
Abstract
Acinetobacter baumannii is one of the ESKAPE pathogen, which causes pneumonia, urinary tract infections, and is linked to high degree of morbidity and mortality. One-way antibiotic and disinfectant resistance is acquired by the activation of RecA-mediated DNA repair (SOS-response) that maintain ROS-dependent DNA damage caused by these anti-bacterial molecules. To increase the efficacy of different anti-microbial, there is a need to design an inhibitor against RecA of A. baumannii. We have performed homology modeling to generate the structure of RecA, followed by model refinement and validation. High-throughput virtual screening of 1,80,313 primary and secondary metabolites against RecA was performed in HTVS, SP, and XP docking modes. The selected 195 compounds were further analyzed for binding free energy by molecular mechanics approach. The selected top two molecules from molecular mechanics approach were further validated by molecular dynamics simulation (MDS). In-silico high-throughput virtual screening and MDS validation identified ZINC01530654 or (+−)-2-((4-((7-Chloro-4-quinolyl)amino)pentyl)ethylamino)ethanol sulfate (or hydroxychloroquine sulfate) as a possible lead molecule binding to RecA protein. We have experimentally determined the mechanism of ZINC01530654 to RecA protein. These findings suggest a strategy to chemically inhibit the vital process controlled by RecA that could be helpful for the development of new antibacterial agents.
Similar content being viewed by others
Introduction
Acinetobacter baumannii is an opportunistic ESKAPE pathogen associated with bacteremia, pneumonia, wound infections, meningitis, and urinary tract infections [1, 2]. Infections caused by Acinetobacter leads to high degree of morbidity, mortality, and increased costs. The major risk factors include prolonged hospitalization, recent invasive procedures, admission in intensive care unit, prolonged antimicrobial agent exposure, use of catheters, respirometers and ventilators, and local colonization pressure on susceptible patients in the hospital setup [3,4,5,6,7]. Carbapenems are one of the most effective antibiotics against A. baumannii and other nosocomial pathogen [8] but Acinetobacter have emerged resistant mechanism to this class [1, 5, 9,10,11,12,13,14,15]. Disinfectants, such as phenol, sodium hypochlorite, ethanol, etc. are used to sterile or clean the hospital environment.
Recently, reduced susceptibility of different disinfectants towards A. baumannii has emerged in the hospital [16, 17] and its reduced susceptibility to biocides is associated with co-resistance to carbapenem [18, 19]. The genome of Acinetobacter also encodes mechanisms to tolerate biocides and desiccation that enhances its persistence in hospital settings [20]. Antibiotic and disinfectant resistance arises due to the maintenance of resistance mutations or genes required for the resistance by activating SOS-mediated DNA repair that involves activation of RecA protein [21, 22].
Different antimicrobials act by inducing ROS production that results in DNA damage and cell death [23,24,25,26]. DNA damage does not always lead to bacterial death, because cell is protected from DNA damage by DNA repair mechanism that involves SOS response [26,27,28]. RecA (inducer) and LexA (repressor) are two proteins involved in SOS response. RecA has the central role in repair response of SOS mutagenesis [29]. Interaction of RecA with single-stranded DNA (or damaged DNA) leads to the autocatalysis of LexA (inhibitor of the SOS response) resulting in the activation of DNA repair [27, 29, 30]. It is reported that RecA is required for survival of A. baumannii after exposure to oxidative stress produced by reactive oxygen species (ROS) and reactive nitrogen intermediate [27, 31]. As the effects of disinfectants and antibiotics could be compromised or reduced in the presence of functional RecA. An inhibitor of RecA protein of A. baumannii can be useful to enhance the efficacy of current antibiotics or disinfectants. There are different approaches that can be used to find a suitable inhibitor [14, 32,33,34,35,36] here; we have used high throughput virtual screening and molecular dynamics simulation (MDS) approach to design an inhibitor against RecA of A. baumannii. The designed inhibitor is expected to block resistance to bactericidal antibiotics and disinfectant act via ROS production.
Methods
Measurement of ROS
The ROS production in bacterial cell was measured based on conversion of nitro blue tetrazolium (NBT) to blue or purple colored complex formazan crystals in the presence of superoxide anion. The published method was used with some modification [37]. In brief, bacterial pellet was re-suspended in 2% NBT solution for 1 h in dark at room temperature, followed by phosphate-buffered saline washing at 10,000 g for 5 min. This step is followed by methanol wash. The pellet was suspended in a solution of 2 M KOH, for disruption of cell membrane. Dimethyl sulfoxide was used to dissolve formazan crystals for 10 min at room temperature. After centrifugation at 10,000 g for 5 min, supernatant was taken for absorbance at 620 nm. ROS production was measured in the presence of imipenem and disinfectants. Similar experiment was also performed in the presence of designed lead molecule and combination of imipenem and disinfectants. The ROS was quantified in the presence and absence of different disinfectants such as phenol, ethanol, hydrogen peroxide, sodium hypochlorite, and carbapenem antibiotics like imipenem.
Isolation of bacterial DNA and its separation using agarose gel electrophoresis
The bacterial pellet (treated and untreated) was suspended in 500 μl of distilled water. This bacterial suspension was incubated at 100 °C in a boiling water bath for 15 min and was immediately transferred to frozen ice. Subsequently, suspension was centrifuged at 3,000 g at 4 °C for 10 min. The supernatant was transferred to a clean 500 μl tube and stored at − 20 °C until analysis [38]. The fragmentation or degradation of DNA isolated from untreated and antibiotic or disinfectant treated (± lead) bacterial samples were checked by 1% agarose gel electrophoresis.
Selection of template
Amino acid sequence of RecA protein of A. baumannii was retrieved from protein database of NCBI (accession no. gb: ALJ87635.1, 349aa). BLASTp (protein-protein BLAST) was used to find templates for homology modeling of RecA. Search of identified sequence templates (from blast) against Protein Data Bank (pdb) database led to the identification of different structural template.
Molecular modeling of RecA
Six BLASTp generated protein templates with highest E-values and % identity were selected and their structures were retrieved from protein data bank. The basic homology modeling was performed using the software MODELERv9.17 as per published protocol [15] . Final models obtained from homology modeling was used for the further refinement using GalaxyWEB refine tool.
Validation of modeled structure
All the five models generated after refinement were further analyzed using Protein Structure Validation Suite [39], which gives information about all the essential validation parameters such as PROCHECK, VERIFY3D [40], and Ramachandran plots. Another validation tool, i.e., ProSA-Web [41] was also used to calculate an overall quality score for a specific input structure. Secondary structures of modeled proteins were determined by PDBSUM software [42]. The best model was chosen and subjected to energy minimization using protein preparation wizard application of Schrödinger suite. The minimized protein structure was used for virtual screening studies.
Active site prediction
Site map was used to determine active site of RecA [43]. The site with the highest site score was selected and active site residue information was obtained using PyMOL. Enrichment calculator with known ligands and decoy were also used to validate the selected active site.
Protein preparation and receptor grid generation
Modeled RecA were prepared using default parameter of protein preparation wizard module in Schrodinger. Receptor grid of RecA was generated for optimized and minimized RecA using selected 51 amino acid residues of predicted binding site.
Selection of metabolite (primary and secondary) from the ZINC database
We have selected Zbc (Zinc Biogenic compounds) database for the screening of suitable inhibitors against RecA protein. This database consists of secondary (commonly called natural products) and primary metabolite (commonly called simple metabolite). This library consists of a total of 1,80,313 compounds and basic information about the compounds such as its molecular weight, structures and activity are available in the database.
Ligand preparation and ADMET analysis
All 1,80,313 compounds were first subjected to LigPrep based ligand preparation. LigPrep module [44] prepares three-dimensional structures for compounds with a high quality and correct chirality [45]. Ionization states for the compounds were generated at pH of 7.0 ± 2.0. The Epik and tautomeric states were generated followed by the desaltation of ligands. An OPLS_2005 force field was used for the minimization of the compounds. A maximum of 32 stereoisomers were generated for each 1,80,313 ligands. The first step of virtual screening involves the evaluation of drug-likeliness of small molecules. Drug-like molecules exhibit favorable absorption, distribution, metabolism, excretion, toxicological (ADMET) parameters. For any small molecule to be considered a likely lead molecule, it must satisfy these (ADME) properties and have a good toxicological profile. The QuikProp application was used for determining various ADMET and physiochemical properties of ligands [46]. A total of 2,68,790 ligands stereoisomers have been selected after ADMET analysis. The selected 2,68,790 ligands were undergone Lipinski’s filtering.
Lead screening using HTVS(high-throughput virtual screening), SP(standard precision), and XP(extra precision) docking
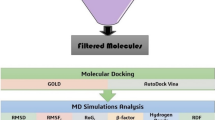
Lead screening workflow was performed using Maestro v11 to screen the metabolite library against the binding site of RecA protein target. We skipped the ligand preparation step included in the virtual screening workflow due to their prior preparation using LigPrep. This step was followed by screening of selected ligands for receptor grid of RecA. The screening was based on docking with default parameters using Glide (Grid-based ligand docking from energetics) program of Schrodinger suite [47]. The scaling factor was kept at default and OPLS_2005 force field was used during the three step of docking process. HTVS ligand docking was followed by SP and XP docking. The XP docking helps in removing the false positives and the scoring function is much stricter than the HTVS. The greater the XP Glide score, the better affinity of the hit to bind to the protein target. By default, virtual screening workflow module retains 10% of the best compounds for RecA target proteins. The steps of lead screening have been shown through a flowchart diagram (Fig. 1).

Flowchart representation of different steps of lead screening method used (Color figure online)
Validation of docking protocol or enrichment calculations
We have used enrichment calculator to check the parameters that decides whether a docking program is able to select active ligands with respect to inactive ligands (decoys) and whether it is able to select these active ligands in the top % of a ranked database. The Schrodinger based standard decoys have been used in this study [47]. The enrichment calculations were done by ROC analysis of XP-docked ligands and active ligands with decoys [48]. ROC curve checks the ability of docking protocol to discriminate between active molecules among the decoy set of compounds.
Molecular mechanics/generalized born surface area calculations of selected library compounds
The estimation of free binding energies for the best hit docked complexes using MM force fields and implicit solvation was done using the molecular mechanics/generalized born surface area (MM-GBSA) method using the prime module of Schrodinger suite v11. The protein–ligand complexes were ranked based on their binding free energy calculation.
Molecular dynamics simulation
MDS was performed using GROMACS version 5.1.4 [49]. It helps in determining the interatomic motions, as well as the dynamics of protein and protein–ligand complex [50]. The MDS is done to validate the structure based on the stability of protein as well as the protein–ligand complex. The RMSD (root mean square deviation) values are monitored under this study along with several other factors such as temperature, pressure, density, potential energy, etc. The pdb structure of our modeled protein was used to construct the topology. This was done using GROMOS96-43a1 force field. Further, this ligand topology was constructed using PRODRG2 server with GROMOS87/GROMOS96 force field [51]. A cubic period box and dodecahedron box setting was used in case of protein (RecA) and RecA-lead molecule complex, respectively, with 1.0 nm distance (minimum) between protein and edge of box. RecA have – 8.99 negative charges; hence, nine sodium ions (positive charge) were added to neutralize the charge of the protein due to the presence of water molecules (solvation) in the system and this was followed by energy minimization of the system. The solvent molecules of the system were minimized using maximum 50,000 steps of steepest descent minimization algorithm, while protein was kept frozen. The maximum force was kept 1000 kJ/mol/nm. This step was followed by equilibration of solvent and ions around the protein. Equilibration was conducted in two phases and first phase was NVT ensemble (constant number of particle, volume, and temperature) or isothermal–isochoric or canonical equilibration for 100 ps with maximum 50,000 steps. This NVT step stabilizes the temperature of the system. This step is followed by NPT ensemble (constant number of particle, pressure, and temperature) or isothermal–isobaric ensemble, for 100 ps with maximum 50,000 steps. Isothermal–isobaric ensemble most closely resembles the experimental conditions. Both above steps equilibrate the system at desired temperature and pressure; hence, MDS was performed for 100 ps (1 ns) timescale of protein as well as protein-inhibitor complex. Each component of system like protein, ligand, water molecules, and ions were coupled in constant temperature and pressure. The MDS was performed for RecA alone and three RecA–ligands complexes including top two ligands (ZINC01530654 and ZINC01532364) and on other isomer or tautomer (T-ZINC01530654) of ZINC01530654 with energy lower than second ligand.
In-vitro validation of designed lead molecules
The designed leads was experimentally validated using disc diffusion assay using our published methods [33]. Disc diffusion assay was performed in the presence of a carbapenem, i.e., imipenem (16 µg/ml), disinfectants (H2O2), ZINC01530654 (16 mg/ml), and in combination. The ROS was also monitored in above conditions.
Results
In the present study, we have used two clinical strains (RS-6694 and RS-307) of A. baumannii. Both these strains showed similar results and the results obtained with only one of the strains, RS-6694 is presented here. This strain was collected from AIIMS, New Delhi, and found to be a multi-drug-resistant strain with high minimum inhibitory concentration (>16 μg/ml) for imipenem (a carbapenem).
Correlation of ROS production and DNA fragmentation
The results showed that the treatment of A. baumannii with disinfectants, and a carbapenem (i.e. imipenem), showed significant overproduction of ROS as compared with their absence (Fig. 2). ROS may lead to the DNA fragmentation. Therefore, we have tested the DNA fragmentation and found that the DNA breakdown is higher in the bacteria after treatment with imipenem or disinfectants (Fig. 3). To overcome DNA damage, bacteria activate SOS response, where RecA protein pays a significant role [27, 31]. This showed that RecA protein of A. baumannii might be important and useful to explore in term of increasing the efficacy of current used anti-bacterials.

Reactive oxygen species (ROS) production in resistant strains (RS-6694) of A. baumannii under the influence of different hospital disinfectants such as phenol, sodium hypochlorite, ethanol and hydrogen peroxide and carbapenem antibiotic such as imipenem

Effect of reactive oxygen species (produced by carbapenem and disinfectant treatment) on the total DNA of RS-6694 strain of A. baumannii
Modeled structure and validation
The BLAST search for potential templates yielded good E-values and greater % identity for 2REC ʻA’, 1N03 ʻA’, IU94ʻA’ and 3CMVʻA’, 5JRJʻA’, and 2ZR0ʻA’ (Supplementary Table ST1). The structural and sequence alignments of these templates with the target protein along with their RMS values were determined using TM align (Supplementary Figure SF-1 and Supplementary Table ST-2). The model was prepared using these multiple templates and undergoes refinement using galaxy refine tool. The characteristic parameters of five different models are listed in Table 1. Model 2 with lowest RMSD, high Ramachandran favorable area among all five models, has been selected for further study. The validation results of this refined model of RecA are provided in Table 2 and Fig. 4. Besides validation analysis, secondary structure of modeled protein was also determined using PUBsum and its wiring diagram are shown in Fig. 5. ProMotiff PUBsum analysis showed that the modeled RecA contain 34.4% alpha helix and 22.3% beta-sheet and remaining 43.3% other amino acid. It contains 3 beta sheets, 11 helices, 2 beta-alpha-beta motifs, 6 beta hairpins, 6 beta bulges, 14 strands, 13 helix-helix interactions, 32 beta-turns and gamma turns.

a Ribbon structure of modeled RecA protein. b Ramachandran plot summary for selected residues from Procheck showing 98.3% residues in most favorable regions, 0.9% in allowed regions and 0.9% in disallowed regions. c Validation of RecA modeled structure using Prosa-web shows Z-score as – 7.73 for modeled RecA. d Knowledge-based energy at different sequence position of modeled RecA (Color figure online)

Secondary structure of amino acid residues of RecA (349 aa) obtained using PDBSum (Color figure online)
Active site prediction
Out of all different sites provided by sitemap for RecA, the site with the highest site score was selected. The active site was made up of 51 residues, namely Val36, Glu37, Ala38, Val39, Ser40, Gly42, Leu44, Asp47, Lys57, Gln77, Ala80, Gln81, Cys82, Lys84, Ser85, Gly87, Lys105, Leu106, Gly107, Asp138, Val230, Glu232, Val237, Gln260, Leu262, Lys265, Gly266, Thr267, Asn268, Gln269, Leu270, Gly271, Leu273, Asn302, Ile305, Arg306, Phe308, Glu309, Thr312, Ala315, Glu316, Glu319, Arg320, Arg323, Gln336, Ile337, Glu338, Asp339, Leu345, Leu346, and Glu348. In the present study, all the residues present within 4 Å surrounding of active site were considered and used for generation of docking grid.
ADME-toxicity analysis and Lipinski’s filter
All conformers of different ligands were initially screened for their ADMET properties via computational approach. This step comes under the virtual screening of leads and is included prior to the docking based filter of compounds in our study. After ligand filtration, on the basis of these properties and toxicity, the filtered 2,68,790 leads were selected. The selected ligands underwent Lipinski’s filtering. Only 1,94,802 ligands conformers have been selected by this filter and used for high throughput virtual screening.
Virtual high-throughput screening
Virtual screening was done to design effective RecA inhibitor by screening of ZINC metabolites compounds on the basis of their affinity for the target protein. This is initiated by HTVS ligand docking of 1,94,802 probable ligands with RecA. This step shortlisted 19,480 compounds. It was followed by SP and XP docking, which filtered number of remaining compounds to 1948 and 195 respectively. Molecular docking output for the XP docked 195 compounds against RecA have been shown (Supplementary data in Table ST-3).
Enrichment calculations of selected compounds
These XP-docked 195 compounds were subjected to enrichment calculations. There are different parameters used to describe the result but Boltzmann-enhanced discrimination of receiver operating characteristics (BEDROC) are most commonly used [52]. The ROC value obtained for the XP docked compounds was 0.90. The BEDROC value came out to be 0.896 as can be deduced from Fig. 6. The area under curve was obtained as 0.9, which was found to be greater than 0.5 that validates our XP docking result.

Representation of ROC curve shows sensitivity and specificity of XP mode of docking (Color figure online)
Binding free energy calculations using MM-GBSA solvation
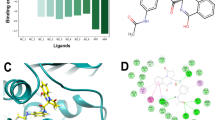
The leads 195 hits were then subjected to binding free energy calculations using MM-GBSA approach. The MM-GBSA analysis of selected 195 compounds is listed in table (Table 2 shows top two ligands and data for all the 195 compounds are listed in supplementary table ST-3). As evident from the Table 3 and Supplementary Table TS-3, ZINC01530654 (– 68.99) and ZINC01532364 (– 66.67) showed lower binding energy among all 195 compounds. The zinc database provides the nomenclature of this compound ZINC01530654 as ( + −)-2-((4-((7-Chloro-4-quinolyl)amino)pentyl)ethylamino)ethanol sulfate (or hydroxychloroquine sulfate) and ZINC01532364 as ( + −)-N-(2-((2-Hydroxy-3-(p-hydroxyphenoxy)propyl)amino)ethyl)-4-morpholinecarboxamide (or Xamoterol hemifumarate). The physical and chemical properties at physiological pH have been shown in Table 4 and Table 5. The interactions of ZINC01530654 and ZINC01532364 with RecA are shown in Fig. 7. A tautomer of top hit ZINC01530654, i.e, ZINC01530654-T (with free energy of − 59.56 kcal/mol) was also selected for MDS analysis along with top two ligands, i.e., ZINC01530654 (− 69.99 kcal/mol) and ZINC01532364 (− 66.67 kcal/mol).

Surface view showing interaction of RecA with ZINC01530654 (a) and ZINC01532364 (c). Similarly, (b) represents the interacting residues of RecA with ZINC01530654, whereas (d) showed the interacting residues of RecA with ZINC01532364 (Color figure online)
MDS analysis
The MDS results (Fig. 8) showed that ZINC01532364 and RecA-ZINC01530654 have good interaction with the RecA protein. The RecA protein showed biphasic stability (40 ps with RMSD 0.23 nm, and 75 ps with RMSD 0.26 nm). RecA-ZINC01532364 complex also showed biphasic stability (RMSD 0.25 nm at 11 ps; RMSD 0.32 nm at 75 ps). The similar pattern was also seen in RecA-ZINC01532364 complex, which also showed biphasic stability (RMSD 0.34 nm at 50 ps; RMSD0.38 nm at 80 ps). The RecA-ZINC01530654-T complex monophasic stability and complex is stabilized at 60 ns with RMSD of 0.43 nm. Result also showed that ZINC01530654 have simulation pattern and stability, which is very similar to the RecA protein; hence ZINC01530654 has been selected for use in-vitro experiment validation.

Result of molecular dynamics simulation showing root mean square deviation plot of RecA as a function of time for modeled RecA protein alone (1), RecA- ZINC1530654 complex (2), RecA- ZINC01532364 complex (3), and RecA-ZINC1530654 tautomer complex (4) as a function of time (Color figure online)
Off-target analysis of lead molecule
Nonspecific targets or off-targets of lead molecule were checked by Swiss target prediction. The result showed that lead does not interact with any bacterial off-target except ion channels that is required for influx of lead molecules. Further studies need to be done to validate its specificity towards RecA of A. baumannii.
Experimental validation of designed lead molecule
ROS production and disc diffusion assay with different combinations of carbapenem, disinfectants, and ZINC01530654 validate the results (Figs. 9 and 10). We have estimated the ROS production in the carbapenem resistant strains (RS-6694 and RS-307) of A. baumannii under different conditions and result shows that ROS production is increased after treatment with ZINC01530654 with combination of carbapenem (e.g., imipenem) or disinfectant (e.g., H2O2) (Fig. 9). The disc diffusion assay (Fig. 10) showed that ZINC01530654 alone produces 0.8 cm clear zone, imipenem alone produces 1.5 cm, whereas imipenem with ZINC01530654 produces 2.5 cm zone. A negative control (Rec-A-deficient Escherichia coli) was used where it has little effects. These experimental results suggest the use of ZINC01530654 in combination with imipenem or disinfectant could be additive or synergistic.

ROS estimation under different combination of carbapenem, disinfectant, and ZINC01530654

Disc diffusion assay for analyzing the effect of ZINC01530654 on efficacy of carbapenem (i.e. imipenem). 1, Carbapenem alone; 2, ZINC01530654 alone; 3, Carbapenem with ZINC01530654 (Color figure online)
Discussion
In a recent study of infections in intensive care unit conducted among 75 countries, A. baumannii was found to be the fifth most common pathogen. The success of A. baumannii is believed to be related to its ability to survive after exposure to antibiotics and disinfectants [53]. Acinetobacter has the extraordinary ability to accumulate a great variety of resistance through different mechanisms, either mutations or acquisition of genetic elements such as plasmids, integrons, transposons, resistant islands, making this microorganism multi- or pan-drug-resistant. ROS production is known to be a response to the action of various disinfectants and bactericidal antibiotics [54, 55]. ROS production is also used by the host against different pathogens [24, 36]. The ability of Acinetobacter to survive in the environment during prolonged periods of time is due to its innate resistance to desiccation and disinfectants, makes A. baumannii very difficult to eradicate from the clinical setting. Antibiotic and disinfectant resistance arises from the maintenance of resistance mutations or genes required for the resistance. Antibiotic and disinfectant resistance is acquired in response to antibacterial molecules by activating SOS-mediated DNA repair that involves activation of RecA [21]. Therefore, an inhibitor of RecA could be helpful and could be used along with antibiotics and disinfectants to enhance the efficacy of these antimicrobials because both act via ROS production and DNA damage.
We employed high-throughput virtual screening to identify novel primary and secondary metabolite-based hits from ZINC database (BioBlocks) as an inhibitor against RecA that can be used to control A. baumannii. The sequence and structural alignment showed that the templates used in our study showed high similarity. Validation parameters such as Ramachandran plot and PROSA-web analysis were also used for the determination of quality of modeled protein. It can be deduced from result that the model has a higher negative value of Z-score and also it has low energy throughout different residues of the model. As observed from results, in our modeled protein, the predicted binding site for docking had a site score more than 1, suggesting that this site is of particular promise in drug binding. A total of 195 compounds were shortlisted using virtual screening against RecA. They were also evaluated based on their binding free energy calculation for further refined ranking. Two of top hits based on lower binding free energies are ZINC01530654 (− 68.99 kcal/mol) and ZINC01532364 (− 66.67 kcal/mol). Both these leads have very similar binding energies therefore both have been selected for the MDS analysis. We have also selected a tautomer of ZINC01530654 named ZINC01530654-T with free energy of – 59.56 kcal/mol. The MD simulation result showed that RecA-ZINC01530654 and RecA-ZINC01532364 follows biphasic simulation pattern but RecA- ZINC01530654-T follow the monophasic stabilization pattern. The RecA-ZINC01530654 simulation pattern is very similar to the RecA protein and it form most stable complex and its stability is close to the RecA protein. Therefore, we have selected this lead compound for in-vitro experimental validation. The disc diffusion assay, ROS experiment, and DNA fragmentation assay also validate in-silico results.
Hence, the identified lead molecule of the RecA have medicinal values that can be applicable in the treatment of infections caused by A. baumannii and may enhance the efficacy of the antibiotics and disinfectants. The rise in antibiotic and disinfectant resistance is a major health concern with the emergence of antibiotic-resistant ‘superbugs’ in the clinic, stressing the need for strategies that prolong the lifetime of antibiotics, as the discovery of new antibiotics is not keeping pace with the development of resistance. Therefore, our finding suggests a strategy to chemically inhibit or block the vital process controlled by RecA and can be used against A. baumannii for better control of infection and development of new therapeutics with novel mechanism of action. The lead compound could be further investigated experimentally for its interaction with purified RecA, its toxicity and its effective concentration that further validate its potentiality as a RecA inhibitor in A. baumannii.
References
Tiwari V, Kapil A, Moganty RR. Carbapenem-hydrolyzing oxacillinase in high resistant strains of Acinetobacter baumannii isolated from India. Microb Pathog. 2012;53:81–86. https://doi.org/10.1016/j.micpath.2012.05.004.
Maragakis LL, Perl TM. Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clin Infect Dis. 2008;46:1254–63. https://doi.org/10.1016/j.micpath.2012.05.004.
Jang TN, Lee SH, Huang CH, Lee CL, Chen WY. Risk factors and impact of nosocomial Acinetobacter baumannii bloodstream infections in the adult intensive care unit: a case-control study. J Hosp Infect. 2009;73:143–50 https://doi.org/10.1016/j.jhin.2009.06.007.
Mahgoub S, Ahmed J, Glatt AE. Underlying characteristics of patients harboring highly resistant Acinetobacter baumannii. Am J Infect Control. 2002;30:386–90.
Roy, R, Tiwari, M, Donelli, G & Tiwari, V Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence, 2017; https://doi.org/10.1080/21505594.2017.1313372.
Tiwari V, Tiwari M. Phosphoproteomics as an emerging weapon to develop new antibiotics against carbapenem resistant strain of Acinetobacter baumannii. J Proteom. 2015;112:336–8. https://doi.org/10.1016/j.jprot.2014.09.008.
Tiwari V, Tiwari M. Quantitative Proteomics to study Carbapenem Resistance in Acinetobacter baumannii. Front Microbiol. 2014;5:512. https://doi.org/10.3389/fmicb.2014.00512.
Zhanel GG, et al. Comparative review of the carbapenems. Drugs. 2007;67:1027–52.
Tiwari V, Moganty RR. Conformational stability of OXA-51 beta-lactamase explains its role in carbapenem resistance of Acinetobacter baumannii. J Biomol Struct Dyn. 2014;32:1406–20. https://doi.org/10.1080/07391102.2013.819789.
Tiwari V, Rajeswari MR. Effect of Iron Availability on the Survival of Carbapenem-Resistant Acinetobacter baumannii: a Proteomic Approach. J Proteom & Bioinforma. 2013;06:125–31. https://doi.org/10.4172/jpb.1000270.
Tiwari V, Vashistt J, Kapil A, Moganty RR. Comparative proteomics of inner membrane fraction from carbapenem-resistant Acinetobacter baumannii with a reference strain. PloS One. 2012;7:e39451. https://doi.org/10.1371/journal.pone.0039451.
Vashist J, Tiwari V, Das R, Kapil A, Rajeswari MR. Analysis of penicillin-binding proteins (PBPs) in carbapenem resistant Acinetobacter baumannii. Indian J Med Res. 2011;133:332–8.
Verma, P, Tiwari, M & Tiwari V. In-silico high throughput virtual screening and molecular dynamics simulation study to identify inhibitor for AdeABC efflux pump of Acinetobacter baumannii. J Biomol Struct Dyn. 2017; 1–13. https://doi.org/10.1080/21505594.2017.1313372.
Tiwari V, Tiwari D, Patel V, Tiwari M. Effect of secondary metabolite of Actinidia deliciosa on the biofilm and extra-cellular matrix components of Acinetobacter baumannii. Microb Pathog. 2017;110:345–51. https://doi.org/10.1016/j.micpath.2017.07.013.
Tiwari V, Patel V, Tiwari M. In-silico screening and experimental validation reveal L-Adrenaline as anti-biofilm molecule against biofilm-associated protein (Bap) producing Acinetobacter baumannii. Int J Biol Macromol. 2018;107(Pt A):1242–52. https://doi.org/10.1016/j.ijbiomac.2017.09.105.
Liu WJ, et al. Frequency of antiseptic resistance genes and reduced susceptibility to biocides in carbapenem-resistant Acinetobacter baumannii. J Med Microbiol. 2017;66:13–17. https://doi.org/10.1099/jmm.0.000403.
Buffet-Bataillon S, Tattevin P, Maillard JY, Bonnaure-Mallet M, Jolivet-Gougeon A. Efflux pump induction by quaternary ammonium compounds and fluoroquinolone resistance in bacteria. Future Microbiol. 2016;11:81–92. https://doi.org/10.2217/fmb.15.131.
Fernandez-Cuenca F, et al. Reduced susceptibility to biocides in Acinetobacter baumannii: association with resistance to antimicrobials, epidemiological behaviour, biological cost and effect on the expression of genes encoding porins and efflux pumps. J Antimicrob Chemother. 2015;70:3222–9. https://doi.org/10.1093/jac/dkv262
Kawamura-Sato K, Wachino J, Kondo T, Ito H, Arakawa Y. Correlation between reduced susceptibility to disinfectants and multidrug resistance among clinical isolates of Acinetobacter species. J Antimicrob Chemother. 2010;65:1975–83. https://doi.org/10.1093/jac/dkq227
Tucker AT, et al. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. MBio. 2014;5:e01313–01314. https://doi.org/10.1128/mBio.01313-14.
Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR. RecA inhibitors potentiate antibiotic activity and block evolution of antibiotic resistance. Cell Chem Biol. 2016;23:381–91. https://doi.org/10.1016/j.chembiol.2016.02.010.
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. https://doi.org/10.1016/j.cell.2007.06.049.
Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755–76. https://doi.org/10.1146/annurev.biochem.77.061606.161055.
Fang, FC. Antimicrobial actions of reactive oxygen species. MBio. 2011 Sep6; 2(5). pii: e00141–11. https://doi.org/10.1128/mBio.00141-11.
Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–9.
Nautiyal A, Patil KN, Muniyappa K. Suramin is a potent and selective inhibitor of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. J Antimicrob Chemother. 2014;69:1834–43. https://doi.org/10.1093/jac/dku080.
Aranda J, et al. Acinetobacter baumannii RecA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J Bacteriol. 2011;193:3740–7. https://doi.org/10.1128/JB.00389-11.
Galhardo RS, Almeida CE, Leitao AC, Cabral-Neto JB. Repair of DNA lesions induced by hydrogen peroxide in the presence of iron chelators in Escherichia coli: participation of endonuclease IV and Fpg. J Bacteriol. 2000;182:1964–8.
Grinholc M, et al. Fine-tuning recA expression in Staphylococcus aureus for antimicrobial photoinactivation: importance of photo-induced DNA damage in the photoinactivation mechanism. Appl Microbiol Biotechnol. 2015;99:9161–76. https://doi.org/10.1007/s00253-015-6863-z.
Cox MM. Molecular Genetics of Recombination. Aguilera A, Rothstein R editors. 135–167. Springer: Berlin, Heidelberg, 2007.
Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2:820–32. https://doi.org/10.1038/nrmicro1004.
Tiwari V, Roy R, Tiwari M. Antimicrobial active herbal compounds against Acinetobacter baumannii and other pathogens. Front Microbiol. 2015;6:618. https://doi.org/10.3389/fmicb.2015.00618.
Tiwari M, Roy R, Tiwari V. Screening of herbal-based bioactive extract against carbapenem-resistant strain of Acinetobacter baumannii. Microb Drug Resist. 2016;22:364–71. https://doi.org/10.1089/mdr.2015.0270.
Tiwari V, Khokar MK, Tiwari M, Barala S, Kumar M. Anti-bacterial activity of polyvinyl pyrrolidone capped silver nanoparticles on the carbapenem resistant strain of Acinetobacter baumannii. J Nanomed Nanotechnol. 2014;5:246. https://doi.org/10.4172/2157-7439.1000246.
Tiwari M, Raghav R, Tiwari V. Comparative anti-bacterial activity of differently capped silver nanomaterial on the carbapenem sensitive and resistant strains of Acinetobacter baumannii. J Nanomed Nanotechnol. 2015;6:314.
Tiwari V, Tiwari M, Solanki V. Polyvinylpyrrolidone-capped silver nanoparticle inhibits infection of carbapenem-resistant strain of Acinetobacter baumannii in the human pulmonary epithelial cell. Front Immunol. 2017;8:973. https://doi.org/10.3389/fimmu.2017.00973.
Singh N, Wadhawan M, Tiwari S, Kumar R, Rathaur S. Inhibition of Setaria cervi protein tyrosine phosphatases by phenylarsine oxide: a proteomic and biochemical study. Acta Trop. 2016;159:20–28. https://doi.org/10.1016/j.actatropica.2016.03.004
Peng X, et al. Comparison of direct boiling method with commercial kits for extracting fecal microbiome DNA by Illumina sequencing of 16S rRNA tags. J Microbiol Methods. 2013;95:455–62. https://doi.org/10.1016/j.mimet.2013.07.015.
Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007; 66:778–95.
Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404.
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–410. https://doi.org/10.1093/nar/gkm290.
Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37:D355–359. https://doi.org/10.1093/nar/gkn860.
Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–89. https://doi.org/10.1021/ci800324m.
Schrödinger. Schrödinger Release 2016-4: LigPrep. https://www.schrodinger.com/ligprep (2016).
Tiwari V, Moganty RR. Structural studies on New Delhi Metallo-beta-lactamase (NDM-2) suggest old beta-lactam, penicillin to be better antibiotic for NDM-2-harbouring Acinetobacter baumanni. J Biomol Struct Dyn. 2013;31:591–601. https://doi.org/10.1080/07391102.2012.706075.
Schrödinger. Schrödinger Release 2016-4: QikProp. https://www.schrodinger.com/qikprop (2016).
Halgren TA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. https://doi.org/10.1021/jm030644s.
Florkowski CM. Sensitivity, specificity, receiver-operating characteristic (ROC) curves and likelihood ratios: communicating the performance of diagnostic tests. Clin Biochem Rev. 2008;29(Suppl 1):S83–87.
Abraham MJ, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. https://doi.org/10.1016/j.softx.2015.06.001.
Tiwari V, Nagpal I, Subbarao N, Moganty RR. In-silico modeling of a novel OXA-51 from beta-lactam-resistant Acinetobacter baumannii and its interaction with various antibiotics. J Mol Model. 2012;18:3351–61. https://doi.org/10.1007/s00894-011-1346-3.
Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–63. https://doi.org/10.1107/S0907444904011679.
Empereur-Mot C, et al. Predictiveness curves in virtual screening. J Chemin-. 2015;7:52. https://doi.org/10.1186/s13321-015-0100-8.
Roca I, Espinal P, Vila-Farres X, Vila J. The Acinetobacter baumannii oxymoron: commensal hospital dweller turned pan-drug-resistant menace. Front Microbiol. 2012;3:148. https://doi.org/10.3389/fmicb.2012.00148.
Belenky P, et al. Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep. 2015;13:968–80. https://doi.org/10.1016/j.celrep.2015.09.059.
Dwyer DJ, et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA. 2014;111:E2100–2109. https://doi.org/10.1073/pnas.1401876111.
Acknowledgements
VT thanks SERB, DST, India for Start Up grant (SB/YS/LS-07/2014). MT thanks Central University of Rajasthan for Ph.D. fellowship.
Author information
Authors and Affiliations
Contributions
VT conceived and designed the experiments, performed the experiments (in-silico modeling and drug designing, MDS, and ROS estimation for imipenem), and analysed the data. MT performed experimental validation of lead. DB performed ROS estimation for disinfectants. VT wrote the manuscript. MT and VT proofread the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Tiwari, V., Tiwari, M. & Biswas, D. Rationale and design of an inhibitor of RecA protein as an inhibitor of Acinetobacter baumannii. J Antibiot 71, 522–534 (2018). https://doi.org/10.1038/s41429-018-0026-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-018-0026-2
This article is cited by
-
Identification of vaccine and drug targets in Shigella dysenteriae sd197 using reverse vaccinology approach
Scientific Reports (2022)
-
Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa
Scientific Reports (2019)
-
Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii
Scientific Reports (2018)
