
Abstract
We have found cyclophane-type adenosine derivatives having p-quinone amide moieties (1 and 2) as weak inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase (CpIMPDH) from the Hokkaido University Chemical Library via the luciferase-based high-throughput screening. To obtain more potent inhibitors, we synthesized four new derivatives free from cyclophane rings (3–6). The N-H derivatives 3 and 5 showed more potent activities (24.4 and 11.1 μM, respectively) in the presence of dithiothreitol (DTT), whereas the N-methyl derivative 4 indicated more potent activity (2.1 μM) without DTT. Conformational analysis of compounds 3 and 4 suggested that N-H amide 3 binds to IMP-binding site in the DTT mediated manner.
Similar content being viewed by others


Introduction
Cryptosporidium parvum is a waterborne pathogen, which causes watery or mucoid diarrhea and abdominal pain in many mammal species, including human. The infection is usually self-limiting in immunocompetent patients. Unfortunately, the infections could be severe in immunocompromised patients, such as children, elderly person, or AIDS patients [1,2,3].
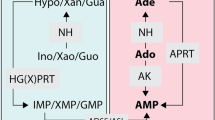
For the prevention of such infections, we have focused on the inhibition of protozoan inosine 5′-monophosphate dehydrogenase (IMPDH) because these microbes rely solely on the IMPDH-mediated guanosine nucleotides biosynthesis. In previous work, we established a luciferase-based high-throughput screening system to identify IMPDH inhibitors from the Pharmakon repositioning library of off-patent US Food and Drug Administration approved compounds (Microsouce Discovery Systems, Inc., CT). The screening of 1600 compounds resulted in the discovery of three irreversible inhibitors: disulfiram, bronopol, and ebselen. In the efficacy experiment against cryptosporidiosis in severe combined immunodeficiency mouse, a decrease in the number of oocyst shed was observed on the oral administration of disulfiram and bronopol [4].
Next, we focused on the chemical library from the Faculty of Pharmacy, Hokkaido University (Hokkaido University Chemical Library). The identified cyclophane-type compounds 1 and 2 as hit compounds in the first screening, showed relatively weak activities. Therefore, four novel derivatives of 1 and 2 were synthesized and three compounds 3–5 showed potent inhibitory activities. In this article, we report synthesis and biological activity of these novel adenosine-derived CpIMPDH inhibitors.
Results and discussion
High-throughput screening of CpIMPDH inhibitors from a chemical library
For the discovery of new protozoan IMPDH inhibitors, a high-throughput screening was performed by using Hokkaido University Chemical Library, which comprises 1600 compounds derived from the Faculty of Pharmacy, Hokkaido University. The screening system providing high accuracy (Z′-factor value of 0.7) [4] has been previously established by the authors based on the detection of NADH consumed by luciferase. In addition, the assay was designed so as to sift out NAD+ mimics as hits by adding the excess amount of NAD+ (1.6 mM), because the cofactor ubiquitously functions in diverse biological systems. After screening, the activities of 1260 were successfully evaluated from 1600 compounds without mechanical errors, and 44 compounds showed comparable activities to mycophenolic acid, a known IMPDH inhibitor. These hits were next subjected to reproducibility test with triplicating assays. The reproducibility was affirmed only in 19 compounds out of 44. Interestingly, 15 of 19 compounds were adenosine derivatives as typified by cyclophane-type 1 and 2 (Fig. 1), which had been originally developed targeting heat shock protein 90 [5, 6]. The ribonucleoside substructures are frequently conserved in IMPDH inhibitors mimicking both IMP and NAD+ [7]. Thus, these compounds could be regarded as the promising lead compounds to develop novel IMPDH inhibitors. On the other hand, the cyclophane skeleton is rarely seen in IMPDH inhibitors, except for naturally occurring halicyclamine A [8,9,10,11,12,13]. Therefore, we next designed new adenosine derivatives 3–6 of which p-quinones were liberated from cyclophane and undertook syntheses of these compounds.

Structures of hit compounds in high-throughput screening of CpIMPDH and designed derivatives
Synthesis of compounds 3–6
Nucleoside unit 12 was first synthesized as shown in Schemes 1. 2′,3′-Hydroxy groups and 5′-hydroxy group of adenosine 7 were protected with isopropylidene and TBS groups, respectively to give 9 [14, 15]. After protecting 6-amino group with trityl group, TBS group was effectively deprotected by tetrabutylammonium fluoride to give 5′-alcohol 11. Dess–Martin oxidation of 11 afforded 5′-aldehyde 12, which was subjected to Horner–Wadsworth–Emmons (HWE) reaction without further purification.

Synthesis of adenosine unit 12
Phosphonate units, 18 and 21 were synthesized as shown in Scheme 2. Methoxyhydroquinone 13 was protected with MOM group followed by nitration [16] with ammonium nitrate in the presence of trifluoroacetic anhydride to give desired 15 and deprotected 16, which was converted to 15 by protection with MOM group. Nitro group of 15 was converted to amine to give 17, which was converted to N-H phosphonate 18 by coupling with diethylphosphonoacetic acid in 95% in two steps [17]. To synthesize N-methyl phosphonate 21, compound 15 was converted to trifluoroacetoamide 19 by catalytic hydrogenation followed by protection with trifluoroacetyl group. Methylation, deprotection of trifluoroacetyl group, and acylation with bromoacetyl chloride afforded 20. Arbuzov reaction of 20 with triethylphosphite gave desired N-methyl phosphonate 21 in 84% yield from 15.

Synthesis of phosphonate units 18 and 21
HWE reaction of aldehyde 12 with N-H phosphonate 18 gave desired compound 22 under the neutral conditions using zinc triflate, triethylamine, and N,N,N′,N″-tetramethylethylenediamine developed by Schauer et al. [18]. Similarly, compound 23 was synthesized from 12 and N-methyl phosphonate 21. Compounds 22 and 23 were converted to 5′-substituted adenosine 3 and 4, respectively. Hydrogenation of compound 3 with Pd/C gave saturated amide 5. Although the same treatment of 4 gave compound 6, the activity was not tested due to the limited amount (Scheme 3).

Synthesis of adenosine derivatives 3–6
Evaluation of IMPDH inhibitory activities
The inhibitory activities of hit compounds 1 and 2, synthetic compounds 3–5 against recombinant CpIMPDH were evaluated. The apparent IC50 values were determined using essentially the same conditions as the luciferase-based high-throughput assay, except that different concentrations were used (Table 1). Interestingly, synthetic compounds 3–5 showed remarkable activities (IC50: 0.7–1.3 μM) compared with hit compounds (>50 μM) as we expected. Next, we determined IC50 values on fluorescence-based low-throughput assay (Ex 340 nm/Em 465 nm) to eliminate the possible effect by luciferase in the activity. As shown in Table 2, compounds 3 and 5 showed inferior activities to those in luciferase-based assay (IC50: 209.8 and >500.0 μM, respectively), whereas 4 retained same activity. The result would reflect partial inhibitions by 3 and 5 against luciferase activity. Computational conformation analyses predicted that N-H amide 3 is in an equilibrium between trans-amide (M3–007 in Fig. 2a) and cis-amide (M3–078) at the approximate ratio 96:4, while N-methyl amide 4 is uniformly present as cis-amide in any possible conformer (M4–015, −003, −009, −011, and −071 in Fig. 2b). It can be speculated that cis conformation of 4 enables selective inhibition against CpIMPDH.

Predicted stable conformers of a compound 3 and b compound 4. The numbers in parentheses are relative energies to the most stable conformers M3–007 and M4–015
In the assay of enzymes that require cysteine as a catalytic residue, reducing agents such as dithiothreitol (DTT) or glutathione are commonly added to protect the residue from inactivators. Also in IMPDH, the cysteine residue positioned near IMP-binding site (e.g. Cys331 in human type II IMPDH) is known to play a critical role in the catalysis. Thus, we next evaluated the activities of 3–5 in the presence of 1.0 mM DTT. These inhibitors showed the IC50 values at 11.1–76.8 μM against CpIMPDH in DTT-supplemented conditions, and the values were similar against human type II IMPDH (Table 2). CpIMPDH is known to have a dramatic difference in NAD+ binding site from that of hIMPDH II. Especially the residues related to the binding of adenine ring, such as His253, Phe282, Thr45, and Gln469, are not conserved in CpIMPDH, which discriminates the specificity to NAD+ versus nicotinamide hypoxanthine dinucleotide [19]. The similar IC50 values between Cp- and hIMPDH II may be due to the less contribution of adenine ring recognition. The addition of reducing agents often attenuates the activity due to the competition or the direct contact with inhibitors [20], and the same phenomena were observed in bronopol, ebselen, and disulfiram, previously discovered Cys-targetting inhibitors [4] (Table 2). Among the synthetic compounds, only N-methyl derivative 4 behaves according to these precedent examples (76.8 μM with DTT), which in turn suggests that cysteine residue is involved in this inhibition, such as via Michael addition to quinone moiety. In contrast, it would be noteworthy that compounds 3 and 5 showed superior activities in the DTT-supplemented conditions. This pronounced difference would stem from the conformational preference of each compound. The “stretched” conformation in compounds 3 and 5 conjure a binding model seen in C2-mycophenolic adenine dinucleotide (C2-MAD), a ligand mimicking NAD+ [21]. Given that 3 and 5 bind in the same manner as C2-MAD, the improved activity may be explained by crosslinking via DTT between Cys and p-quinone moieties, because they are to be positioned away by 5–10 Å. Meanwhile, these compounds can possibly adopt a different binding manner from C2-MAD considering the screening system set to eliminate NAD+ mimics and the above-discussed similarity in IC50 values with hIMPDH. Obviously, this is true also for compound 4. Although further investigations, such as X-ray crystallography or detailed docking simulation are required, each N-H and N-methyl amide was proved to have clearly different mode of binding against CpIMPDH. This variance may provide the optional application of these compounds, such as combined or selective use depending on different physiological conditions.
Conclusion
By the use of high-throughput screening system, cyclophane-type adenosine derivatives furnishing p-quinone moieties (1 and 2) were discovered as weak inhibitors of CpIMPDH from the Hokkaido University Chemical Library. To ameliorate the activities, we designed and synthesized four new derivatives of which p-quinones were liberated from cyclophane (3–6). All tested compounds (3–5) showed more potent activities at the absence of DTT than the hit compounds (1 and 2) in luciferase-based high-throughput assay. Interestingly, the supplement of DTT in fluorescence-based low-throughput assay differentiated the activity of N-methyl amide 4 from those of N-H amides 3 and 5, which would stem from the conformational difference between cis- and trans-amides. These insights raised a significance of impact by reducing agents in the IMPDH assay and will be helpful for the development of novel IMPDH inhibitors.
Experimental procedures
Chemistry
Organic solvents were purified by normal methods. Unless otherwise stated, all commercially available chemicals of the highest purity were used without further purification. Thin layer and column chromatography were performed with Merck Silica Gel 60 (0.063–0.020 mm) and Merck Silica Gel 60 (0.040–0.063 mm), respectively. All NMR spectra were measured with JNM-ECA-500, JNM-AL-400, or JNM-ECX-400P (JEOL, Tokyo, Japan). Chemical shifts are defined using tetramethylsilane as the internal standard, except for the measurement with DMSO-d6 in which solvent peaks (2.49 ppm for 1H and 39.7 ppm for 13C) were used. Coupling constants (J) are given in Hz. Mass spectra were acquired with ESI techniques using JMS-HX 110, JMS-700TZ, or JMS-T100LP (JEOL, Tokyo, Japan).
2′,3′-O-Isoprppylideneadenosine (8)
Adenosine (20 g, 75 mmol) was dissolved in 800 mL of acetone–DMF (3:1, v/v) and to the mixture was added 14 g of TsOH monohydrate (75 mmol). After 4 h stirring, 19 mL of 2,2-dimethoxypropane (150 mmol) was added and the reaction was allowed to stir for another 6 h. The mixture was partially evaporated and partitioned between EtOAc (200 mL) and sat. Na2CO3 aq. The water layer was washed with CHCl3 and CH2Cl2. Combined organic layer was then washed with brine (100 mL) and dried over anhydrous Na2SO4. After the removal of solvent in vacuo, 2′,3′-O-isoprppylideneadenosine (8) was recovered by trituration with EtOAc–hexane as colorless crystals (8.0 g, 35%). 1H NMR (400 MHz, DMSO-d6) δ 8.32 (1H, s, H, H-2), 8.14 (1H, s, H-8), 7.34 (2H, s, H-6), 6.10 (1H, d, J = 2.8 Hz, H-1′), 5.33 (1H, dd, J = 6.4, 3.2 Hz, H-2′), 4.95 (1H, dd, J = 6.2, 2.5 Hz, H-3′), 4.20 (1H, m, H-4′), 3.52 (2H, m, H-5′), 1.53 (3H, s, Me), 1.31 (3H, s, Me).
5′-O-tert-Butyldimethylsilyl-2′,3′-O-isopropylideneadenosine (9)
Imidazole (5.4 g, 79 mmol) and 7 were dissolved in 50 mL of DMF and tert-butyldimethylchlorosilane (6.0 g, 40 mmol) was added to the solution at 0 °C under argon atmosphere. The reaction was quenched with MeOH in 1 h and was partitioned between EtOAc (300 mL) and sat. Na2CO3 aq. (250 mL). The organic layer was washed with water (150 mL × 3) and brine (100 mL), dried over anhydrous Na2SO4 and evaporated in vacuo. The product 9 was recovered as colorless crystals by filtration, and also from the filtrate after evaporation and trituration with hexane (14 g, quant.). 1H NMR (400 MHz, DMSO-d6) δ 8.39 (lH, s, H-2), 8.05 (1 H, s, H-8), 6.17 (1 H, d, J = 2.8 Hz, H-1′), 5.48 (2 H, br s, H-6), 5.27 (lH, dd, J = 6.4, 2.3 Hz, H-2′), 4.94 (1 H, dd, J = 6.4, 2.3 Hz, H-3′), 4.43 (1 H, dd, J = 6.9, 4.1 Hz, H-4′), 3.88 (1 H, dd, J = 11.4, 4.1 Hz, H-5′a), 3.76 (1 H, dd, J = 11.4, 4.1 Hz, H-5′b), l.64 (3 H, s, Me), 1.41 (3 H, s, Me), 0.84 (9 H, s, t-Bu), 0.02 and 0.01 (6 H, s, Me).
5′-O-tert-Butyldimethylsilyl-2′,3′-O-isopropylide-6-N-triphenylmethyladenosine (10)
To a solution of 9 (10 g, 24 mmol) and triethylamine (7.9 mL, 57 mmol) in 100 mL of CH2Cl2, 7.8 g of trityl chloride (28 mmol) was added at 0 °C. The reaction was allowed to stir at room temperature then 50 °C under argon atmosphere. After complication of the reaction, sat. Na2CO3 aq. was added and resulting white precipitate was filtered off. The filtrate was evaporated and dissolved in 200 mL of EtOAc. The organic layer was washed with 200 mL of water, 0.1 M HCl aq., sat. Na2CO3 aq. and brine. The EtOAc layer was dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was purified with silica gel column chromatography eluting with EtOAc:hexane = 1:10–3:1 then MeOH:CHCl3 = 1:4 to give 11 g of 10 (71%) and 2.8 g of starting material 9 (20%). 1H NMR (400 MHz, CDCl3) δ 8.06 (1H, s, H-2), 7.97 (1H, s, H-8), 7.30 (15H, m, Tr), 6.92 (1H, s, H-6), 6.12 (1H, d, J = 2.7 Hz, H-1′), 5.28 (1H, dd, J = 6.2, 2.7 Hz, H-2′), 4.93 (1H, dd, J = 6.4, 4.1 Hz, H-4′), 3.84 (1H, dd, J = 11.5, 4.1 Hz, H-5′a), 3.75 (1H, dd, J = 11.5, 4.1 Hz, H-5′b), 1.61 (3H, s, Me), 1.38 (3H, s, Me), 0.83 (9H, s, t-Bu), 0.00 and –0.02(6H, s, Me).
2′,3′-O-Isopropylidene-6-N-triphenylmethyladenosine (11)
To a solution of 10 in THF (150 mL) was added 17 mL of tetra-n-butylammonium bromide 1 M solution at 0 °C under argon atmosphere. After 40 min, the mixture was evaporated and directly subjected to a silica gel column chromatography (EtOAc:hexane = 1:1–5:1) to isolate 11 as a yellow foam (9.1 g, quant.). 1H NMR (400 MHz, CDCl3) δ 7.98 (1H, s, H-2), 7.79 (1H, s, H-8), 7.30 (15H, m, Tr), 7.05 (1H, s, H-6), 5.83 (1H, d, J = 5.0 Hz, H-1′), 5.17 (1H, t, J = 5.7 Hz, H-2′), 5.08 (1H, d, J = 6.0 Hz, H-3′), 4.52 (1H, s, H-4′), 3.93 (1H, dt, J = 12.8, 1.8 Hz, H-5′a), 3.75 (1H, dt, J = 12.8, 1.8Hz, H-5′b), 1.64 (3H, s, Me), 1.36 (3H, s, Me).
(E)-N-(3-Methyl-1,4-bismethoxymethylphenyl)-3-{(3aR,4R,6R,6aR)-6-(6-triphenylmethylamino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-enamide (22)
Alcohol 4 (550 mg, 1.0 mmol) was dissolved in CH2Cl2 (10 mL) and excess amount (>4.0 eq.) of Dess–Martin periodinane was added at 0 °C under argon atmosphere. After complication of the reaction, sat. Na2S2O3 aq. and sat. Na2CO3 aq. were added. To the mixture was added 20 mL of water and extracted with EtOAc (30 mL). The organic layer was washed with sat. Na2CO3 aq. (20 mL) and brine (20 mL), dried over anhydrous Na2SO4 and evaporated in vacuo. Resulting crude product (330 mg) was used in next step without further purification.
The aldehyde 18, Zn(OTf)2 (440 mg, 1.2 mmol), triethylamine (560 μL, 4.0 mmol), and TMEDA (180 μL, 1.2 mmol) were added to 3.0 mL of THF. A solution of 11 in THF (12 mL) was added to the mixture, which was allowed to stir for 15 under argon atmosphere. The mixture was evaporated and partitioned between EtOAc (40 mL) and water (40 mL). The organic layer was washed with 20 mL of water, 0.1 M HCl aq., sat. Na2CO3 aq., and brine. The EtOAc layer was dried over anhydrous Na2SO4 and evaporated in vacuo. Subsequent silica gel column chromatography eluting with EtOAc:hexane = 2:3–2:1 gave 14 as brown foam (420 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 8.05 (1H, s, purinyl H-2), 7.85 (1H, s, purinyl H-8), 7.58 (1H, s, phenyl), 7.30 (15H, m, Tr), 6.99 (1H, dd, J = 15.6, 5.0 Hz, olefinic H-3), 6.95 (1H, s, purinyl H-6), 6.77 (1H, s, phenyl), 6.13 (1H, dd, J = 15.6, 1.4 Hz, olefinic H-2), 6.10 (1H, d, J = 6.1 Hz, H-6), 5.49 (1H, dd, J = 6.1, 2.5 Hz, H-6a), 5.18 (2H, s, OCH2OCH3), 5.05 (3H, m, H-3a and OCH2OCH3), 4.83 (1H, t, J = 3.4 Hz, H-4), 3.83 (3H, s, OMe), 3.53 (3H, s, OCH2OCH3), 3.41 (3H, s, OCH2OCH3), 1.63 (3H, s, Me), 1.38 (3H, s, Me); LR-ESI-MS m/z = 814.33 [M]+.
(E)-N-methyl-N-(3-Methyl-1,4-bismethoxymethylphenyl)-3-{(3aR,4R,6R,6aR)-6-(6-triphenylmethylamino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-enamide (23)
Following the same procedure as synthesis of 22, N-methyl product 23 was prepared (1.7 g, 57%) from the reaction between crude 12 and 1.6 g of 21 (3.6 mmol).
1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 8.05 (1H, s, purinyl H-2), 8.03 (1H, s, purinyl H-2), 7.73 (1H, s, purinyl H-8), 7.71 (1H, s, purinyl H-8), 7.30 (30H, m, Tr), 6.90 (6H, m, purinyl H-6, olefinic H-3 and phenyl), 6.79 (1H, s, phenyl), 6.78 (1H, s, phenyl), 6.02 (4H, m, H-6 and olefinic H-2), 5.33 (1H, dd, J = 6.4, 2.8Hz, H-6a), 5.26 (1H, dd, J = 6.4, 2.8 Hz, H-6a), 5.12 (8H, m, OCH2OCH3), 4.88 (2H, m, H-3a), 4.59 (2H, m, H-4), 3.87 (6H, s, OMe), 3.49 (3H, s, OCH2OCH3), 3.46 (3H, s, OCH2OCH3), 3.39 (3H, s, OCH2OCH3), 3.35 (3H, s, OCH2OCH3), 3.22 (3H, s, NMe), 3.21 (3H, s, NMe), 1.56 (3H, s, Me), 1.55 (3H, s, Me), 1.32 (6H, s, Me); ESI-MS m/z = 828 .35 [M]+.
(E)-N-(4-Methoxy-p-benzoquinonyl)-3-{(3aR,4R,6R,6aR)-6-(6-triphenylmethylamino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-enamide (3)
A precursor 22 was dissolved in 80% trifluoroacetic acid aqueous solution. After stirring for 25 min, dilution with EtOH and evaporation was repeated several times. The concentrate was dissolved in 15 mL of MeOH and Pd/C (Pd 10%) (31 mg) was added to the solution. The suspension was allowed to stir at O2 atmosphere. After 25 min, Pd/C was removed by celite filtration and solvent was removed in vacuo. Subsequent recrystallization from MeOH gave 82 mg of 3 as yellow crystals (73%).
1H NMR (400 MHz, DMSO-d6) δ 8.49 (1H, s, purinyl H-2), 8.26 (1H, s, purinyl H-8), 7.80 (2H, br s, purinyl H-6), 7.42 (1H, s, quinonyl), 7.01 (1H, dd, J = 16.0, 5.5 Hz, olefinic H-3), 6.82 (1H, dd, J = 16.0, 1.5 Hz, olefinic H-2), 6.15 (1H, s, quinonyl), 5.99 (1H, d, J = 5.0 Hz, H-6), 4.62 (1H, t, J = 5.0 Hz, H-6a), 4.53 (1 H, t, J = 5.5 Hz, H-4), 4.22 (1 H, t, J = 5.0 Hz, H-3a), 3.79 (3H, s, OMe); 13C NMR (125 MHz, DMSO-d6) δ 183.3, 182.8, 165.7, 159.9, 158.9, 153.5, 149.4, 144.1, 142.0, 140.7, 124.8, 119.6, 113.0, 105.8, 88.6, 83.5, 74.2, 73 .7, 57.3; ESI-MS m/z = 442.12 [M]+.
(E)-N-(4-Methoxy-p-benzoquinonyl)-3-{(3aR,4R,6R,6aR)-6-(6-triphenylmethylamino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl}propanamide (5)
Amide 3 (15 mg, 27 μmol) was dissolved in 6.0 mL of MeOH and to the solution was added 10 mg of Pd/C (Pd 10%). The reaction was allowed to stir under H2 atmosphere for 8 h followed by O2 atmosphere for another 70 min. Pd/C was removed by celite filtration and solvent was evaporated in vacuo. Recrystallization with the mixed solvent of MeOH, EtOAc, and hexane gave propanamide 5 (8.1 mg, 54%) as yellow crystals.
1H NMR (400 MHz, DMSO-d6) δ 8.45 (1H, d, J = 3.6 Hz, purinyl H-2), 8.24 (1H, d, J = 3.2 Hz, purinyl H-8), 7.86 (2H, br s, purinyl H-6), 7.24 (1H, s, quinonyl), 6.09 (1H, s, quinonyl), 5.85 (1H, d, J = 5.6 Hz, H-6), 4.63 (1H, t, J = 5.0 Hz, H-6a), 4.05 (1H, t, J = 5.0 Hz, H-3a), 3.87 (1H, m, H-4), 3.79 (3H, s, OMe), 2.63 (2H, t, J = 7.5 Hz, olefinic H-2), 1.93 (2H, m, olefinic H-3); 13C NMR (125 MHz, DMSO-d6) δ 182.7, 182.2, 173.8, 159.2, 156.1, 152.7, 149.4, 139.8, 139.8, 119.2, 111.6, 105.1, 87.4, 83.0, 73 .1, 73.0, 56.6, 32.9, 28.4, 22.1; ESI-MS m/z = 467.13 [M + Na]+.
(E)-N-Methyl-N-(4-methoxy-p-benzoquinonyl)-3-{(3aR,4R,6R,6aR)-6-(6-triphenylmethylamino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-enamide (4)
Following the same procedure as synthesis of 3, N-methyl product 4 was prepared in 77% yield. Recrystallization was performed with the combination of MeOH and EtOAc. 1H NMR (500 MHz, DMSO-d6) δ 8.35 (1H, s, purinyl H-2), 8.20 (1H, s, purinyl H-8), 7.75 (2H, br s, purinyl H-6), 6.83 (1H, dd, J = 14.9, 6.3 Hz, olefinic H-3), 6.78 (1H, s, quinonyl), 6.16 (1H, dd, J = 14.9, 1.5 Hz, olefinic H-2), 6.12 (1H, s, quinonyl), 5.92 (1H, d, J = 5.2 Hz, H-6), 4.64 (1H, t, J = 5.2 Hz, H-6a), 4.46 (1H, t, J = 6.3 Hz, H-4), 4.15 (1H, t, J = 5.2 Hz, H-3a), 3.77 (3H, s, OMe), 3.12 (3H, s, NMe); 13C NMR (100 MHz, DMSO-d6) δ 182.9, 181.5, 165.3, 159.1, 153.9, 150.0, 149.0, 147.2, 141.6, 140.9, 127.1, 123.5, 119.1, 106.7, 87.8, 83.0, 73.7, 72.8, 56.8, 36.4; ESI-MS m/z = 479.13 [M + Na]+.
Computational conformation analysis
All calculations were performed using the Spartan’16 (Wavefunction Inc., Irvine, CA, USA) in gas conditions. The structures of compounds 3 and 4 were built and the conformer distributions were first analyzed by MMFF. For the given conformers of 3 and 4, IDs were assigned as “M3-NNN” or “M4-NNN”, respectively, where the numbers after prefixes are the order of relative energies after molecular mechanics calculation. The conformers within relative energies of 40 kJ/mol were geometrically optimized by Hartree–Fock/3–21G, and energies of resulting conformers within 40 kJ/mol were evaluated using density functional (DF)/ωB97 X-D/6–31G* calculation. The conformers with >15 kJ/mol relative energies were discarded and geometry optimization was again performed using DF/ωB97 X-D/6–31G*. This series of DF calculations returned M3–007 and M4–015 as the lowest-energy conformers together with the conformers within 10 kJ/mol relative energies (one conformer for M3–007 and four conformers for M4–015; see Fig. 2). Finally, energies and Boltzmann distribution at 297 K of these conformers were calculated using DF/ωB97 X-V/6–311 + G (2df, 2p).
Biology
Expression and purification of recombinant CpIMPDH and human IMPDH II
The PCR product coding CpIMPDH amplified with the primer sets 5′-TTTTGGATCCTCAAACATGGGTACA-3′ and 5′-TTTTGAATTCCTATTTACT-ATAATT-3′ was cloned into pCR2.1-TOPO vector (Invitrogen Japan KK, Tokyo, Japan). The target gene was digested by BamHI and EcoRI, and inserted into a pGEX-6P-2 plasmid (GE Healthcare Bio-Science Corp., UK). The plasmid was then transformed to ECOS E. coli BL21 (DE3) (Wako Pure Chemical Ind., Ltd., Japan). The cells were grown overnight at 30 °C in 50 mL 2× YT broth containing 100 μg/mL ampicillin. Afterwards, the broth was subcultured to 700 ml of medium containing a final concentration of 1.0 mM isopropyl-1-thio-β-galactopyranoside (Sigma-Aldrich Japan, Tokyo, Japan) and 100 μg/mL ampicillin. After 5 h incubation at 25 °C, cells were harvested by centrifugation, washed with PBS solution, and stored frozen at –80 °C until usage.
hIMPDH II plasmid was a generous gift from Prof. Lizbeth Hedstrom, Brandeis University, USA. The plasmid was transformed in the same manner as CpIMPDH.
High-throughput screening of IMPDH inhibitors
The HTS assay was established using the NAD(P)H-Glo Assay Kit (Promega, Madison, WI, USA). Briefly, 10 μL of substrate solution containing 50 mM Tris-HCl pH 8.0, 200 mM KCl, 0.1 mg/mL BSA, 1.6 mM β-NAD+ (Oriental Yeast Co., Tokyo, Japan), 100 μM IMP (Sigma-Aldrich Japan, Tokyo, Japan), and assay kit was added to white 384-well plates. Mycophenolic acid (APAC Pharmaceutical LLC, Hangzhou, China), a known IMPDH inhibitor, or the chemical library from the Faculty of Pharmacy, Hokkaido University was also added to the wells in concentration of 10 μM. Reaction was started by addition of 10 μL enzyme solution containing 50 mM Tris-HCl pH 8.0, 0.1 mg/mL BSA, and CpIMPDH. Reaction was carried out at 30 °C for 30 min in dark. Solution handlings was carried out using Mosquito LCP (TTP LabTech Ltd., Melbourn, UK) and Multidrop Combi (Thermo Scientific, Waltham, MA, USA). The production of NADH was monitored by the measurement of luminescence (RLU) using either VERITAS Microplate Luminometer (Turner Biosystems, Sunnyvale, CA, USA) or EnSpire Multimode Reader (Perkin-Elmer, Waltham, MA, USA). The experiments were performed without repetition in first screening, and in triplicate in other assays.
Determination of IC50 values of hit and synthetic compounds
Standard IMPDH assay solution containing 50 mM Tris-HCl pH 8.0, 100 mM KCl, 3 mM EDTA, 0.1 mg/mL BSA, 1.0 mM DTT, and appropriate amounts of inhibitors with a total volume of 270 μL. Substrate concentrations in the solution were 500 μM NAD+ and 250 μM IMP for CpIMPDH. Reaction was started by the addition of 30 μL enzyme solution (CpIMPDH: 0.15 μg/mL; hIMPDH: 0.75 μg/mL). The activity of the enzyme was measured by monitoring NADH production in either absorbance at 340 nm or fluorescence emissions at 465 nm. IC50 values were calculated by plotting the recorded NADH yield against log of compound concentration in GraFit ver. 7 (Erithacus Software, UK). All screening assays in this study was conducted with 0.5% DMSO as vehicle and library compound concentration of 10 μM unless stated otherwise.
References
DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. The infectivity of Cryptosporidium parvum in healthy volunteers. N Engl J Med. 1995;332:855–9.
Hunter PR, Nichols G. Epidemiology and clinical features of Cryptosporidium infection in immunocompromised patients. Clin Microbiol Rev. 2002;15:145–54.
Fayer R. Cryptosporidium. A wter-borne zoonotic parasite. Vet Parasitol. 2004;126:37–56.
Sarwono AEY, Mitsuhashi S, Kabir MHB, Shigetomi K, Okada T, Ohsaka F, Otsuguro S, Maenaka K, Igarashi M, Kato K, Ubukata M. Repurposing existing drugs: identification of irreversible IMPDH inhibitors by high-throughput screening. J Enzym Inhib Med Chem. 2019;34:171–8.
Muranaka K, Ichikawa S, Matsuda A. Development of the carboxamide protecting group, 4-(tert-butyldimethylsiloxy)-2-methoxybenzyl. J Org Chem. 2011;76:93.
Muranaka K, Ichikawa S, Matsuda A. Design and synthesis of 3′,5′-ansa-adenosines as potential Hsp90 inhibitors. Tetrahedron Lett. 2009;50:5102–6.
Sintchak MD, Nimmersgern E. The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacol. 2000;47:163–84.
Jaspars M, Pasupathy V, Crews P. A tetracyclic diamine alkaloid, halicyclamine A, from the marine sponge Haliclona sp. J Org Chem. 1994;59:3253–5.
Felczak K, Vince R, Pankiewicz KW. NAD-based inhibitors with anticancer potential. Bioorg Med Chem Lett. 2014;24:332–6.
Franchetti P, Cappellacci L, Pasqualini M, Petrelli R, Jayaprakasan V, Jayaram HN, Boyd DB, Jain MD, Grifantini M. Synthesis, conformational analysis, and biological activity of new analogues of thiazole-4-carboxamide adenine dinucleotide (TAD) as IMP dehydrogenase inhibitors. Bioorg Med Chem. 2005;13:2045–53.
Chen L, Wilson DJ, Xu Y, Aldrich CC, Felczak K, Sham YY, Pankiewicz KW. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J Med Chem. 2010;53:4768–78.
Franchetti P, Cappellacci L, Perlini P, Jayaram HN, Butler A, Schneider BP, Collart FR, Huberman E, Grifantini M. Isosteric analogues of nicotinamide adenine dinucleotide derived from furanfurin, thiophenfurin, and selenophenfurin as mammalian inosine monophosphate dehydrogenase (type I and II) inhibitors. J Med Chem. 1998;41:1702–7.
Digits JA, Hedstrom L. Drug selectivity is determined by coupling across the NAD+ site of IMP dehydrogenase. Biochemistry. 2000;39:1771–7.
Townsend AP, Roth S, Williams HE, Stylianou E, Thomas NR. New S-adenosyl-L-methionine analogues: synthesis and reactivity studies. Org Lett. 2009;11:2976–9.
Enomoto K, Nagasaki T, Yamauchi A, Onoda J, Sakai K, Yoshida T, Maekawa K, Kinoshita Y, Nishino I, Kikuoka S, Fukunaga T, Kawamoto K, Numata Y, Takemoto H, Nagata K. Development of high-throughput spermidine synthase activity assay using homogeneous time-resolved fluorescence. Anal Biochem. 2006;351:229–40.
Andrus MB, Hicken EJ, Meredith EL, Simmons BL, Cannon JF. Selective synthesis of the para-quinone region of geldanamycin. Org Lett. 2003;5:3859–62.
Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6:4459–62.
Schauer DJ, Helquist P. Mild zinc-promoted Horner–Wadsworth–Emmons reactions of diprotic phosphonate reagents. Synthesis. 2006;21:3654–60.
Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: identification of functional, structural, and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–7.
Lee H, Torres J, Truong L, Chaudhuri R, Mittal A, Johnson ME. Reducing agents affect inhibitory activities of compounds: results from multiple drug targets. Anal Biochem. 2012;423:46–53.
Risal D, Strickler MD, Goldstein BM. Crystal structure of human inosine monophosphate dehydrogenase type II complexed with the MPA/NAD analog C2-MAD. PDB ID: 1NF7. https://doi.org/10.2210/pdb1NF7/pdb. Released 27 Jan 2004.
Acknowledgements
The preliminary screening study was partly supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number 18am0101093j0002, Hokkaido University, Global Facility Center (GFC), Pharma Science Open Unit (PSOU), funded by MEXT under “Support Program for Implementation of New Equipment Sharing System” and Takeda Science Foundation. We sincerely thank the Institute for Fermentation, Osaka (IFO) Japan for the financial support. This work was supported in part by Grant-in-Aid for Challenging Exploratory Research (JSPS No. 25660051).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is dedicated to the 88th anniversary of the birth of Dr. Kiyoshi Isono.
Rights and permissions
About this article

Cite this article
Shigetomi, K., Sarwono, A.E., Ichikawa, S. et al. Novel adenosine-derived inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J Antibiot 72, 934–942 (2019). https://doi.org/10.1038/s41429-019-0199-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0199-3
This article is cited by
-
An update on Cryptosporidium biology and therapeutic avenues
Journal of Parasitic Diseases (2022)
