
Abstract
Five new anthraquinones, morakotins A–E (1–5), together with seven known compounds, lunatin (6), rheoemodin (7), YM187781 (8), bislunatin (9), 6-(1-hydroxypentyl)-4-methoxypyran-2-one, 9,11-dehydoergrosterol peroxide, and cerevisterol, were isolated from the insect pathogenic fungus Cordyceps morakotii BCC 56811. The morakotin structures were elucidated from NMR spectroscopic and mass spectrometric data. The absolute configurations of bianthraquinone compounds, morakotins C–E (3–5), were determined by application of the exciton chirality method. Compounds 3, 7, 8, and 9 showed weak to moderate antimycobacterial and antifungal activities. Compounds 4 and 8 exhibited antibacterial activity against both Bacillus cereus and Staphylococcus aureus (MIC 3.13–25 µg ml−1), whereas compounds 3 and 9 were active against B. cereus (MIC 12.5 and 3.13 µg ml−1, respectively), and compound 7 was active against Acinetobacter baumannii (MIC 12.5 µg ml−1).
Similar content being viewed by others
Introduction
Anthraquinones and their derivatives comprise a large group of natural quinoid compounds. The chemical diversity of these compounds and their biological properties have attracted the attention of the pharmaceutical, textile, dyeing, and food colorant industries. Anthraquinones exhibit a wide range of biological activities including antibacterial, antifungal, antiviral, insecticidal, and anticancer [1, 2]. Anthraquinones are widespread in fungi, especially endophytic fungi [3,4,5,6].
Insect pathogenic fungi are known to be potential sources of novel bioactive substances; however, only a few species of ant pathogens have been chemically explored [7, 8]. As part of our ongoing program on bioactive compounds from insect pathogenic fungi, we selected an ant pathogen (Cordyceps morakotii, strain BCC 56811) for chemical investigation, since the crude extract exhibited antibacterial activity against Bacillus cereus (IC50 3.13 µg ml−1) and HPLC and 1H NMR spectra showed profiles suggestive of novel compounds. Nine anthraquinone compounds were isolated, including new morakotins A–E (1–5), lunatin (6) [9], rheoemodin (7) [10], YM187781 (8) [11], and bislunatin (9) [12] (Fig. 1), together with (1′S)-6-(1-hydroxypentyl)-4-methoxypyran-2-one [13], 9,11-dehydroergosterol peroxide [14], and cerevisterol [15]. We report here the isolation, structure elucidation, and biological activities of these compounds.

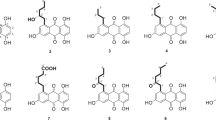
Chemical structures of compounds 1–9
Results and discussion
Morakotin A (1) was obtained as an orange solid. The molecular formula was established as C16H12O7 by HRESIMS, in combination with 13C NMR spectroscopy. The UV spectrum showed the typical absorption pattern of an anthraquinone chromophore at 233, 286, 320, and 443 nm. The IR spectrum exhibited major absorption bands for hydroxyl (3261 cm−1), non-chelated quinone carboxyl (1622 cm−1), and chelated quinone carbonyl (1606 cm−1). The 1H and 13C NMR spectra indicated the presence of two D2O exchangeable protons, one singlet aromatic proton, two meta-coupling aromatic protons, two methoxy groups, five oxygenated quaternary carbons, and two carbonyl groups (Table 1). The downfield hydroxyl protons at δH 12.17 and 12.28 indicated H-bonding with a carbonyl, which corresponded to the low absorption frequency of a chelated quinone carbonyl group at 1606 cm−1 in the IR spectrum. The HMBC spectrum showed correlations from H-4 to C-2/C-3/C-4a/C-9a/C-10, H-5 to C-7/C-10, H-7 to C-5/C-8, 2-OCH3 to C-2, and 3-OCH3 to C-3. These data revealed the structure of morakotin A as depicted in Fig. 1. The correlations from 3-OCH3 to H-4 and 2-OCH3 in NOESY spectrum also supported the position of the methoxy groups at C-2 and C-3. Therefore, morakotin A (1) was determined to be 1,6,8-trihydroxy-2,3-dimethoxyanthracene-9,10-dione.
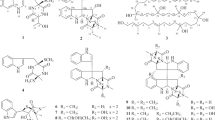
The UV and IR spectra of morakotin B (2) were almost identical to those of morakotin A (1). The 1H NMR spectra of these two compounds were also similar except for the presence of one more methoxy signal in morakotin B (2). HRESIMS gave the molecular formula C17H14O7, 14 amu higher than that of morakotin A (1), which indicated that morakotin B (2) has one additional methoxy group. The HMBC correlation from these methoxy protons (δH 3.93) to C-6 (Fig. 2) led to the deduction that the hydroxyl group at C-6 in morakotin A (1) is replaced by a methoxy group in morakotin B (2). This assignment was further confirmed by the correlations from 6-OCH3 to H-5 and H-7 in the NOESY spectrum. Consequently, morakotin B (2) was identified as 1,8-dihydroxy-2,3,6-trimethoxyanthracene-9,10-dione.

Selected HMBC, and NOESY correlations of compounds 2 and 5
The presence of 15 carbon signals in the 13C NMR spectrum and the quasimolecular ion peak in the HRESIMS (m/z 601.0632 [M−H]−, calcd. for C30H17O14 601.0624) led to the conclusion that morakotin C (3) was a symmetrical anthraquinone dimer. When compared with that of the known co-metabolite bislunatin (9) [12], the 1H NMR spectrum of morakotin C (3) showed the absence of one of the meta-coupled aromatic protons and the presence of one more carbonyl chelated hydroxyl group (δH 13.04) per monomeric unit. The IR spectrum exhibited only a chelated quinone carbonyl band at 1598 cm−1. In combination with the molecular formula, which indicated the presence of two more oxygen atoms than that of bislunatin, the planar structure of morakotin C (3) was established as shown in Fig. 1. The HMBC correlations from H-2 to C-4/C-9a, H-7 to C-5/C-8a, 1-OH to C-2/C-1/C-9a, 5-OH to C-5/C-6/C-10a, 8-OH to C-7/C-8/C-8a, and 6-OCH3 to C-6 also supported the structural features of morakotin C (3) as described above. Morakotin C (3) was, thus, determined as 1,1′,3,3′,5,5′,8,8′-octahydroxy-6,6′-dimethoxy-4,4′-bianthracene-9,9′,10,10′-tetraone.
The HRESIMS data established the molecular formula of morakotin D (4) as C29H16O12. The UV and IR spectra were similar to those of the known co-metabolite bislunatin (9) [11]. The NMR spectroscopic data revealed the presence of four hydroxyl groups, two pairs of meta-coupling aromatic protons, two singlet aromatic protons, one methoxy group, and four carbonyl groups (Table 2), indicating an unsymmetrical dimeric anthraquinone. The correlations from H-2 to C-4/C-9a, H-5 to C-7/C-10/C-8a, H-7 to C-5/C-8a, 1-OH to C-1/C-2/C-9a, 8-OH to C-7/C-8/C-8a, and OCH3 to C-6 in the HMBC spectrum and the correlations from methoxy protons to H-5 and H-7 in the NOESY spectrum established one of the monomer units as the known co-metabolite lunatin (6) [9]. The other part was found to resemble that of the known co-metabolite rheoemodin (7) [10], which is joined to another unit at C-4 and C-4′ positions, as deduced by the HMBC correlations from H-2′ to C-4′ /C-9a′, H-5′ to C-7′/C-10′/C-8a′, H-7′ to C-5′/C-8a′, 1′-OH to C-1′/C-2′/C-9a′, 8′-OH to C-7′/C-8′/C-8a′. The chemical shifts at C-4 and C-4′ were also consistent with those of other 4,4′ coupled bianthraquinone derivatives [11, 12]. Therefore, morakotin D (4) was identified as 1,1′,3,3′,6′,8,8′-heptahydroxy-6-methoxy-4,4′-bianthracene-9,9′,10,10′-tetraone.
Morakotin E (5) gave the same molecular formula to that of morakotin D (4), i.e., C29H16O12, as deduced from HRESIMS. Comparison of the 1H and 13C NMR spectra between these two compounds revealed the significant shift of two singlet aromatic protons (from δH 6.68, 6.69 in compound 4 to 7.28, 7.29 in compound 5) and two quaternary carbons (from δC 123.6, 124.0 in compound 4 to 113.8, 113.9 in compound 5). The HMBC correlations from H-4 to C-2/C-9a/C-10, H-5 to C-7/C-10, H-7 to C-5/C-8a, 1-OH to C-1/C-9a, 8-OH to C-7/C-8, 6-OCH3 to C-6, H-4′ to C-2′/C-9a′/C-10′, H-5′ to C-8a′/C-10′, H-7′ to C-5′ and the NOESY correlations from 6-OCH3 to H-5 and H-7 led to the deduction that Morakotin E (5) has the same monomer units as those of morakotin D (4) with the connection at C-2 and C-2′. The chemical shifts at the junction (C-2 and C-2′) were in agreement with those obtained from other 2,2′ coupled bianthraquinone compounds [16]. Morakotin E (5) was, thus, determined to be 1,1′,3,3′,6′,8,8′-heptahydroxy-6-methoxy-2,2′-bianthracene-9,9′,10,10′-tetraone.
The absolute configurations of bianthraquinone compounds were determined by application of the exciton chirality method [17,18,19,20]. For morakotin C (3), CD coupling was found with strong negative first and positive second Cotton effect due to the long axis of the anthraquinone moieties, indicating the two chromophores were twisted in an anticlockwise manner. Therefore, the absolute configuration was suggested to be R. The opposite CD cotton curve, positive first and negative second Cotton effect, was observed for the known co-metabolite bislunatin (9), which showed that the anthraquinone chromophores were twisted in a clockwise manner; however, the optical rotation value was low \(([\alpha]^{25}_{\mathrm{D}} + 44, c \,{\mathrm{0.61}} \,{\mathrm{dioxane}})\) when compared with that reported in the literature (\([\alpha]^{26}_{\mathrm{D}} \)+1690, c 0.25, EtOH) [12], suggesting the non-equivalent mixture of atropisomers slightly rich in S-isomer. Morakotins D (4), E (5), and compound 8 exhibited low values of optical rotations and weak CD signals, which suggested that these compounds were also non-equivalent mixtures of atropisomers.
The seven known compounds were identified as lunatin (6) [9], rheoemodin (7) [10], YM187781 (8) [11], bislunatin (9) [12], (1′S)-6-(1-hydroxypentyl)-4-methoxypyran-2-one [13], 9,11-dehydroergosterol peroxide [14], and cerevisterol [15] by analysis of their spectroscopic data and their physical properties, which were identical to those reported in the literature data.
Compounds 3, 4, and 6–9 were assayed for antimicrobial activity against various bacterial and fungal strains (Mycobacterium tuberculosis H37Ra, B. cereus, Staphylococcus aureus, Acinetobacter baumannii, Pseudomonas aeruginosa, and Candia albicans) and cytotoxicity against mammalian cancer cell lines (MCF-7, and NCI-H187) and noncancerous Vero cells (Table 3). Compounds 1, 2, and 5 were not tested owing to insufficient sample. Compounds 3, 7, 8, and 9 exhibited weak to moderate anti-TB and antifungal activities. For antibacterial activity, compounds 4 and 8 were active against both B. cereus and S. aureus (MIC 3.13–25 µg ml−1), whereas compounds 3 and 9 were active only against B. cereus (MIC 12.5 and 3.13 µg ml−1, respectively). Only compound 7 was active against A. baumannii (MIC 12.5 µg ml−1). All compounds showed cytotocixity against all tested cell-lines (IC50 5.37–47.97 µg ml−1), except that compound 4 was inactive against Vero cells.
Materials and methods
General experimental procedures
Melting points were measured using a Metler MP90 melting point apparatus and are uncorrected. Optical rotation measurements were conducted by using a JASCO P-1030 digital polarimeter. UV and FT-IR spectra were recorded on an Analytic Jena Spekol 1200 spectrophotometer and a Bruker Alpha spectrometer. The CD spectra were recorded on a JASCO J-180 spectropolarimeter. NMR spectra were recorded on Bruker Advance III 400 and Bruker Advance 500 spectrometers. ESITOF MS data were obtained on a Bruker micrOTOF mass spectrometer. Column chromatography was performed on silica gel 60 (70–230 Mesh ASTM, Merck). HPLC experiments were performed using Dionex-Ultimate 3000 series equipped with a binary pump, an autosampler and a diode array detector.
Fungal material
The fungus used in this study was isolated from an ant (Hymenoptera), collected from the evergreen forest in Chiang Mai Province, Thailand. The organism was deposited in the BIOTEC Culture Collection (BCC) as BCC 56811 on 16 November 2012. This fungus was identified as C. morakotii of the Class Sordariomycetes, order Hypocreales, family Cordycipitaceae, on the basis of morphology, by Miss Kanoksri Tasanathai, BIOTEC, and was confirmed by the molecular identification method using the rDNA internal transcribed spacer region (ITS rDNA: GenBank accession number MH828333), analyzed by Dr. Nattawut Boonyuen, BIOTEC.
Fermentation, extraction, and isolation
C. morakotii BCC 56811 was cultured on potato dextrose agar at 25 °C for 30 days. The agar was cut into pieces (1 × 1 cm) and inoculated into 8 × 250-ml Erlenmeyer flasks containing 25 ml of potato dextrose broth (PDB medium contained potato starch 4.0 g and dextrose 20.0 g in 1.0 l distilled water). After incubation at 25 °C for 7 days on a rotary shaker (200 rpm), each primary culture was transferred into a 1-l Erlenmeyer flask containing 250 ml of the same medium and incubated under the same conditions for 7 days. Each 25 ml portion of the secondary culture was transferred into 80 × 1-l Erlenmeyer flasks containing 250 ml of PDB and was statically incubated at 25 °C for 53 days.
After filtration of the culture, the cells were macerated in MeOH (1 l) for 3 days, and then in CH2Cl2 (1 l) for 3 days. The MeOH and CH2Cl2 extracts were combined and evaporated under reduced pressure. Water (800 ml) was added and the mixture was extracted with hexanes (3 × 800 ml), followed by EtOAc (3 × 800 ml). Trituration of the dark brown solid (3.05 g) obtained from the EtOAc extract with MeOH followed by filtration gave a dark red solid (1.35 g), which was further purified by preparative HPLC using a reverse phase column (SunFire C18 OBD, 10 μm, 19 × 150 mm, step gradient elution with 40−70% MeCN/H2O) to furnish compounds 2 (0.6 mg), 3 (38.3 mg), 4 (6.6 mg), 6 (25.4 mg), 7 (3.9 mg), 8 (18.9 mg), and 9 (17.2 mg). The filtrate (1.69 g) was fractionated using Sephadex LH 20 (4.0 × 36 cm) and eluted with 100% MeOH to provide 8 fractions (A1–A8). Compounds 6 (49.6 mg) and 7 (43.4 mg) were obtained from fractions A5 and A6, respectively. Further purification of fraction A2 by preparative HPLC (step gradient elution with 20−100% MeCN/H2O) afforded 6-(1-hydroxypentyl)-4-methoxypyran-2-one (7.9 mg). Fraction A3 was subjected to preparative HPLC (step gradient elution with 70−100% MeCN/H2O) to give compounds 2 (1.3 mg) and 9 (0.5 mg). Compounds 1 (0.6 mg) and 6 (2.1 mg) were obtained from fraction A4 after purification by preparative HPLC (step gradient elution with 45−75% MeCN/H2O). Purification of fraction A7 by preparative HPLC (step gradient elution with 55−75% MeCN/H2O) yielded compound 8 (0.7 mg) and 9 (0.3 mg). Fraction A8 was purified by preparative HPLC (step gradient elution with 40−80% MeCN/H2O) to afford compounds 4 (5.4 mg), 5 (1.4 mg), 8 (1.6 mg), and 9 (0.3 mg).
The hexanes extract of the mycelium (19.49 g) was subjected to silica gel column chromatography (7.3 × 22.5 cm), using 2% MeOH/CH2Cl2 as an eluent, to give six fractions (B1–B6). Compound 2 (3.3 mg) and cerevisterol (63.9 mg) were obtained from fraction B2 and B6, respectively, after trituration with MeOH followed by filtration. Fraction B4 was fractionated by Sephadex LH 20 (4.5 × 60 cm) and eluted with 100% MeOH to provide 4 subfractions (B4-1–B4-4). Further purification of subfraction B4-2 by preparative HPLC (step gradient elution with 55−75% MeCN/H2O) yielded 9,11-dehydroergosterol peroxide (12.6 mg). Trituration of subfraction B4-4 with MeOH followed by filtration furnished compound 6 (25.0 mg) as an orange solid. Consecutive purification of fraction B5 by Sephadex LH 20, eluted with 100% MeOH, followed by preparative HPLC (step gradient elution with 35−70% MeCN/H2O) gave compound 7 (1.8 mg) and 9 (0.9 mg).
The culture broth was extracted with EtOAc (3 × 20 l) and evaporated to dryness, leaving a brown gum (7.69 g). The crude extract was consecutively purified by Sephadex LH 20, eluted with 100% MeOH, followed by preparative HPLC to provide compounds 3 (16.0 mg), 6 (9.2 mg), and 7 (13.1 mg).
Morakotin A (1): Orange solid; UV (MeOH) λmax (log ε) 233 (3.96), 286 (4.13), 320 (3.85), 443 (3.79) nm; IR (ATR) νmax 3261, 2924, 1622, 1606, 1571, 1452, 1400, 1364, 1325, 1274, 1167, 1136 cm-1; 1H and 13C NMR spectroscopic data in acetone-d6, Table 1; HRMS (ESITOF) m/z 339.0480 [M + Na]+ (calcd. for C16H12O7+Na, 339.0475).
Morakotin B (2): Orange solid; UV (MeOH) λmax (log ε) 233 (3.78), 283 (3.91), 319 (3.63), 439 (3.65) nm; IR (ATR) νmax 3451, 2925, 1627, 1604, 1461, 1415, 1368, 1270, 1186, 1164, 1137 cm-1; 1H and 13C NMR spectroscopic data in DMSO-d6, Table 1; HRMS (ESITOF) m/z 353.0616 [M + Na]+ (calcd. for C17H14O7+Na, 353.0632).
Morakotin C (3): Dark red solid; m.p. (dec.) 200.0–202.0°C; \([\alpha]^{25}_{\mathrm{D}}\)−184 (c 0.22, dioxane); UV (dioxane) λmax (log ε) 271 (4.24), 284 (4.25), 312 (4.29), 487 (4.27), 504 (4.28), 539 (4.20) nm; CD (dioxane) [θ] (nm) 0 (276), −161200 (264), 0 (255), +19300 (251), 0 (249); IR (ATR) νmax 3434, 2924, 1598, 1466, 1403, 1307, 1271, 1227 cm-1; 1H and 13C NMR spectroscopic data in DMSO-d6, Table 1; HRMS (ESITOF) m/z 601.0632 [M − H]− (calcd. for C30H17O14, 601.0624).
Morakotin D (4): Red solid; \([\alpha]^{25}_{\mathrm{D}}\)+11 (c 0.19, dioxane); UV (dioxane) λmax (log ε) 272 (3.99), 298 (4.14), 457 (3.98), 485 (3.93) nm; IR (ATR) νmax 3407, 2924, 1622, 1601, 1461, 1380, 1240 cm-1; 1H and 13C NMR spectroscopic data in DMSO-d6, Table 2; HRMS (ESITOF) m/z 579.0531 [M + Na]+ (calcd. for C29H16O12+Na, 579.0534).
Morakotin E (5): Orange solid; \([\alpha]^{25}_{\mathrm{D}}\)−22 (c 0.16, dioxane); UV (dioxane) λmax (log ε) 262 (4.07), 295 (4.37), 321 (4.07), 451 (4.09) nm; IR (ATR) νmax 3363, 2924, 1620, 1597, 1440, 1383, 1313, 1253 cm-1; 1H and 13C NMR spectroscopic data in DMSO-d6, Table 2; HRMS (ESITOF) m/z 555.0561 [M − H]− (calcd. for C29H15O12, 555.0569).
Biological assays
Growth inhibitory activity against M. tuberculosis H37Ra and cytotoxicity to Vero cells (African green monkey kidney fibroblast, ATCC CCL-81) was performed by using the green fluorescent protein (GFP) based method [21, 22]. The resazurin microplate assay was used to evaluate anti-B. cereus (ATCC 11778), anti-C. albicans (ATCC 90028) and cytotoxicity against MCF-7 cells (human breast cancer, ATCC HTC-22) and NCI-H187 cells (human small-cell lung cancer, ATCC CRL-5804) [23]. Antibacterial activity against S. aureus (ATCC 29213), A. baumannii (ATCC 19606) combined with PAβN, and P. aeruginosa (K 2733) was evaluated by using the standard protocols published by the Clinical and Laboratory Standard Institute [24, 25].
References
Fouillaud M, Venkatachalam M, Girard-Valenciennes E, Caro Y, Dufossé L. Anthraquinones and derivatives from marine-derived fungi: Structural diversity and selected biological activities. Mar Drugs. 2016;14:64.
Gessler NN, Egorova AS, Belozerskaya TA. Fungal anthraquinones. Appl Biochem Microbiol. 2013;49:85–99.
Aly AH, Debbab A, Kjer J, Proksch P. Fungal endophytes from higher plants: a prolific source of phytochemicals and other bioactive natural products. Fungal Divers. 2010;41:1–16.
Borges WS, Pupo MT. Novel anthraquinone derivatives produced by Phoma sorghina, an endophyte found in association with the medicinal plant Tithonia diversifolia (Asteraceae). J Braz Chem Soc. 2006;17:929–34.
Huang W-Y, Cai Y-Z, Xing J, Corke H, Sun M. A potential antioxidant resource: Endophytic fungi from medicinal plants. Econ Bot. 2007;61:14–30.
Xu A, Wang Y, Wen J, Liu P, Liu Z, Li Z. Fungal community associated with fermentation and storage of Fuzhuan brick-tea. Int J Food Microbiol. 2011;146:14–22.
Isaka M, Kittakoop P, Kirtikara K, Hywel-Jones NL, Thebtaranonth Y. Bioactive substances from insect pathogenic fungi. Acc Chem Res. 2005;38:813–23.
Kittakoop P, et al. Bioactive naphthoquinones from Cordyceps unilateralis. Phytochemistry. 1999;52:453–7.
Jadulco R, et al. New metabolites from sponge-derived fungi Curvularia lunata and Cladosporium herbarum. J Nat Prod. 2002;65:730–3.
Huang R, Wang T, Xie X-S, Ma K-X, Fang X-W, Wu S-H. Secondary metabolites from an endophytic fungus Nigrospora sp. Chem Nat Compd. 2016;52:697–9.
Koichi T, Masato W, Koji N, Nami N, Akito Y, JP 2000239216 (A) (2000).
Agusta A, Ohashi K, Shibuya H. Bisanthraquinone metabolites produced by the endophytic fungus Diaporthe sp. Chem Pharm Bull. 2006;54:579–82.
Ayer WA, Trifonov LS, Hutchison LJ, Chakravarty P. Metabolites from a wood-inhabiting cup fungus. Urnula craterium Nat Prod Lett. 2000;14:405–10.
Gunatilaka AAL, Gopichand Y, Schmitz FJ, Djerassi C. Minor and trace sterols in marine invertebrates. 26. Isolation and structure elucidation of nine new 5.alpha.,8.alpha -epidoxy sterols from four marine organisms. J Org Chem. 1981;46:3860–6.
Jinming G, Lin H, Jikai L. A novel sterol from Chinese truffles Tuber indicum. Steroids. 2001;66:771–5.
Koyama J, Morita I, Tagahara K, Aqil M. Bianthraquinones from Cassia siamea. Phytochemistry. 2001;56:849–51.
Harada N, Suzuki S, Uda H, Nakanishi K. Circular dichroism studies of dimeric fungal pigments, (-)-luteoskyrin and (+)-rugulosin. Chem Lett. 1972;1:67–70.
Koyama K, Aida S, Natori S. Supplemental observations on atropisomerism of fungal bis(naphtho-γ-pyrone)s. Chem Pharm Bull. 1990;38:2259–61.
Koyama K, Natori S, Iitaka Y. Absolute configurations of chaetochromin A and related bis (naphtho-γ-pyrone) mold metabolites. Chem Pharm Bull. 1987;35:4049–55.
Mason SF, Seal RH, Roberts DR. Optical activity in the biaryl series. Tetrahedron. 1974;30:1671–82.
Changsen C, Franzblau SG, Palittapongarnpim P. Improved green fluorescent protein reporter gene-based microplate screening for antituberculosis compounds by utilizing an acetamidase promoter. Antimicrob Agents Chemother. 2003;47:3682–7.
Hunt L, Jordan M, De Jesus M, Wurm FM. GFP-expressing mammalian cells for fast, sensitive, noninvasive cell growth assessment in a kinetic mode. Biotechnol Bioeng. 1999;65:201–5.
O'Brien J, Wilson I, Orton T, Pongnan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–6.
CLSI. Methods for dilution antimicrobial susceptibility test for bacteria that growth aerobically; 7th Approve Standard. Wayne PA: Clinical and Laboratory Standards Institute; 2006. CLSI document M7-A7
CLSI. Performance standards for antimicrobial susceptibility testing; 16th Informational Supplement. Wayne PA: Clinical and Laboratory Standards Institute; 2006. CLSI document M100-S16
Acknowledgements
This work was supported by National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency (NSTDA), and Joint research grant from Thailand Research Fund (grant No. DBG5980002) and National Natural Science Foundation of China (grant No. 81561148013). We thank Dr. Philip J. Shaw for manuscript editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher’s note:
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, M., Kornsakulkarn, J., Srichomthong, K. et al. Antimicrobial anthraquinones from cultures of the ant pathogenic fungus Cordyceps morakotii BCC 56811. J Antibiot 72, 141–147 (2019). https://doi.org/10.1038/s41429-018-0135-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0135-y
This article is cited by
-
Two new diketopiperazines from the Cordyceps fungus Samsoniella sp. XY4
The Journal of Antibiotics (2023)
-
Geosmithia—widespread and abundant but long ignored bark beetle symbionts
Mycological Progress (2023)
-
Production of antimicrobial metabolites against pathogenic bacteria and yeasts by Fusarium oxysporum in submerged culture processes
Bioprocess and Biosystems Engineering (2021)
-
Antibacterial and cytotoxic metabolites of termite-associated Streptomyces sp. BYF63
The Journal of Antibiotics (2020)
