
Abstract
The kedarcidin chromophore is a formidible target for total synthesis. Herein, we describe a viable synthesis of this highly unstable natural product. This entailed the early introduction and gram-scale synthesis of 2-deoxysugar conjugates of both l-mycarose and l-kedarosamine. Key advances include: (1) stereoselective allenylzinc keto-addition to form an epoxyalkyne; (2) α-selective glycosylations with 2-deoxy thioglycosides (AgPF6/DTBMP) and Schmidt donors (TiCl4); (3) Mitsunobu aryl etherification to install a hindered 1,2-cis-configuration; (4) atropselective and convergent Sonogashira-Shiina cyclization sequence; (5) Ohfune-based amidation protocol for naphthoic acid; (6) Ce(III)-mediated nine-membered enediyne cyclization and ester/mesylate derivatisation; (7) SmI2-based reductive olefination and global HF-deprotection end-game. The longest linear sequence from gram-scale intermediates is 17-steps, and HRMS data of the synthetic natural product was obtained for the first time.
Similar content being viewed by others
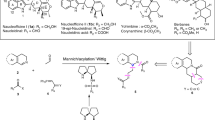

Introduction
Total synthesis is a challenging field. Even more so, if the natural product is complex in structure and non-obvious in construction. The ensuing challenge reaches unprecedented levels when the natural product is highly unstable. Even more so, if late-stage synthetic precursors are equally unstable. Very few natural products have been tackled under such criteria. Outstanding cases in the antitumor antibiotic field include the ten-membered and nine-membered cyclic enediynes [1,2,3,4,5,6]. Complexity aside, the latter enediynes are arguably more challenging to make because of increased ring-strain [7,8,9,10,11,12,13]. A case in point is the kedarcidin chromophore (1, Fig. 1). This nine-membered cyclic enediyne exists kinetically stabilized in nature, as part of its chromoprotein complex, kedarcidin [14, 15]. The enediyne 1, for example, decomposes within 1–2 h at room temperature once separated from its non-covalently bound apoprotein, even in aprotic solvents. Notably, nine-membered bicyclic enediynes like 1 readily undergo both spontaneous and nucleophile-induced cycloaromatizations via highly reactive p-benzyne diradical species to give aromatized benzenoid products [16,17,18], some of which are more readily isolated and synthesized in stable cyclized forms on the bench [19,20,21,22,23].

Structural revisions and numbering system of the kedarcidin chromophore (1)
Kedarcidin itself was first discovered in 1990 by Bristol-Myers Squibb. It was identified as a cytotoxic product from the supernatant of an unkown microbe cultured from a soil sample collected in the Maharasta State of India. In 1991, the company disclosed the product (kedarcidin) to be a new potent, chromoprotein antitumor antibiotic [24, 25]. The producing organism was eventually designated to be an actinomycete strain L585–6 of uncertain taxonomy. Today, the genus is likely to be Streptoalloteichus sp. ATCC 53650 (not Saccharothrix). This particular species has recently been shown to produce kedarcidin. Gene sequencing has also shown ATCC 53650 to contain all of the biosynthetic machinery necessary to construct the kedarcidin chromophore (1) [26].
During 1992–1994, the bioactivities and structures of the isolated chromophore (1) and apoprotein of kedarcidin were further elucidated by Leet and colleagues within Bristol-Myers Squibb [27,28,29]. Like other chromoprotein antitumor antibiotics, kedarcidin elicits an extraordinary ability to drive an astonishing sequence of histone/DNA recognition and peptide/nucleotide cleavage events [1,2,3,4,5,6,7,8,9,10,11,12]. The acidic apoprotein of kedarcidin is proposed to first associate and enzymatically cleave the basic histone-coiled proteins [30]. Subsequent exposure of chromosomal DNA, release of the enediyne core (1), naphthyl-based DNA intercalation, 5′-TCCTN-3′ sequence recognition, and Masamune-Bergman cycloaromatization of 1, thereby generates a p-benzyne diradical that is transiently and non-covalently bound to DNA. This latter species then initiates DNA-strand breaking and crosslinking events via hydrogen abstraction of the deoxyribose backbone. These oxidative events consequently trigger cell death via the generation of carbon-centered radicals and radical oxygen species (ROS). Despite this non-trivial sequence of events, kedarcidin still elicits potent, yet selective in vivo antitumor activity against P388 leukemia and B16 melanoma cells.
Equally eventful and non-trivial has been the structural elucidation of the kedarcidin chromophore (1). To date, extensive NMR, MS/MS, chemical degradation, derivatization, reductive, radical-trapping, biosynthetic, and total synthesis studies have provided convincing evidence for the enediyne structure 1. In 1993, Leet et al. described in full their seminal characterization studies of the chromophore structure [28]. They first proposed an azatyrosyl α-amino motif about the ansa-macrolide bridge (Fig. 1). In 1997, we updated the whole structure to be antipodal and demonstrated the chromophore to be a β-amino acid derived ansa-macrolide (1′) [31]. It is noteworthy that the amino-mutase to achieve such a β-amino motif has only recently been characterized [32]. In 2007, Myers and coworkers completed an impressive total synthesis of this 1997-structure 1′ [33]. Comparison of natural and synthetic 1H NMR data, nevertheless, indicated the C10-α-epimeric stereoconfiguration of 1′ should be inverted to 1.
In 2009, we provided strong NMR spectroscopic evidence for Myers’ C10-β-epimer 1 through the synthesis of the complete aglycon 2 of the kedarcidin chromophore in protected form [34]. The currently accepted target for synthesis is thus Myers’ structure 1. Herein, we report a detailed account of our early-stage incorporation of both kedarcidin sugars (as elaborate O-protecting groups) and the convergent construction of the multicyclic, fully functionalized cyclic epoxyenediyne core. Collectively, our efforts have led to the development of a viable total synthesis of 1 as characterized by HRMS. Product instabilities have, however, prevented clean NMR characterization of the cyclic enediyne material in unprotected form.
Results and discussion
In previous studies to the kedarcidin aglycon 2, we secured several enantioselective routes to gram quantities of all key fragments: epoxy-iodocyclopentene 3, aza-β-tyrosine 4, alkyne-polyol 5, and naphthoic acid 6 (Scheme 1) [31, 34,35,36,37,38]. We also determined practical methods to synthesize the 2-deoxy sugars, l-mycarose 7 and l-kedarosamine 8 [39]. Not only this, but we developed and achieved the direct α-selective glycosylation of several advanced C10-α-epimeric aglycon precursors to 1′ [33, 40, 41]. The key question now was when to incorporate the kedarcidin sugars into our general synthesis plan (Scheme 1). The C4/C5-dioxy, epoxybicyclo[7.3.0]-dodecenediyne frameworks like 10 and 11 are known to be exceedingly unstable [34]. Among other decomposition possibilities, such frameworks are prone to undergo facile oxy-Cope ring openings to afford bis-allenyl species [37]. The question thus came down to incorporating the sugars at an early or late stage en route to making 9. Importantly, these glycosylation events should be executed before cyclization into a highly labile, nine-membered ring system like 10. In either case, the efficiency and α-stereoselectivity of our current glycosylation protocols [40, 41] needed to be tested on newly functionalized substrates of unknown reactivity (cf. 3, 5, and 9).

General total synthesis plan for the kedarcidin chromophore (1)
At first, a late-stage glycosylation strategy was investigated. The ansa-macrolides 18 and 19 (akin to 9) were thus targeted as suitable l-mycarose and l-kedarosamine acceptors, respectively (Scheme 2). Treatment of 5 with 2,2-dimethoxypropane and acetyl deprotection afforded the acetonido-alkyne 12 in 76% yield, 2 steps. Sonogashira coupling of 12 with the known iodo-cyclopentene 13 [34] in degassed DMF under Pd2(dba)3·CHCl3/CuI catalysis, followed by selective protodesilylation of the TMS-C-acetylene, gave the ansa-macrolide precursor 14 in 61% yield, 2 steps. Saponification of 14 afforded the corresponding carboxylic acid. This acid was immediately subjected to Shiina macrolactonization conditions with 2-methyl-6-nitrobenzoic anhydride (MNBA) [42, 43]. These conditions gave the macrolide 15 as a single atropisomer in 62% yield, 2 steps. Mild and selective N-Boc deprotection of 15 (via an O-TBS carbamate) [44] and HOAt-mediated [45] condensation of the free amine 16 with the known naphthoic acid 6 (R=H) [31] gave the amide 17. Final treatment of 17 with TBAF, dual C-trimethylsilylation and O-trimethylsilylation, and chemoselective C10-O-desilylation, gave the l-mycarose C10-O-acceptor 18 with its terminal acetylene suitably C-protected (thereby minimizing known complications via Ag(I)-complexation) [40]. The alternative treatment of amide 17 with TFA/H2O (1:2) gave the l-kedarosamine C13-O-acceptor 19 with the C4-OH free (thereby improving known reactivity issues) [41].

Synthesis of late-stage, C10-α-epimeric aglycon acceptors 18 (for l-mycarose) and 19 (for l-kedarosamine)
Having the desired macrocyclic glycosyl acceptors in hand, we first examined the reactivity of 18 with l-mycarose (Scheme 3). The C10/C11-cis acceptor 18 under established α-selective conditions (AgPF6/DTBMP) with the thioglycoside 7 failed to yield any 2-deoxypyranoside (20). This result could not be overturned and was in contrast to the reactivity of the known C10/C11-trans acceptor 21 to give 22 [40], as well as the success of the AgPF6/DTBMP glycosylation method during the advanced stages of the total synthesis of the C10-epimer 1′ by the group of Myers [33]. Clearly, the cis-facial proximity of the chloropyridyl unit sterically prevented the glycosylation event.

Glycosylation of C10-α-epimeric alcohol 18 with l-mycarose (7)
Next, the glycosylation of the l-kedarosamine Schmidt donor 8 with the C4/C13-diol acceptor 19 was examined (Scheme 4). Initially, our reported α-selective conditions were found unsuccessful, for example, by using BF3 or TiCl4 at low or ambient temperatures in chlorinated solvents [41]. Eventually, we succeeded with BF3·Et2O in dichloroethane (DCE) at an elevated temperature (40 °C). This gave the desired 2°-α-pyranoside 23 as the minor product (19% isolated yield) in a 1:2 ratio with the 3°-glycoside 24. As found previously, no glycosylation occurred when the C4-OH group was protected. Such results do not fair well for a total synthesis.

Glycosylation of C4/C13-diol acceptor 19 with l-kedarosamine (8)
According to these findings, both glycosylations would be better performed at an early-stage of the synthesis (cf. Scheme 1). Such timings would allow for steric hindrances to be minimized (cf. 3 and 18; 5 and 19). In effect, the 2-deoxy-α-pyranoside sugar functionalities may be viewed as elaborate THP protecting groups (Sg1, Sg2) en route to constructing a bis-glycosylated enediyne cyclization precursor (e.g., 9). Although more risky, this strategy offers a more convergent total synthesis of 1. The acid lability, free hydroxyl and amino functionality, and extra steric potentials of the 2-deoxypyranosides, were thus considered to present additional synthetic challenges (vide infra).
Undeterred by such challenges, we elected to prepare gram quantities of the C10 and C13 O-glycosylated versions of 3 and 5, respectively (Scheme 1). These fragments would be used later for azatyrosine (4) incorporation and Sonogashira coupling studies (vide infra). We first targeted the propargyl oxirane moiety 3 as a suitable C10/C11-trans glycosyl acceptor (Scheme 5). After a few modifications to established procedures, the iodo-cyclopentenone 25 was prepared as its C10-OTES silyl ether (not as its TBS ether) [36]. Similar to the protocols of Chemla and Caddick [46, 47], the allenyl zinc species of 3-chloro-1-trimethylsilylpropyne (prepared at –78 °C) was reacted with the ketone 25 at –18 °C overnight. The crude chlorohydrin 26 was then treated with DBU in dichloromethane to afford the epoxyalkyne 27 stereoselectively in 70% yield, 2 steps. This latter step avoided the use of potassium carbonate [34], so that the TMS-C-protected alkyne 27 could be formed directly. Unlike its C10-OTBS counterpart [34], the TES ether of 27 could also be removed chemoselectively under Brønsted acid conditions to give the desired C10-OH acceptor 3. Gratifyingly, the thioglycoside 7 reacted smoothly with 2 equivalents of the 2°-alcohol 3 in the presence of AgPF6/DTBMP [40]. This furnished the C10/C11-trans α-pyranoside 28 exclusively in 81% yield. The excess alcohol 3 was then recovered and recycled. Gram quantities of pure 28 were produced in this manner.

Synthesis of C10-OH acceptor 3 and α-glycosylation with l-mycarose (7)
Next, the gram-scale, α-selective glycosylation of the C13/C4-diol acceptor 5 was pursued with various l-kedarosamine donors 8 (Scheme 6). Due to no silyl acetylene protection, AgPF6/DTBMP conditions were incompatible with 5 [41]. We thus chose NIS/TfOH to activate the thioglycoside of 8 [48]. This afforded the 2°-α-pyranoside 29 in a maximum yield of 26%. Coupling with the alternative glycosyl fluoride of 8 under Cp2HfCl2/AgClO4 conditions did not improve yields (15% at best) [49, 50]. Eventually, we found TiCl4 to be superior to BF3·Et2O in coupling the Schmidt donor 8 and diol 5 under our reported conditions [41]. For scale-up purposes, two-equivalents of diol 5 were used relative to 8, whereby 0.5 equivalents of TiCl4 were added under the gentle reflux of CH2Cl2. This rapidly gave the desired 2°-α-pyranoside 29 in a 40% isolated yield. Excess 5 was also recovered (ca. one-equivalent) and all cases produced minor amounts of the 3°-α-pyranoside 30 (R=H) as an inseparable mixture with 29. Here again, it is emphasized that no glycosylation occurred at all when the C4-OH group was protected. Gratifyingly, all pyranosides 29/30 were found to be α-anomeric (J = 4.0 Hz coupling constants). This is consistent with high kinetic control, presumably by virtue of the axial NMe2 group within an oxocarbenium conformation (8→29).

Glycosylation of diol 5 with l-kedarosamine (8) and origin of α-selectivity
Having gram quantities of the l-mycarose and l-kedarosamine fragments 28 and 29 in hand, azatyrosine incorporation of 4 and the search for suitable Sonogashira coupling substrates were explored (Scheme 7). Low temperature, reductive deprotection of the benzoate 28, by using DIBAL in toluene, thus provided 32. The cis-relative C10/C11-stereochemistry was next achieved by phenolic Mitsunobu inversion [51] of the allylic C11-β-alcohol 32 by the β-amino-2-chloroazatyrosine 4. For scale-up purposes, the use of DMEAD was found preferable to DEAD [52]. Triethylsilyl (TES) protection of the tertiary alcohol on l-mycarose then gave the l-mycarose fragment 34. Initial attempts at Sonogashira coupling between the iodoalkene 34 and the alkyne 29 or its diol 31 were, however, unsuccessful. These attempts were in contrast to previous successful studies with a C13-OMOM equivalent of the bulky l-kedarosamine fragment 29 [34]. We therefore decided to explore alternative substrates to achieve this key Sonogashira coupling step and minimize potential steric effects.

Mitsunobu installation of azatyrosine 4 to afford C10/C11-trans fragment 33 and attempted Sonagashira coupling between the sugar bearing fragments 34 and 29
Additional steric and conformational effects by the kedarosamine moiety were considered the primary causes for the unproductive iodoalkene-alkyne coupling between 29 and 34. We thus prepared various cylic diol-protected versions of 29. These modified substrates 36a–c proved to be successful under our established Pd(0)/Cu(I) Sonogashira conditions (Scheme 8) [35,36,37,38]. The orthoester 36c was selected as the optimal substrate for subsequent hydrolytic, ansa-macrolactonization studies. This minimized the loss of acid labile 2-deoxypyranoside moieties during the methanolysis of 37c to its free diol 38 (82%). The alternative cyclic acetals 37a/b could not be deprotected under those mild acidic conditions [53, 54]. Final saponification of 38 and Shiina macrolactonization [42, 43] generated the atropisomeric ansa-macrolide 39 exclusively in 52%, two steps. ROESY NMR analysis between the protons of the pyridyl C4′ and epoxy C8 of 39 confirmed its structure. We thus secured a viable route to bis-glycosylated cyclization precursors like 9 (Scheme 1).

Sonogashira coupling and saponification-macrolactonization study
Before progressing forward with 39 and attaching the naphthamide moiety 6, we became concerned at our protecting group strategy to 1 (cf. Scheme 1). Thus far, relatively strong O-TBS protected 2-deoxysugar fragments 34 and 36 were selected. Although useful in establishing the chemistry to advanced ansa-macrolides, a final global deprotection sequence to 1 needed to be both rapid and mild due to enediyne instabilities (cf. 10 and 11). We thus directed our attention to adjusting the protecting groups on the l-mycarose (7), l-kedarosamine (8) and naphthamide (6) moieties. Model substrates 40, 42, and 44 [14] were thus prepared and subjected to excess TBAF/o-nitrophenol and HF∙Et3N according to Myers’ established deprotection sequence to 1′ (Scheme 9) [33]. This study demonstrated the clear need for TES protection of the sugar moieties 40 (for R1) and 42 (for R3) during the end-game of a total synthesis, as well as the need for pivaloyl (Piv) phenolic protection for the naphthamide (44). In all these cases, deprotection could be achieved cleanly within 10–30 min In contrast, the TBS ethers of 40 (for R1) and of 42 (for R3) remained intact even after 3 h. Bis-TMS protection (R1, R2) of the mycarose 40 was also found acceptable, but other silyl combinations were not.

Protecting group selection under Myers’ global deprotection conditions [33]
Armed with this information and the experience gained in preparing 39, we turned our attention to assembling a suitably protected version of the advanced intermediate 9 for subsequent enediyne cyclisation studies. After several trials, we settled on making the bis-glycosylated ansa-macrolide 50 according to Scheme 10. In this particuar case, we began with the TES-protected kedarosamine fragment 46 (freshly prepared) and the TBS-protected mycarose fragment 33 (3°-OH free). After Sonogashira coupling and orthoester methanolysis to diol 47, the TES ether proximal to NMe2 was found to cleave during the Shiina macrolactonization step. This generated the ansa-macrolide 48. After TES ether re-protection of the l-kedarosamine moiety of 48, a chemoselective one-pot amidation procedure was developed. This entailed the sequential addition of TMSOTf/2,6-lutidine, akin to Ohfune’s NH-Boc deprotection conditions [44], followed by saturated aqueous sodium bicarbonate solution and the one-pot addition of a preformed CH2Cl2 solution of the HOBt-activated naphthoate ester 6. This afforded the fully protected ansa-macrolide 50 in 63% yield from 48.

Reliable assembly of a fully protected, storable ansa-macrolide (50)
Under this scheme, we could reliably prepare 30–90 mg quantities of 50. Here, samples could be safely stored as dilute CH2Cl2 solutions at –20 °C over a couple of months. When required, a suitably protected aldehyde substrate would thus be prepared for immediate enediyne cyclization studies. Preparations of the mono-TES (54) and bis-TMS (55) protected aldehydes are given in Scheme 11. Indeed, we found the silylation strategy to be critical during the final deoxygenation-deprotection sequence in order to successfully form the enediyne chromophore 1 in its fully fledged free form (vide infra). After complete TBAF deprotection of 50 to its unstable pentaol, care was needed to achieve the differential Piv, TES, and TMS O-protection pattern as achieved in 51 and 52. In one-pot operations, mono-pivalation of the naphtholic group was first effected at –78 °C with PivCl. Next, mono-triethylsilylation of the kedarsoamine moiety was effected at 0 °C. This was followed by either mono-TES or bis-TMS silyation of the mycarose moiety in the presence of cat. DMAP. Ultimately, in the same pot, the C4-OH was protected as its TMS ether. Subsequent treatment of 51/52 with DDQ resulted in the N-demethylated alcohol 53. Although the oxidative N-methyl cleavage process could not be circumvented, crude 53 was readily N-methylated under reductive amination conditions using formalin and NaBH3CN. Dess-Martin periodinane (DMPI) oxidation of the primary alcohol then delivered the aldehydes 54 and 55 in good overall yields (75–90% over 3 steps from 51/52).

Preparation of aldehyde enediyne cyclisation precursors (54/55)
The formidable challenges to transform multicyclic alkyne-aldehydes like 54/55 into fully fledged, epoxybicyclo[7.3.0]-dodecadienediyne cores (e.g., 54→56, 55→57) should not be underestimated by any means. Whilst the aldehydes themselves are considered unstable in traditional senses, once the nine-membered enediyne cores are forged, our experience dictated that all subsequent synthetic operations and characterization studies should be ideally performed within 16 h, especially for kedarcidin-based chromophores. All reagents, methods and work-up operations need to be mild, streamlined, and rapid in both chemical and practical senses. After considerable experimentation and refinement of reaction timings and bench skills, a 6-step sequence to 1 was eventually shown to be viable over a total time period of 12-h from 55 (Scheme 12). An overview of the substrates prepared and studied along similar lines to Schemes 11 and 12 (bearing differing protecting groups and leaving groups, R1–R4) is given in Table 1.

Successful nine-membered enediyne cyclisation of 54/55 and end-game sequence to 1 (see Table 1 for other substrates studied)
Specifically, the nine-membered epoxy-diyne cyclization of 55 using CeCl3/LiHMDS to give the C4→C5 O-migrated TMS product 57 necessitated careful quenching with phosphate buffer (pH 7) at –78 °C (Scheme 12) [55,56,57,58,59]. The resulting product 57 was treated with TBAF carefully at –78 °C to remove the C5-O-TMS group. For the cis-C4,C5-diol mesylate derivative 62, desilylation was immediately followed by mesylate formation and then esterification with p-trifluoromethylbenzoyl (TFBz) chloride. The bis-OTMS, bis-OTFBz substrate 63 was also prepared by omitting the mesylation step. This proved to be more time economical, but 63 was found to be more unstable than its C5-OMs counterpart (62). It should be noted that for diyne systems, electron withdrawing C4,C5-diol substituents marginally reduce the propensity of nine-membered cores from undergoing oxy-Cope like sigmatropic rearrangments (e.g., 1H NMR data was obtained for 59) [35, 37] and systems with differing stereochemistries and oxygenation patterns, which are less prone to such rearrangements and side-reactions, can be purified and clean NMR data acquired [33,34,35]. For the cases in hand, the epoxy diyne cores 58–63 remained highly unstable to all silica gel chromotography techniques and all work-up operations. Nevertheless, we were able to reliably obtain high resolution mass spectroscopic (HRMS) data for all substrates studied in Scheme 11, Scheme 12 and Table 1.
Further discussion is necessary for the final olefination-deprotection studies of these diyne substrates (see Table 1). All freshly cyclized C4,C5-diol mesylate derivatives 58–62 were first subjected to reductive olefination by SmI2 at –20 °C to afford the fully-fledged epoxydienediynes 64–68 [38]. After HRMS data collection, these were immediately subjected to the established global deptrotection conditions, namely, by brief exposure with TBAF/o-nitrophenol and then exposure to HF-Et3N over differing time scales (Scheme 9). The TBS-protected mesylate derivatives 59–61 conferred the greatest stabilities and could not be transformed to 1. The protected and cycloaromatized forms of 1 were, however, detected by HRMS analysis of 67 before and after treatment with cyclohexa-1,4-diene in THF (Table 1). Interestingly, the more successful derivatives 61–63 all featured bis-silyl ether protection on the mycarose moiety. These derivatives all gave accurate HRMS data correlations after SmI2 olefination to 67–68. We thus suspected complexation/activation issues from samarium(II/III)-salts, but additives like pyridine and 2,6-lutidine during work-up procedures (prior to filtration through Celite) did not improve the results. Ultimately, after exhaustive use of the advanced precursor 50, the bis-TMS ethers 62 or 63 gave an accurate match of the HRMS data patterns for 1, albeit in relatively low percentages. A viable total synthesis route to the kedarcidin chromophore was thus identified for the first time in our laboratories.
Conclusion
Herein, we have disclosed our concerted efforts towards securing a total synthesis of the latest revised structure of the kedarcidin chromophore 1 (Scheme 1) [33, 34]. Initial glycosylation studies demonstrated the poor reactivity of late-stage aglycon acceptors like 18 and 19 (Schemes 2–4). Consequently, pre-α-glycosylated fragments of the epoxy-iodoalkene 33 and alkyne-orthester 46 were prepared on gram scales by reworking previously developed chemistry (Schemes 5–8) [34,35,36,37,38]. These fragments were then assembled after optimization of Sonogashira coupling [60], Shiina macrolactonization [61], and mixed-anhydride amidation protocols [45]. These efforts eventually furnished the ansa-macrolide 50 as a storable substrate that is fully-adorned with all the components of the kedarcidin chromophore (Scheme 10).
During latter enediyne cyclization studies, our protecting group strategy was assessed for its potential to succeed at the last step of the synthesis. This highlighted the need for either mono-TES or bis-TMS ether protection of the 2-deoxysugar moieties (Scheme 9). The alkyne-aldehyde cyclization precursors 54/55 were thus prepared in appropriately protected forms (Scheme 11). The subsequent development of a streamlined cyclisation-derivatisation-deprotection sequence to the fully-fledged, nine-membered enediyne proved to be extraordinarily challenging on the bench (Scheme 12). After exhaustive trials and tribulations, the bis-OTMS ether 55 (freshly prepared from 50) was first cyclized to 56/57 under Ce(III)-amide mediation, then derivatized as its C4-O-trifluorobenzoate (TFBz) ester 62 or 63, deoxygenated by SmI2 to its olefin 68, and finally deprotected under buffered hydrogen fluoride conditions to afford the kedarcidin chromophore (1), as inferred by HRMS analysis (Table 1).
To close this paper, we note that the early introduction of 2-deoxy-α-pyranosides as elaborate THP protecting groups offered a convergent route to 1. Accordingly, a viable total synthesis strategy was founded in only 17-steps via the equally convergent synthesis of suitably protected l-α-mycaroside (33) and l-α-kedarosaminide (46) fragments. This result is meaningful for a target of this complexity and fragility, and was achieved in spite of the additional challenges imposed by free hydroxyl/amino-groups and extra bulky/labile-functionality. At the root of our tactical and evolutionary pursuit of this formidable natural product was the development of several powerful, yet chemoselective methods. Over 20-years since kedarcidin was isolated and first characterized [24,25,26,27,28,29,30], several new synthetic organic methods may now be highlighted, namely: Myers’ anionic transannular cyclization [33], stereoselective epoxyalkyne formation [34, 35], atropselective Pd/Cu-Sonogashira coupling [36,37,38, 60], 2-deoxy-α-glycosylation [39,40,41], atropselective Shiina macrolactonization [42, 43, 61], CeX3-mediated enediyne cyclisation [55,56,57,58,59], and SmI2-based reductive olefination [62, 63]. Further application of some of these key methods to the synthesis of the putative biomimetic C1027 enediyne-precursors of the fijiolides will be reported in due course [21].
References
Danishefsky SJ, Shair MD. Observations in the chemistry and biology of cyclic enediyne antibiotics: total syntheses of calicheamicin γ1I and dynemicin A. J Org Chem. 1996;61:16–44.
Smith AL, Nicolaou KC. The enediyne antibiotics. J Med Chem. 1996;39:2103–17.
Xi Z, Goldberg IH. DNA damaging enediyne compounds. In Barton DHR, Nakanishi K, editors. Comprehensive natural product chemistry. Vol. 7 Oxford: Pergamon; 1999. p. 553.
Wolkenberg SE, Boger DL. Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem Rev. 2002;102:2477–596.
Glam U, et al. Antitumor antibiotics: bleomycin, enediynes, and mitomycin. Chem Rev. 2005;105:739–58.
Hamann PR, Upeslacis J, Borders DB. Enediynes. In Cragg GM, Kingston GGI, Newman DJ, editors. Anticancers agents from natural products. 2nd ed. Boca Raton: CRC Press; 2011. p. 575–621.
Hirama M. Design and synthesis of neocarzinostatin analogues. J Synth Org Chem Jpn. 1991;49:1032–42.
Nicolaou KC, Dai W-M. Chemistry and biology of the enediyne anticancer antibiotics. Angew Chem Int Ed Engl. 1991;30:1387–416.
Lhermitte H, Grierson DS. The enediyne and dienediyne based antitumour antibiotics. Methodology and strategies for total synthesis and construction of bioactive analogues. Part 2. Contemp Org Synth. 1996;3:93–124.
Grissom JW, Gunawardena GU, Klingberg D, Huang D. The chemistry of enediynes, enyne allenes and related compounds. Tetrahedron. 1996;52:6453–518.
Brückner R, Suffert J. The bis (enol triflate) route to dienediyne models of the biradical forming and DNA-cleaving natural product neocarzinostatin chromophore. Synlett. 1999;657–79.
Sato I, Hirama M. Recent advances in the synthetic studies of nine-membered enediyne antitumor antibiotics. J Synth Org Chem Jpn. 2010;68:1123–31.
Kobayashi S, et al. The first total synthesis of N1999-A2: absolute stereochemistry and stereochemical implications into DNA cleavage. J Am Chem Soc. 2001;123:11294–5.
Iida K, Hirama M. Synthesis and characterization of nine-membered cyclic enediynes, models of the C-1027 and kedarcidin chromophores: Equilibration with a p-benzyne biradical and kinetic stabilization. J Am Chem Soc. 1995;117:8875–6.
Hirama M. Synthesis and chemistry of chromoprotein antitumor antibiotics: nine-membered enediynes are equilibrated with p-benzyne type biradicals. Pure Appl Chem. 1997;69:525–30.
Myers AG, Hurd AR, Hogan PC. Evidence for facile atropisomerism and simple (non-nucleophilic) biradical-forming cycloaromatization within kedarcidin chromophore aglycon. J Am Chem Soc. 2002;124:4583–5.
Usuki T, et al. Spin trapping of 13C-labeled p-benzynes generated by Masamune–Bergman cyclization of bicyclic nine-membered enediynes. Angew Chem Int Ed. 2004;43:5249–53.
Hirama M, et al. Direct observation of ESR spectra of bicyclic nine-membered enediynes at ambient temperature. Heterocycles. 2006;69:83–89.
Jean M, Tomas S, van de Weghe P. When the nine-membered enediynes play hide and seek. Org Biomol Chem. 2012;10:7453–6.
Nicolaou KC, Tang Y, Wang J. Total synthesis of sporolide B. Angew Chem Int Ed. 2009;48:3449–553.
Nam S-J, et al. Fijiolides A and B, inhibitors of TNF-α-induced NFκB activation, from a marine-derived sediment bacterium of the genus Nocardiopsis. J Nat Prod. 2010;73:1080–6.
Oh D-C, Williams PG, Kauffman CA, Jensen PR, Fenical W. Cyanosporasides A and B, chloro- and cyano-cyclopenta[a]indene glycosides from the marine actinomycete “Salinispora pacifica”. Org Lett. 2006;8:1021–4.
Yamada K, et al. Biomimetic total synthesis of cyanosporaside aglycons from a single enediyne precursor through site-selective p-benzyne hydrochlorination. Angew Chem Int Ed. 2014;53:13902–6.
Lam S, et al. Kedarcidin, a new chromoprotein antitumor antibiotic. I. Taxonomy of producing organism, fermentation and biological activity. J Antibiot. 1991;44:472–8.
Hofstend SJ, Matson JA, Malacko AR, Marquardt H. Kedarcidin, a new chromoprotein antitumor antibiotic II. Isolation, purification and physico-chemical properties. J Antibiot. 1992;45:1250–4.
Lohman JR, et al. Cloning and sequencing of the kedarcidin biosynthetic gene cluster from Streptoalloteichus sp. ATCC 53650 revealing new insights into biosynthesis of the enediyne family of antitumor antibiotics. Mol BioSyst. 2013;9:478–91.
Leet JE, et al. Kedarcidin, a new chromoprotein antitumor antibiotic: structure elucidation of kedarcidin chromophore. J Am Chem Soc. 1992;114:7946–8.
Leet JE, et al. Chemistry and structure elucidation of the kedarcidin chromophore. J Am Chem Soc. 1993;115:8432–43.
Constantine KL, et al. Sequential 1H, 13C, and 15N-NMR assignments and solution conformation of apokedarcidin. Biochemistry. 1994;33:11438–52.
Zein N, et al. Selective proteolytic activity of the antitumor agent kedarcidin. Proc Natl Acad Sci USA. 1993;90:8009–12.
Kawata S, Ashizawa S, Hirama M. Synthetic study of kedarcidin chromophore: revised structure. J Am Chem Soc. 1997;119:12012–3.
Huanga S-X, Lohmana JR, Huanga T, Shen B. A new member of the 4-methylideneimidazole-5-one-containing aminomutase family from the enediyne kedarcidin biosynthetic pathway. Proc Nat Acad Sci. 2013;110:8069–74.
Ren F, Hogan PC, Anderson AJ, Myers AG. Kedarcidin chromophore: synthesis of its proposed structure and evidence for a stereochemical revision. J Am Chem Soc. 2007;129:5381–3.
Ogawa K, Koyama Y, Ohashi I, Sato I, Hirama M. Total synthesis of a protected aglycon of the kedarcidin chromophore. Angew Chem Int Ed. 2009;48:1110–3.
Das P, Mita T, Lear MJ, Hirama M. Synthesis of 13C-labelled, bicyclic mimetics of natural enediynes. Chem Commun. 2002;2624–5.
Koyama Y, et al. Efficient construction of the kedarcidin chromophore ansamacrolide. Org Lett. 2005;7:267–70.
Yoshimura F, Lear MJ, Ohashi I, Koyama Y, Hirama M. Synthesis of the entire carbon framework of the kedarcidin chromophore. Chem Commun. 2007;3057–9.
Ogawa K, Koyama Y, Ohashi I, Sato I Hirama M. Secure route to the epoxybicyclo[7.3.0]dodecadienediyne core of the kedarcidin chromophore. Chem Commun. 2008;6327–9.
Lear MJ, Hirama M. Convenient route to derivatives of the 2-deoxysugar subunits of the kedarcidin chromophore: L-mycarose and L-kedarosamine. Tetrahedron Lett. 1999;40:4897–900.
Lear MJ, Yoshimura F, Hirama M. A direct and efficient α‐selective glycosylation protocol for the kedarcidin sugar, L-mycarose: AgPF6 as a remarkable activator of 2-deoxythioglycosides. Angew Chem Int Ed. 2001;40:946–9.
Ohashi I, Lear MJ, Yoshimura F, Hirama M. Use of polystyrene-supported DBU in the synthesis and α-selective glycosylation study of the unstable schmidt donor of L-kedarosamine. Org Lett. 2004;6:719–22.
Shiina I, Ibuka R, Kubota M. A new condensation reaction for the synthesis of carboxylic esters from nearly equimolar amounts of carboxylic acids and alcohols using 2-methyl-6-nitrobenzoic anhydride. Chem Lett. 2002;286.
Shiina I, Kubota M, Ibuka R. A novel and efficient macrolactonization of ω-hydroxycarboxylic acids using 2-methyl-6-nitrobenzoic anhydride (MNBA). Tetrahedron Lett. 2002;43:7535–9.
Sakaitani M, Ohfune Y. Syntheses and reactions of silyl carbamates. 1. Chemoselective transformation of amino protecting groups via tert-butyldimethylsilyl carbamates. J Org Chem. 1990;55:870–6.
Carpino LA. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J Am Chem Soc. 1993;115:4397–8.
Chemla F, Bernard N, Ferreira F, Normant JF. Preparation of propargylic carbenoids and reactions with carbonyl compounds—a stereoselective synthesis of propargylic halohydrins and oxiranes. Eur J Org Chem. 2001;3295–300.
Baker JR, Thominet O, Britton H, Caddick S. An efficient synthesis of epoxydiynes and a key fragment of neocarzinostatin chromophore. Org Lett. 2007;9:45–8.
Veeneman GH, van Leeuwen SH, van Boom JH. Iodonium ion promoted reactions at the anomeric centre. II. An efficient thioglycoside mediated approach toward the formation of 1,2-trans linked glycosides and glycosidic esters. Tetrahedron Lett. 1990;31:1331–4.
Matsumoto T, Maeta H, Suzuki K, Tsuchihashi G. New glycosidation reaction 1: combinational use of Cp2ZrCl2-AgClO4 for activation of glycosyl fluorides and application to highly β-selective gylcosidation of D-mycinose. Tetrahedron Lett. 1988;29:3567–70.
Suzuki K, Maeta H, Matsumoto T, Tsuchihashi G. New glycosidation reaction 2. Preparation of 1-fluoro-d-desosamine derivative and its efficient glycosidation by the use of Cp2HfCl2-AgClO4 as the activator. Tetrahedron Lett. 1988;29:3571–4.
Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis. 1981;1–28.
Sugimura T, Hagiya K. Di-2-methoxyethyl azodicarboxylate (DMEAD): an inexpensive and separation-friendly alternative reagent for the Mitsunobu reaction. Chem Lett. 2007;36:566–7.
Kaburagi Y, Osajima H, Shimada K, Tokuyama H, Fukuyama T. 4-(tert-Butyldimethylsilyloxy) benzylidene acetal: a novel benzylidene-type protecting group for 1, 2-diols. Tetrahedron Lett. 2004;19:3817–21.
Jobron L, Hindsgaul O. Novel para-substituted benzyl ethers for hydroxyl group protection. J Am Chem Soc. 1999;121:5835–6.
Imamoto T, Kusumoto T, Tawarayama Y, Sugiura Y, Mita T, Hatanaka Y, Yokoyama M. Carbon-carbon bond-forming reactions using cerium metal or organocerium(III) reagents. J Org Chem. 1984;49:3904–12.
Imamoto T, Takiyama N, Nakamura K, Hatajima T, Kamiya Y. Reactions of carbonyl compounds with Grignard reagents in the presence of cerium chloride. J Am Chem Soc. 1989;111:4392–8.
Nishikawa T, Isobe M, Goto T. Synthetic studies on the bicyclo[7.3.1]tridecenediyne system in an antitumor antibiotic, dynemicin A. Synlett. 1991;393–4.
Myers AG, Harrington PM, Kuo EY. Enantioselective synthesis of the epoxy diyne core of neocarzinostatin chromophore. J Am Chem Soc. 1991;113:694–5.
Iida K-i, Hirama M. Efficient route to the nine-membered cyclic diyne system: tuning of the extremely facile Cope rearrangment of 1,5-diyne. J Am Chem Soc. 1994;116:10310–1.
Sonogashira K, Tohda Y, Hagihara N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975;16:4467–70.
Shiina I, Kubota M, Oshiumi H, Hashizume M. An effective use of benzoic anhydride and its derivatives for the synthesis of carboxylic esters and lactones: a powerful and convenient mixed anhydride method promoted by basic catalysts. J Org Chem. 2004;69:1822–30.
Komano K, Shimamura S, Inoue M, Hirama M. Total synthesis of the maduropeptin chromophore aglycon. J Am Chem Soc. 2007;129:14184–6.
Inoue M, Ohashi I, Kawaguchi T, Hirama M. Total synthesis of the C-1027 chromophore core: extremely facile enediyne formation through Sml2-mediated 1,2-elimination. Angew Chem Int Ed. 2008;47:1777–9.
Acknowledgements
This work represents over 20-years of collective effort (1997–2017) and was latterly supported by a Grant-in-Aid for Specially Promoted Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), SORST (Japan), as well as the Science and Technology Agency (JST), Japanese Society for the Promotion of Science, Fellowship and Multidimensional Materials Science Leaders Program (to MJL). We are especially grateful to Dr. John E. Leet at Bristol-Myers Squibb for kindly providing original chromoprotein material and the 1H NMR spectra of the natural kedarcidin chromophore.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dedication: Dedicated to Professor Samuel J. Danishefsky for his outstanding contributions to the total synthesis of highly complex and biologically important natural products.
Supplementary information
Rights and permissions
About this article

Cite this article
Lear, M.J., Hirai, K., Ogawa, K. et al. A convergent total synthesis of the kedarcidin chromophore: 20-years in the making. J Antibiot 72, 350–363 (2019). https://doi.org/10.1038/s41429-019-0175-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0175-y
