
Abstract
Liposidomycin is a uridyl liponucleoside antibiotic isolated from Streptomyces griseosporeus RK-1061. It was discovered by Isono in 1985, who had previously isolated and developed a related peptidyl nucleoside antibiotic, polyoxin, a specific inhibitor of chitin synthases, as a pesticide. He subsequently isolated liposidomycin, a specific inhibitor of bacterial peptidoglycan biosynthesis from actinomycetes, using a similar approach to the discovery of polyoxin. Liposidomycin has no cytotoxicity against BALB/3T3 cells but has antimicrobial activity against Mycobacterium spp. through inhibition of MraY (MurX) [phospho-N-acetylmuramoyl-pentapeptide transferase (translocase I, EC 2.7.8.13)]. Since the discovery of liposidomycin, several liposidomycin-type antibiotics, including caprazamycin, A-90289, and muraminomycin, have been reported, and their total synthesis and/or biosynthetic cluster genes have been studied. Most advanced, a semisynthetic compound derived from caprazamycin, CPZEN-45, is being developed as an antituberculosis agent. Translocase I is an interesting and tractable molecular target for new antituberculosis and antibiotic drug discovery against multidrug-resistant bacteria. This review is dedicated to Dr Isono on the occasion of his 88th birthday to recognize his role in the study of nucleoside antibiotics.
Similar content being viewed by others
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, has long been considered as one of the most serious infectious diseases. Worldwide, TB is one of the top ten causes of death, and millions of people continue to become ill with TB every year. In 2017, TB caused an estimated 1.3 million deaths among HIV-negative people. Globally, the best estimate is that 10 million people developed TB in 2017: 5.8 million men, 3.2 million women, and 1.0 million children [1]. The first-line antituberculosis drugs for adults recommended by the World Health Organization (WHO) are isoniazid, rifampicin, pyrazinamide, ethambutol, and streptomycin. TB resistance to first-line therapy has increased in recent years, due in-part to long treatment times and poor patient compliance increasing the need for new therapeutic options [2]. In 2017, WHO published its first ever list of antibiotic-resistant, “priority pathogens,” a catalog of 12 bacterial families that pose the greatest threat to human health. They include well-known gram-positive organisms such as Enterococcus faecium and methicillin-resistant Staphylococcus aureus. Thus, it is important to identify novel biological targets and develop new compounds for clinical use against TB and/or against multidrug-resistant (MDR) bacteria [3].
Isono and his coworkers (Antibiotic Laboratory, RIKEN), who isolated and developed the nucleoside pesticide polyoxin in 1965 [4], studied and published comprehensive reviews on nucleoside antibiotics [5, 6]. Dr Isono has also explored the isolation of specific nucleoside inhibitors against bacterial peptidoglycan, modeled after polyoxin, using as substrates— Escherichia coli Y-10 particulate enzyme, UDP-MurNAc pentapeptide from Bacillus cereus, and 14C-UDP-GlcNAc. In 1985, he and his collaborators, including myself, reported the first specific nucleoside inhibitor against peptidoglycan biosynthesis that acted through inhibition of MraY [phospho-N-acetylmuramoyl-pentapeptide-transferase (translocase I, EC 2.7.8.13)], and named it liposidomycin based on its structure [7,8,9,10,11].
This review summarizes the history of peptidoglycan inhibitors, with the discovery and study of liposidomycin and related compounds that are liposidomycin-type liponucleosides that target MraY. Dr Isono’s vision and goal may be realized through this work, because the liposidomycin analog caprazamycin [12] has new antibiotic potential against M. tuberculosis, and CPZEN-45 [13], a new derivative that overcomes the disadvantage of caprazamycin, is a clinical candidate for anti-TB use in the near future.
Bacterial peptidoglycan biosynthesis and peptidoglycan inhibitor molecules
Bacterial peptidoglycan biosynthesis has been a promising therapeutic target for antibiotics ever since the discovery of penicillin in 1929 [14], because no counterpart to bacterial peptidoglycan exists in eukaryotic cells [10, 11, 15]. Bacterial peptidoglycan comprises a β-1,4-linked glycan of alternating N-acetyl-glucosamine (GlcNAc) and N-acetyl-muramic acid (MurNAc) sugars. UDP-GlcNAc is used for the assembly of both peptidoglycan in bacteria and chitin in fungi. UDP-MurNAc is biosynthesized from UDP-GlcNAc by the addition of an enolpyruvyl group to the 3’-hydroxyl, via transferase MurA (UDP-GlcNAc enol pyruvate transferase) [16], followed by reduction to a lactyl sidechain by reductase MurB (UDP-N-acetylglucosamine-enolpyruvate reductase) [17]. Fosphomycin (formerly, phosphonomycin) [18] inhibits Mur A (Fig. 1a).

Bacterial peptidoglycan biosynthesis (a) and MraY (tranlocase I) reaction (b)
Assembly of the cytoplasmic peptidoglycan precursor UDP-MurNAc-L-Ala-D-Glu-L-Lys is accomplished using a series of ATP-dependent respective amino acid ligases (MurC, MurD, and MurE) that add l-alanine, d-glutamic acid, and l-lys sequentially to the lactyl side chain of UDP-MurNAc. The final two amino acids (D-Ala-D-Ala) are synthesized by D-Ala-D-Ala ligase (Ddl), and this completes the synthesis of UDP-MurNAc-L-Ala-D-Glu-L-Lys-D-Ala-D-Ala (Park’s nucleotide) through the action of the UDP-MurNAc-L-Ala-D-Glu-L-Lys (or meso-DAP):D-Ala-D-Ala (MurF). D-Cycloserine (D-4-amino-3-isoxazolidone) [19] inhibits both Ddl and alanine racemase (Alr), which is an enzyme catalyzing the conversion of l-alanine to d-alanine [20] (Fig. 1a).
The first step in the intramembrane reactions is the transfer of phospho-MurNAc-pentapeptide from UDP-MurNAc-pentapeptide to undecaprenyl phosphate (C55-P), catalyzed by translocase I (MraY) (Fig. 1a, b). The reaction is reversible, requires the presence of Mg2+ ions, and leads to the formation of undecaprenyl-diphospho-MurNAc-pentapeptide (lipid I) [21]. Bacterial genome sequencing revealed that only one copy of the mraY gene exists in E. coli. Uridyl liponucleoside antibiotics, such as tunicamycin [22], liposidomycin [7], and caprazamycin [12], inhibit this enzyme. Similar liponucleoside antibiotics, such as A-84830A [23], A-97065 [24] A-94964 [25], A-90289 [26], and muraminomycin [27], have also been reported. Uridyl peptide antibiotics, such as capuramycin [28], mureidomicin [29], pacidamycin [30], napsamycin [31], muraymycin [32], A-102395 [33], and sansanmycin [34], are also known as translocase I inhibitors. Although amphomycin [35] is not a nucleoside antibiotic, it was reported to inhibit MraY [36].
A residue of GlcNAc from UDP-GlcNAc is then attached to the undecaprenyl-diphospho-MurNAc-pentapeptide (lipid I). This reaction is catalyzed by a glycosyl transferase enzyme known as translocase II (Mur G), yielding lipid II. Two highly modified cyclopeptides composed of 17 amino acids, enduracidin (or enramycin) [37] and ramoplanin [38], bind to lipid II competitively (Fig. 1a). After the assembly of lipid II is flipped from the cytoplasmic face of the membrane to the external face by MurJ (flippase) [39] (Fig. 1a), it is then transformed into peptidoglycan by penicillin-binding proteins that have transglycosylation and transpeptidation functions. Vancomycin [40] acts by binding the terminal amino acid residue L-Lys-D-Ala-D-Ala-COOH, and β-lactams such as penicillin [14] inhibit the transpeptidase. The cross-linking provides the structural rigidity of a mature peptidoglycan, which is required to maintain the cell shape and prevent cell lysis. Bacitracin [41] inhibits the dephosphorylation of lipid pyrophosphate in the late step of the lipid cycle after completing the biosynthesis of peptidoglycan (Fig. 1a).
Gram-positive bacteria are encompassed by a thick peptidoglycan cell wall. However, gram-negative bacteria are encompassed by an additional outer membrane. Mycobacterium spp. such as M. tuberculosis exhibit cell wall structures with unusual complexity. The mycolyl-arabinogalactan-peptidoglycan (mAGP) complex represents the cell wall core structure creating a lipophilic pseudo-outer membrane [2, 10, 42, 43] (Fig. 2). Small molecules can penetrate the cell membrane of gram-positive bacteria. However, few antibiotics are available against gram-negative bacteria and/or Mycobacterium spp. because of the limited permeability of antibiotics through their outer membranes. In the biosynthetic pathway of peptidoglycan and mycolyl-arabinogalactan in M. tuberculosis, the membrane proteins translocase I (MurX: the ortholog of MraY in various bacteria) and WecA (polyprenyl phosphate-N-acetylglucosamine-1-phosphate transferase; the ortholog of TagO in Bacillus subtilis) are involved and are important drug targets [43]. Caprazamycin inhibits the former enzyme and CPZEN-45 inhibits the latter enzyme, as described in Section “Caprazamycin, A-90289A, and muraminomycin as liposidomycin analogs with specific MraY inhibition activity” (Fig. 2).

Cell wall structure of Mycobacterium tuberculosis
Although bacterial peptidoglycan biosynthesis is a well-proven target for antibiotic action, it is surprising that no clinical antibiotics are available that target translocase I (Fig. 1a, b). In the case of mureidomycin A, a translocase I inhibitor, the intrinsic resistance of bacteria is due to low permeability [44]. TB drug resistance has emphasized the need to identify new targets for antibiotic action that lack cross resistance to existing therapies, such as inhibitors of translocase I [10, 11, 15, 43].
Structural features of polyoxin as a chitin synthase inhibitor and tunicamycin as an MraY inhibitor
Chitin is a linear homopolymer of β-(1,4)-linked GlcNAc, formed biosynthetically from UDP-GlcNAc by chitin synthases (chs) 1, chs 2, and chs 3, which were discovered in studies of Saccharomyces cerevisiae mutants with disrupted chitin synthase genes [45]. The absence of all three chs is uniformly lethal. These chitin-containing structures affect the viability of fungi and are not present in mammalian cells. Chs is located in the plasma membrane, requiring that inhibitors be transported into the cell. Isono et al. isolated and developed a peptidyl nucleoside antibiotic, named polyoxin [4,5,6], as a pesticide that inhibits chitin to combat rice blast disease caused by Piricularia oryzae. Compared to the structure with uridyl liponucleoside antibiotics, it mimics only UDP-GlcNAc, a substrate for chs, and is a competitive inhibitor of chs [46] (Fig. 3a).

Structures of polyoxin (a), tunicamycin (b), liposidomycin (c), caprazamycin (d), A-90289 (e) and muraminomycin (f)
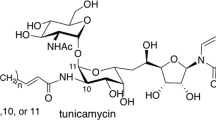
Tunicamycin was isolated from Streptomyces lysosuperificus (later Streptomyces chartreusis) as an antiviral compound by Tamura and Takatsuki [22]. The process of its discovery and various biological activities of tunicamycin have been reported and reviewed in a book entitled Tunicamycin [47]. Briefly, the structure is composed of uracil, fatty acid, and the two sugars of GlcNAc and undecadialdose (Fig. 3b). Gram-positive bacteria such as the Bacillus genus are sensitive to tunicamycin (MIC: 0.1–20 μg/mL) through inhibition of MraY. It has since been found that tunicamycin inhibits the human enzyme UDP-GlcNAc:dolichyl-phosphate GlcNAc phosphotransferase [GlcNAc-1-P-transferase (GPT or DPAGT1) (EC 2.7.8.15)], which catalyzes the first and committed step of N-linked protein glycosylation in the endoplasmic reticulum membrane. Thus, tunicamycin showed severe toxicity in eukaryotic cells, and is used to study the endoplasmic reticulum stress response as a bioprobe [48]. The recent studies have compared of the structures of tunicamycin bound to DPAGT1 and MraY enabling the design of analogues with altered lipid side chains imparting selectivity for MraY over DPAGT1 [49,50,51]. Chemically modifying tunicamycin, hydrogenating the N-acetyl double bond or hydrogenating both the N-acetyl and uridyl double bonds have also been reported to impart less toxic to eukaryotic cells, while retaining their antibacterial activity [52].
Liposidomycin as the first specific MraY inhibitor in bacterial peptidoglycan biosynthesis
Dr Isono has attempted to isolate a specific nucleoside inhibitor like polyoxin against peptidoglycan biosynthesis using enzyme assays. I had the chance to study natural product chemistry in RIKEN for 2 years as a visiting researcher from Snow Brand Milk Products., Co. Ltd (now Meg Milk Snow Brand Co. Ltd) after graduating from a master’s program in the laboratory of biological chemistry (Profs. Shimura and Mizuno) at Tohoku University in 1984. Initially, Dr Isono provided ten kinds of actinomycete strains that had inhibitory activity against peptidoglycan, mannan, and β-1,3-glucan biosynthesis in order to isolate each inhibitor. To isolate new compounds, a well-designed screening system relying upon basic studies and unique microorganisms (natural sources) is vital, as described by Dr Omura, who won the 2015 Nobel Prize for discoveries concerning a novel therapy against infections caused by roundworm parasites [53]. One of the strains, a liposidomycin producer, called the RK-1061 strain, was later identified as Streptomyces griseosporeus [7]. The RK-1061 strain produces more than ten kinds of liposidomycins that show peaks during high-performance liquid chromatography (HPLC); fortunately, three kinds of new antibiotics, liposidomycins A, B, and C, were isolated in 1985; their structures were elucidated by Ubukata et al. [8]. They each possessed 5′-substituted uridine, 5-amino-5-deoxyribose-2-sulfate, and perhydro-1,4-diazepine moieties but differed in the structure of the lipid side chains, representing one of the most bizarre structures for nucleoside antibiotics [54, 55] (Fig. 3c). The Mycobacterium genus is sensitive to liposidomycin [Mycobacterium phlei: minimum inhibitory concentration (MIC) = 1.6 μg/mL] [7]. The discovery of new liposidomycin compounds, supervised by Dr Isono, led me to the isolation of new biologically active compounds (bioprobes) and opened the door to my work in chemical biology.
The molecular target of liposidomycin C was found to be MraY (translocase I), via the traditional method using 14C-UDP-MurNAc-pentapeptide without UDP-GlcNAc [9]. Liposidomycin B weakly inhibited the formation of Dol-p-p-GlcNAc (IC50, 20 μg/mL) but did not inhibit dolichyl phosphoryl glucose or dolichyl phosphoryl mannose, the precursors for mammalian glycoprotein synthesis, even at 400 μg/mL. By contrast, tunicamycin strongly inhibited the formation of Dol-p-p-GlcNAc (IC50, 0.03 μg/mL) but only weakly inhibited the formation of bacterial lipid intermediate I (IC50, 44 μg/mL) [56]. This was the first specific MraY inhibitor that had no cytotoxicity or inhibitory activity against a similar reaction in eukaryotes. Thereafter, several types of MraY inhibitors were isolated from actinomycetes, and the organic synthetic and/or biosynthetic studies have continued progressing [10, 11].
The molecular mechanism of the inhibition of translocase I by liposidomycin B has been investigated. Using a continuous fluorescent assay, Brandish and Bugg determined that liposidomycin B was noncompetitive versus the UDPMurNAc-dansyl-pentapeptide soluble substrate but competitive against the dodecaprenyl phosphate lipid substrate, which differs from the mechanism of tunicamycin [57].
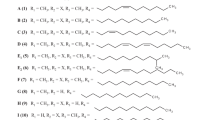
Each of the original three liposidomycins (A, B, and C) has a sulfate moiety at the hydrophilic 2″-aminopentose. Although they showed potent inhibition activity against peptidoglycan biosynthesis, their antimicrobial activities were weak. Therefore, we tried to produce nonsulfated liposidomycins at 2″-aminopentose by changing the fermentation conditions of the RK-1061 strain [58]. Four new liposidomycins were classified as types I–IV based on their structures. The type I compound is the original liposidomycin that has both the sulfate and 3-methylglutaric acid moieties. The type II compound has the sulfate moiety but not the 3-methylglutaric acid moiety. The type III compound has the 3-methylglutaric acid moiety but not the sulfate moiety. The type IV compound has neither moiety [59] (Figs. 3 and 4, Table 1). Although all liposidomycins have the same inhibition activity against peptidoglycan biosynthesis, only types III and IV liposidomycins showed antimicrobial activity against Mycobacterium phlei (Table 1). Because type III and IV compounds lacking the sulfate moiety are more lipophilic than types I and II, they probably penetrate the lipophilic mycobacterial outer membrane better and inhibit MraY, which exists in the inner membrane [60] (Fig. 1). When the effects of liposidomycin A of types I–IV and tunicamycin on the growth of mammalian BALB/3T3 cells were compared, all liposidomycins showed no cytotoxicity even at 25 μg/ml, whereas tunicamycin inhibited 50% of the growth at 0.05 µg/ml. Two types of liposidomycins, A-III and A-IV, without the sulfate moiety at 2″ are potently active against Mycobacterium spp. and show increased antimicrobial activity against Escherichia coli BE1186 and Bacillus subtilis IFO 3513 [60]. HIV-infected and/or cancer patients have been threatened by the opportunistic infections caused by the mycobacterium avium complex (MAC). Liposidomycin C-III, one of the abundant components, has antimicrobial activity against several types of MACs (MIC = 1.2–12.5 μg/mL) [61].

Comparison of the biosynthetic pathway (a) and genes (b) of liposidomycin and caprazamycin
To identify novel antimicrobial agents, it is also important to quickly exclude the known analogs, such as liposidomycin and caprazamycin, by tandem mass spectrometry (MS) and the application of in-line reverse-phase HPLC-electrospray MS (LC-ESI-MS) [62, 63]. A similar strategy could be applied to identify other related antibiotics.
The inhibition activity of deacyl liposidomycin isolated from the fermentation broth (the same as caprazene, which is obtained by the acid hydrolysis of caprazamycins) against translocase I decreased about 200-fold compared with that of the original liposidomycin (Fig. 5). At that time, we did not consider the potential to synthesize simple analogs of liposidomycin such as CPZEN-45 using deacyl liposidomycin without a hydroxyl group.

Structures of each degradation product of liposidomycin and caprazamycin and its analogue CPZEN-45
Because the isolation of liposidomycin is laborious and the recovered quantity is low, precursor-directed biosynthesis is a useful method for the production of a single liposidomycin. Exogenously supplied myristic acid or palmitic acid results in the almost exclusive production of liposidomycin C-III and/or M-III [64]. The four isolated types of liposidomycins are consistent with the subsequent analysis of the biosynthetic genes of uridyl liponucleoside antibiotics [65] (Fig. 4).
Caprazamycin, A-90289A, and muraminomycin as liposidomycin analogs with specific MraY inhibition activity
The liposidomycin-type liponucleoside antibiotic caprazamycin was isolated from Streptomyces sp. MK730-62F2 by Igarashi et al. at the Institute of Microbial Chemistry (BIKAKEN) [12] (Fig. 3d). Takahashi et al. then developed CPZEN-45 as a derivative of caprazamycin as an antibiotic against M. tuberculosis from a core structure of caprazamycin, caprazene (deacyl liposidomycin) [13, 66] (Fig. 5b, c). Igarashi, who won the Sumiki–Umezawa Memorial Award in 2018 from the Japan Antibiotics Research Association with me [67], has summarized the study of caprazamycin, and Takahashi has described the first structure–activity relationship (SAR) of CRZEN-45 in this issue [68, 69]. In addition, two reviews involving caprazamycin and the SARs of the derivatives were published recently in quick succession [65, 70]. They revealed the essential pharmacophores present in the natural caprazamycin scaffold. Caprazamycins differ from liposidomycins in the absence of a sulfate group at the 2″-position of the aminoribose and in the presence of a permethylated l-rhamnose α-glycosidically linked to the 3-methylglutaryl moiety (Figs. 3d, 4, and Table 1).
A-90289A consists of a β-hydroxy palmitic acid moiety and has a structure identical to that of caprazamycin A but also contains a sulfate group, which is characteristic of liposidomycins. However, the sulfate group in A-90289A is attached at the 2’-hydroxy group, as opposed to the 2″-hydroxy group where it is attached in liposidomycins [26] (Fig. 3e and Table 1). The differences in the structures among liposidomycin, caprazamycin, and A-90289A were confirmed theoretically after each gene cluster of biosynthesis was clarified (Fig. 4). The sites of sulfation that differ between liposidomycin and A-90289A may depend on the substrate specificity of the unusual arylsulfate sulfotransferase (ASST) [71].
Muraminomycins have slightly different structures and antimicrobial activities compared to other liponucleoside antibiotics. They possess a 2′-dexyuridine moiety, a 5-amino-2,5-dideoxyribose moiety, a diazepanone ring system, a fatty acid, 3-methylglutaric acid, and succinic-acid-attached 6-methyl-α-rhamnose. The most characteristic structural difference between muraminomycins and liposidomycins, caprazamycins, or A90289A was the loss of hydroxyl groups at C-2′ and C-2″ (Fig. 3f, Table 1) [27, 72].
The primary target of CPZEN-45 in Bacillus subtilis is undecaprenyl phosphate-GlcNAc-1-phosphate transferase (TagO: an ortholog of WecA and involved in the biosynthesis of teichoic acid), which is a different target from that of the parent caprazamycin (MraY). CPZEN-45 also inhibited WecA, which is involved in mycolyl-arabinogalactane biosynthesis in M. tuberculosis and is the ortholog of TagO (Fig. 2) [42, 43, 73], and this is an important result in terms of facilitating the development of new antituberculosis antibiotics.
Organic synthesis and biosynthesis of liponucleoside antibiotics
Two reviews of the organic synthesis and biosynthesis involving nucleoside antibiotics and MraY inhibitors were published in 2003 and 2010 [10, 11]. Since then, organic synthesis involving the total synthesis and biosynthesis of uridyl liponucleoside antibiotics has continued progressing. Tunicamycin V is readily accessible via the longest linear sequence of 24 synthetic steps from uridine and is commercially available from simple materials, with an overall chemical yield of 3.9%. Replacing the GlcNAc and lipid moieties could provide a range of new analogs of tunicamycins [74]. Recently, truncated analogs of tunicamycin V in a series were synthesized, and their MraY inhibition activities were investigated by Yamamoto and Ichikawa. Unfortunately, the MraY inhibition activity of GlcNAc-, lipid-, and uridine-truncated analogs all decreased by more than 100-fold [75]. The complex structure with their binding to MraY from Clostridium bolteae and with tunicamycin-GPT (GPAGT1) will provide new information for seeking potent and/or specific inhibitors against MraY and/or GPT [48,49,50, 76]. Their co-crystal structures indicate that the GlcNAc moiety recognized in tunicamycin is quite different, which shows that the GlcNAc-modified analog is crucial for the specificity between MraY and GPT and/or for potency. Chemically modified tunicamycin with less toxic to eukaryotic cells, but which retain their antibacterial activity were synthesized [52].
The total synthesis of caprazamycin A has been reported and was accomplished in 23 steps. It will guide the synthesis of related uridyl liponucleoside antibiotics such as liposidomycin and A-90289 [77, 78]. Although an analog of caprazamycin, CPZEN-45, has been anticipated as a new antituberculosis drug, the co-crystal structures of caprazamycin analogs to MraY and WecA will also provide information about other new potential drugs (Fig. 6).

Perspective on new antibiotics
The caprazamycin gene cluster was reported in 2009 as the first cluster of a translocase I inhibitor [79]. Since then, the gene clusters of liposidomycin, A-90289, and muraminomycin were also described (Fig. 4) [27, 80, 81]. Gene disruption and heterologous expression experiments of the gene clusters allow the generation of novel biologically active derivatives via pathway engineering and will also yield new analog compounds. In addition, a new type of ASST that had low homology with previous ASSTs was identified as Cpz4 during a study on the biosynthetic cluster genes of caprazamycin and liposidomycin [71]. Surprisingly, similar genes are located adjacent to the caprazamycin gene cluster (Fig. 4). Each biosynthetic gene for liponucleoside antibiotics showed the production of various liposidomycin-type liponucleoside antibiotics (Fig. 4).
Polymerase chain reaction screening was performed using a library of ~2500 strains and degenerate primers of a pyridoxal-5-phosphate-dependent l-Thr:uridine-5′-aldehyde transaldolase by Funabashi et al. [82] and a new nucleoside MraY inhibitor, named sphaerimicin, was discovered in 2013.
Perspective
Since the discovery of the transpeptidase inhibitor penicillin, many types of antibiotics targeting peptidoglycan biosynthesis have been developed mainly from microorganisms. Synthetic and semisynthetic technologies of the natural products have also produced clinically effective antimicrobial agents. In addition, biosynthetic technology and identified cluster genes are expected to yield new structures and/or targets for clinical agents. The biological targets of antimicrobial agents include the cell wall, DNA, protein biosynthesis, and cell membranes. Of these, cell wall biosynthesis is a promising target for antibiotics because there is no cell wall in human cells, providing an opportunity for selective toxicity of the antibiotic [10, 11, 15]. In fact, almost all the targets in peptidoglycan biosynthesis serve as clinical antibiotics, except translocase I (MraY) (Fig. 1). Although no clinical antibiotic against translocase I has been available up to this point, the crystal structure of MraY in complex with muraymycin D2 [32, 83, 84] and tunicamycin can inform the design of new inhibitors targeting MraY and/or WecA [22, 46, 48,49,50, 76]. In addition, the total synthesis and gene cluster of tunicamycin, liposidomycin, caprazamycin, A-90289, and muraminomycin open the door to optimal MraY inhibitors for clinical use, inhibitors like CPZEN-45 (Fig. 6).
Simple, specific, and improved biochemical assay systems continue to be critical to isolating and evaluating new MraY and/or WecA inhibitors from natural sources and/or chemical libraries [85,86,87]. Novel structures of MraY inhibitors isolated from natural sources will provide new information for drug-like related compounds using organic synthesis and biosynthetic genes. Tunicamycin and caprazamycin (including liposidomycin and A-90289) have already become candidates for total synthesis and gene cluster biosynthesis, allowing easy examination of the SAR [88,89,90,91]. Amplifications of the entire biosynthetic gene cluster may improve the production of antibiotics. Precursor-directed biosynthesis and mutasynthesis have become useful tools to generate new antibiotic derivatives. The semisynthetic drug candidate CPZEN-45, which is a WecA (TagO) and MurX (MraY) inhibitor in M. tuberculosis, and specific MraY inhibitors against MDR bacteria may become clinically approved in the near future. The combination of organic synthesis and biosynthesis, aided by newly enabled structural studies of liposidomycin/caprazamycin–MraY and/or CPZEN-45–WecA membrane proteins-drug complexes, can be expected to yield new types of antibiotics in the future (Fig. 6) [92, 93].
The vision of Dr Isono, to develop new clinical antibiotics through the discovery of specific nucleoside antibiotics, with polyoxin for antifungals and liposidomycin for antibacterials, is now being realized.
References
Global tuberculosis report 2018, World Health Organization, ISBN 978-92-4-156564-6.
Hoagland DT, Liu J, Lee RB, Lee RE. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv Drug Deliv Rev. 2016;102:55–72.
Igarashi M, Ishizaki Y, Takahashi Y. New antituberculous drugs derived from natural products: current perspectives and issues in antituberculous drug development. J Antibiot. 2018;71:15–25.
Suzuki S, et al. A new antibiotic polyoxin A. J Antibiot. 1965;18:131.
Isono K. Nucleoside antibiotics: streuture, biological activity, and biosynthesis. J Antibiot. 1988;41:1711–39.
Isono K. Current progress on nucleoside antibiotics. Phramacol Ther. 1991;52:269–86.
Isono K, et al. Liposidomycins: novel nucleoside antibiotics which inhibit bacterial peptidoglycan synthesis. J Antibiot. 1985;38:1617–21.
Ubukata M, Isono K, Kimura K, Nelson CC, McCloskey JA. The structure of liposidomycin B, an inhibitor of bacterial peptidoglycan synthesis. J Am Chem Soc. 1988;110:4416–7.
Kimura K, et al. Liposidomycin C inhibits phospho-N-acetylmuramyl-pentapeptide transferase in peptidoglycan synthesis of Escherichia coli Y-10. Agric Biol Chem. 1989;53:1811–5.
Kimura K, Bugg TDH. Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat Prod Rep. 2003;20:252–73.
Winn M, Goss RJM, Kimura K, Bugg TDH. Antimicrobial nucleoside antibiotics targetting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat Prod Rep. 2010;27:279–304.
Igarashi M, et al. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J Antibiot. 2003;56:580–3.
Takahashi Y, et al. Novel semisynthetic antibiotics from caprazamycins A-G: caprazene derivatives and their antibacterial activity. J Antibiot. 2013;66:171–8.
Fleming A. On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzæ. Br J Exp Pathol. 1929;10:226–36.
Bugg TD, Braddick D, Dowson CG, Roper DI. Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol 2011;29:167–73.
Brown ED, Vivas EI, Walsh CT, Kolter R. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J Bacteriol. 1995;177:4194–7.
Benson TE, Walsh CT, Hogle JM. Crystallization and preliminary X-ray crystallographic studies of UDP-N-acetylenolpyruvylglucosamine reductase. Protein Sci. 1994;3:1125–7.
Hendlin D, et al. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science. 1969;166:122–3.
Harned RL, Hidyph PH, Labaw EK. Cycloserine. I. A preliminary report. Antibiot Chemother. 1955;5:204–5.
Azam MA, Jayaram U. Inhibitors of alanine racemase enzyme: a review. J Enzym Inhib Med Chem. 2016;31:517–26.
Ikeda M, Wachi M, Jung HK, Ishino F, Matsuhashi M. The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide: undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J Bacteriol. 1991;173:1021–6.
Takatsuki A, Arima K, Tamura G. Tunicamycin, a new antibiotic. I. Isolation and characterization of tunicamycin. J Antibiot. 1971;24:215–23.
Aoyagi A, Murakami A. New antibiotic A-84830A. published unexamined Japanese patent application, JP2005-247725.
Fujita Y, Kizuka M, Murakami A. Novel A-97065 substances, their fermentative manufacture, and use as pharmaceuticals. published unexamined Japanese patent application, JP 2006-252237.
Murakami R, et al. A-94964, a novel inhibitor of bacterial translocase I, produced by Streptomyces sp. SANK 60404. I. Taxonomy, isolation and biological activity. J Antibiot. 2008;61:537–44.
Fujita Y, et al. A-90289 A and B, new inhibitors of bacterial translocase I, produced by Streptomyces sp. SANK 60405. J Antibiot. 2011;64:495–501.
Chi Xl, et al. The muraminomicin biosynthetic gene cluster and enzymatic formation of the 2-deoxyaminoribosyl appendage. Medchemcomm. 2013;4:239–43.
Yamaguchi H, et al. Capuramycin, a new nucleoside antibiotic. Taxonomy, fermentation, isolation and characterization. J Antibiot. 1986;39:1047–53.
Inukai M, et al. Mureidomycins A-D, novel peptidylnucleoside antibiotics with spheroplast forming activity. I. Taxonomy, fermentation, isolation and physico-chemical properties. J Antibiot. 1989;42:662–6.
Karwowski JP, et al. Pacidamycins, a novel series of antibiotics with anti-Pseudomonas aeruginosa activity. I. Taxonomy of the producing organism and fermentation. J Antibiot. 1989;42:506–11.
Chatterjee S, et al. Napsamycins, new Pseudomonas active antibiotics of the mureidomycin family from Streptomyces sp. HIL Y-82,11372. J Antibiot. 1994;47:595–8.
Lin YI, et al. Muraymycins, novel peptidoglycan biosynthesis inhibitors: semisynthesis and SAR of their derivatives. Bioorg Med Chem Lett. 2002;12:2341–4.
Murakami R, et al. A-102395, a new inhibitor of bacterial translocase I, produced by Amycolatopsis sp. SANK 60206 . J Antibiot. 2007;60:690–5.
Xie Y, Chen R, Si S, Sun C, Xu H. A new nucleosidyl-peptide antibiotic, sansanmycin. J Antibiot. 2007;60:158–61.
Heinemann B, Kaplan MA, Muir RD, Hooper IR. Amphomycin, a new antibiotic. Antibiot Chemother. 1953;3:1239–42.
Tanaka H, Oiwa R, Matsukura S, Omura S. Amphomycin inhibits phospho-N-acetylmuramyl-pentapeptide translocase in peptidoglycan synthesis of Bacillus. Biochem Biophys Res Commun. 1979;86:902–8.
Higashide E, Hatano K, Shibata M, Nakazawa K. Enduracidin, a new antibiotic. I. Streptomyces fungicidicus No. B5477, an enduracidin producing organism. J Antibiot. 1968;21:126–37.
Cavalleri B, Pagani H, Volpe G, Selva E, Parenti F. A-16686, a new antibiotic from Actinoplanes. I. Fermentation, isolation and preliminary physico-chemical characteristics. J Antibiot. 1984;37:309–17.
Sham LT, et al. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–2.
Mccormick MH, Mcguire JM, Pittenger GE, Pittenger RC, Stark WM. Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot Annu. 1955;3:606–11. 1956
Johnson BA, Anker H, Meleney FL. Bacitracin: a new antibiotic produced by a member of the B. subtilis group. Science. 1945;102:376–7.
Alderwick LJ, Harrison J, Lloyd GS, Birch HL. The mycobacterial cell wall-peptidoglycan and arabinogalactan. Cold Spring Harb Perspect Med. 2015;5:a021113.
Huszár S, et al. N-Acetylglucosamine-1-phosphate transferase, WecA, as a validated drug target in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2017;61:e01310–17.
Isono F, Inukai M. Mureidomycin A, a new inhibitor of bacterial peptidoglycan synthesis. Antimicrob Agents Chemother. 1991;35:234–6.
Cabib E, Mol PC, Shaw JA, Choi WJ. Biosynthesis of cell wall and septum during yeast growth. Arch Med Res. 1993;24:301–3.
Chaudhary PM, Tupe SG, Deshpande MV. Chitin synthase inhibitors as antifungal agents. Mini Rev Med Chem. 2013;13:222–36.
Tamura G. Tunicamycin. Tokyo: Japan Scientific Press; 1982.
Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect. 2007;4:e00211.
Hakulinen JK, et al. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat Chem Biol. 2017;13:265–7.
Yoo J, et al. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol. 2018;25:217–24.
Dong YY, et al. Structures of DPAGT1 explain glycosylation disease mechanisms and advance TB antibiotic design. Cell. 2018;175:1045–1058.
Price NP, et al. Modified tunicamycins with reduced eukaryotic toxicity that enhance the antibacterial activity of β-lactams. J Antibiot. 2017;70:1070–1077.
Omura S. Philosophy of new drug discovery. Microbiol Rev. 1986;50:259–79.
Ubukata M, et al. Structure elucidation of liposidomycins, a class of complex lipid nucleoside antibiotics. J Org Chem. 1992;57:6392–403.
Ubukata M. The logic of biologically active small molecules: amazing ability of microorganisms. Biosci Biotechnol Biochem. 2018;82:1063–72.
Muroi M, Kimura K, Osada H, Inukai M, Takatsuki A. Liposidomycin B inhibits in vitro formation of polyprenyl (pyro)phosphate N-acetylglucosamine, an intermediate in glycoconjugate biosynthesis. J Antibiot. 1997;50:103–4.
Brandish PE, et al. Modes of action of tunicamycin, liposidomycin B, and mureidomycin A: inhibition of phospho-N-acetylmuramyl-pentapeptide translocase from Escherichia coli. Antimicrob Agents Chemother. 1996;40:1640–4.
Kimura K, et al. New types of liposidomycins that inhibit bacterial peptidoglycan synthesis and are produced by Streptomyces. I. Producing organism and medium components. J Antibiot. 1998;51:640–6.
Kimura K, et al. New types of liposidomycins that inhibit bacterial peptidoglycan synthesis and are produced by Streptomyces. II. Isolation and structure elucidation. J Antibiot. 1998;51:647–54.
Kimura K, et al. Selective inhibition of the bacterial peptidoglycan biosynthesis by the new types of liposidomycins. J Antibiot. 1998;51:1099–104.
Suzuki K. unpublished data.
Esumi Y, et al. New types of liposidomycins produced by Streptomyces that inhibit bacterial peptidoglycan synthesis. Structure elucidation of fatty acid components by tandem mass spectrometry. J Antibiot. 1999;52:281–7.
Tsvetanova BC, Price NP. Liquid chromatography-electrospray mass spectrometry of tunicamycin-type antibiotics. Anal Biochem. 2001;289:147–56.
Kagami S, Esumi Y, Nakakoshi M, Yoshihama M, Kimura K. Control of liposidomycin production through precursor-directed biosynthesis. J Antibiot. 2003;56:552–6.
Wiker F, Hauck N, Grond S, Gust B. Caprazamycins: biosynthesis and structure activity relationship studies. Int J Med Microbiol. 2019;309:319–324.
Pstragowski M, Zbrzezna M, Bujalska-Zadrozny M. Advances in pharmacotherapy of tuberculosis. Acta Pol Pharm. 2017;74:3–11.
Kimura K. Studies of novel bioprobes isolated from rare natural sources using mutant yeasts. J Antibiot. 2019;72:579–89.
Igarashi M. New natural products to meet the antibiotic crisis: a personal journey. J Antibiot. 2019. https://doi.org/10.1038/s41429-019-0224-6.
Ishizaki Y, et al. Synthesis and biological activity of CPZEN-45, a novel anti-tuberculosis drug. J Antibiot. 2019. https://doi.org/10.1038/s41429-019-0225-5.
Patel B, et al. Caprazamycins: promising lead structures acting on a novel antibacterial target MraY. Eur J Med Chem. 2019;171:462–74.
Kaysser L, et al. A new arylsulfate sulfotransferase involved in liponucleoside antibiotic biosynthesis in streptomycetes. J Biol Chem. 2010;285:12684–94.
Fujita Y, et al. Muraminomicins, new lipo-nucleoside antibiotics from Streptosporangium sp. SANK 60501-structure elucidations of muraminomicins and supply of the core component for derivatization. J Antibiot. 2019. https://doi.org/10.1038/s41429-019-0215-7.
Ishizaki Y, et al. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem. 2013;288:30309–19.
Yamamoto K, Yakushiji F, Matsumaru T, Ichikawa S. Total synthesis of tunicamycin V. Org Lett. 2018;20:256–9.
Yamamoto K, Katsuyama A, Ichikawa S. Structural requirement of tunicamycin V for MraY inhibition. Bioorg Med Chem. 2019;27:1714–9.
Herling J, Dunevall E, Ek M, Brändén G. Structural basis for selective inhibition of antibacterial target MraY, a membrane-bound enzyme involved in peptidoglycan synthesis. Drug Discov Today. 2018;23:1426–35.
Nakamura H, et al. Total synthesis of (-)-caprazamycin A. Angew Chem Int Ed Engl. 2015;54:3136–9.
Nakamura H, et al. Total synthesis of caprazamycin A: practical and scalable synthesis of syn-β-hydroxyamino acids and introduction of a fatty acid side chain to 1,4-diazepanone. J Am Chem Soc. 2019;141:8527–40.
Kaysser L, et al. Identification and manipulation of the caprazamycin gene cluster lead to new simplified liponucleoside antibiotics and give insights into the biosynthetic pathway. J Biol Chem. 2009;284:14987–96.
Kaysser L, Siebenberg S, Kammerer B, Gust B. Analysis of the liposidomycin gene cluster leads to the identification of new caprazamycin derivatives. ChemBioChem. 2010;11:191–6.
Funabashi M, et al. The biosynthesis of liposidomycin-like A-90289 antibiotics featuring a new type of sulfotransferase. Chembiochem. 2010;11:184–90.
Funabashi M, et al. Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew Chem Int Ed Engl. 2013;52:11607–11.
Chung BC, et al. Structural insights into inhibition on lipid I production in bacterial cell wall synthesis. Nature. 2016; 533. https://doi.org/10.1038/nature17636.
Divya MB, Abdullah M, Saxena S, Guruprasad L. Inhibitor binding studies of Mycobacterium tuberculosis MraY (Rv2156c): insights from molecular modeling, docking and simulation studies. J Biomol Struct Dyn. 2018:1–33. https://doi.org/10.1080/07391102.2018.1526715.
Mihalyi A, Jamshidi S, Slikas J, Bugg TD. Identification of novel inhibitors of phospho-MurNAc-pentapeptide translocase MraY from library screening: Isoquinoline alkaloid michellamine B and xanthene dye phloxine B. Bioorg Med Chem. 2014;22:4566–71.
Siricilla S, Mitachi K, Skorupinska-Tudek K, Swiezewska E, Kurosu M. Biosynthesis of a water-soluble lipid I analogue and a convenient assay for translocase I. Anal Biochem. 2014;461:36–45.
Mitachi K, et al. Fluorescence-based assay for polyprenyl phosphate-GlcNAc-1-phosphate transferase (WecA) and identification of novel antimycobacterial WecA inhibitors. Anal Biochem. 2016;512:78–90.
Yamamoto K, Ichikawa S. Tunicamycin: chemical synthesis and biosynthesis. J. Antibiot. 2019. https://doi.org/10.1038/s41429-019-0200-1.
Fer MJ, et al. Bacterial transferase MraY, a source of inspiration towards new antibiotics. Curr Med Chem. 2018;25:6013–29.
Niu G, Tan H. Nucleoside antibiotics: biosynthesis, regulation, and biotechnology. Trends Microbiol. 2015;23:110–9.
Chen S, Kinney WA, Lanen SV. Nature’s combinatorial biosynthesis and recently engineered production of nucleoside antibiotics in Streptomyces. World J Microbiol Biotechnol. 2017;33:66.
Walsh CT, Wencewicz TA. Prospects for new antibiotics: a molecule-centered perspective. J Antibiot. 2014;67:7–22.
Mashalidis EH, et al. Chemical logic of MraY inhibition by antibacterial nucleoside natural products. Nat Commun. 2019;10:2917.
Acknowledgements
The research presented herein was conducted in the Antibiotic Laboratory of RIKEN, Applied Microbiology Group of Snow Brand Milk Products. Co. Ltd., and the Chemical Biology Laboratory of Iwate University. I thank all my students, collaborators, and colleagues. I especially express sincere thanks to Drs. Kiyoshi Isono, Masakazu Uramoto, Makoto Ubukata and Hiroyuki Osada for guidance in chemical biology research, and to Drs Nobuo Miyata and Makoto Yoshihama for the opportunity to conduct research in the company (Snow Brand). I would like to thank to Prof. Tim Bugg, Warwick University for my stay in his Lab., and to Emeritus Professor Don R Phillips, La Trobe University and Enago (www.enago.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kimura, Ki. Liposidomycin, the first reported nucleoside antibiotic inhibitor of peptidoglycan biosynthesis translocase I: The discovery of liposidomycin and related compounds with a perspective on their application to new antibiotics. J Antibiot 72, 877–889 (2019). https://doi.org/10.1038/s41429-019-0241-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0241-5
This article is cited by
-
Therapeutic applications of carbohydrate-based compounds: a sweet solution for medical advancement
Molecular Diversity (2024)
-
A review of current antibiotic resistance and promising antibiotics with novel modes of action to combat antibiotic resistance
Archives of Microbiology (2023)
-
Origin of the 3-methylglutaryl moiety in caprazamycin biosynthesis
Microbial Cell Factories (2022)
