of neoepitopes in cancer. Nat Biotechnol. 2018;36:1056–8.
Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell. 2018;34:211–24.e6.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–21.
Pardi N, Hogan MJ, Weissman D. Recent advances in mRNA vaccine technology. Curr Opin Immunol. 2020;65:14–20.
Darrigrand R, Pierson A, Rouillon M, Renko D, Boulpicante M, Bouyssie D, et al. Isoginkgetin derivative IP2 enhances the adaptive immune response against tumor antigens. Commun. Biol. 2021;4:269.
Iwata M, Ozawa Y, Uenaka T, Shimizu H, Nijima J, Kanada RM, et al. E7107, a new 7-urethane derivate of pladienolide D, diplays curative effect against several human tumor xenografts. Experimental and Molecular Therapeutics 27: Natural Products: Proc Amer Assoc Cancer Res; 2004. p. Supplement.
Eskens FA, Ramos FJ, Burger H, O’Brien JP, Piera A, de Jonge MJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013;19:6296–304.
Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig N Drugs. 2014;32:436–44.
Praveen Sridhar SC, Ying0Jie Lock, Fabio Petrocca. Abstract 420: Preclinical evaluation of te SF3B1 inhibitor E7107 in triple negative breast cancer. AACR Annual Meeting 2017. Washington, DC: Cancer Research: American Association for Cancer Research; 2017.
Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS, et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood. 2019;134:673.
Owa T, Yoshino H, Okauchi T, Yoshimatsu K, Ozawa Y, Sugi NH, et al. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J Med Chem. 1999;42:3789–99.
Fukuoka K, Usuda J, Iwamoto Y, Fukumoto H, Nakamura T, Yoneda T, et al. Mechanisms of action of the novel sulfonamide anticancer agent E7070 on cell cycle progression in human non-small cell lung cancer cells. Investig N. Drugs. 2001;19:219–27.
Ozawa Y, Sugi NH, Nagasu T, Owa T, Watanabe T, Koyanagi N, et al. E7070, a novel sulphonamide agent with potent antitumour activity in vitro and in vivo. Eur J Cancer. 2001;37:2275–82.
Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356:eaal3755.
Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol. 2017;13:675–80.
Huang G, Zhou Z, Wang H, Kleinerman ES. CAPER-alpha alternative splicing regulates the expression of vascular endothelial growth factor(1)(6)(5) in Ewing sarcoma cells. Cancer. 2012;118:2106–16.
Dowhan DH, Hong EP, Auboeuf D, Dennis AP, Wilson MM, Berget SM, et al. Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERalpha and CAPERbeta. Mol Cell. 2005;17:429–39.
Assi R, Kantarjian HM, Kadia TM, Pemmaraju N, Jabbour E, Jain N, et al. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer. 2018;124:2758–65.
Campagne S, Boigner S, Rudisser S, Moursy A, Gillioz L, Knorlein A, et al. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat Chem Biol. 2019;15:1191–8.
Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11:511–7.
Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–48.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl J Med. 2017;377:1713–22.
Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J Med Chem. 2018;61:6501–17.
Slaugenhaupt SA, Mull J, Leyne M, Cuajungco MP, Gill SP, Hims MM, et al. Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum Mol Genet. 2004;13:429–36.
Boussaad I, Obermaier CD, Hanss Z, Bobbili DR, Bolognin S, Glaab E, et al. A patient-based model of RNA mis-splicing uncovers treatment targets in Parkinson’s disease. Sci Transl Med. 2020;12:eaau3960.
Yoshida M, Kataoka N, Miyauchi K, Ohe K, Iida K, Yoshida S, et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc Natl Acad Sci USA. 2015;112:2764–9.
Sako Y, Ninomiya K, Okuno Y, Toyomoto M, Nishida A, Koike Y, et al. Development of an orally available inhibitor of CLK1 for skipping a mutated dystrophin exon in Duchenne muscular dystrophy. Sci Rep. 2017;7:46126.
Shibata S, Ajiro M, Hagiwara M. Mechanism-Based Personalized Medicine for Cystic Fibrosis by Suppressing Pseudo Exon Inclusion. Cell Chem Biol. 2020;27:1472–82.e6.
Ohe K, Hagiwara M. Modulation of alternative splicing with chemical compounds in new therapeutics for human diseases. ACS Chem Biol. 2015;10:914–24.
Fedorova O, Jagdmann GE Jr, Adams RL, Yuan L, Van Zandt MC. Pyle AM. Small molecules that target group II introns are potent antifungal agents. Nat Chem Biol. 2018;14:1073–8.
Haselbach D, Komarov I, Agafonov DE, Hartmuth K, Graf B, Dybkov O, et al. Structure and Conformational Dynamics of the Human Spliceosomal B(act) Complex. Cell. 2018;172:454–64.e11.
Acknowledgements
MY was supported in part by a Grant-in-Aid for Scientific Research (S) (JP19H05640) from the Japan Society for the Promotion of Science (JSPS) and a Grant-in-Aid for Scientific Research on Innovative Areas (JP18H05503) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). JKCS was supported by a Grant-in-Aid for Early-Career Scientists (JP20K15420) from JSPS. This work was inspired by the JSPS Asian Chemical Biology Initiative.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schneider-Poetsch, T., Chhipi-Shrestha, J.K. & Yoshida, M. Splicing modulators: on the way from nature to clinic. J Antibiot 74, 603–616 (2021). https://doi.org/10.1038/s41429-021-00450-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-021-00450-1
This article is cited by
-
Alternative splicing in lung influences COVID-19 severity and respiratory diseases
Nature Communications (2023)
-
Non-canonical functions of spliceosome components in cancer progression
Cell Death & Disease (2023)
-
CDK11 regulates pre-mRNA splicing by phosphorylation of SF3B1
Nature (2022)
