
Abstract
For a long time, water has been speculated to play an essential role in the interactions of proteins and cells with artificial biocompatible materials. The current question is how water molecules at the interfaces affect the adsorption of proteins and the adhesion of cells. To answer this question, we introduce recent works that investigated the interfacial behavior of water near self-assembled monolayers (SAMs) by different types of analytical techniques. By combining these findings, we discuss how interfacial water affects the protein and cell resistance of various bioinert SAMs.
Similar content being viewed by others
Bioinert self-assembled monolayers
Interaction of artificial materials with biomolecules and cells
The importance of the interfaces of artificial materials with biomolecules, cells, and tissues is rapidly becoming clearer in light of the increasing needs for biosensors, artificial organs and tissues, and regenerative medicines in our aging society [1, 2]. The essential issue in this field is how materials can precisely induce the desired responses of the biomolecules and cells in contact with the materials [1, 3,4,5]. Such desired responses include adsorption, determination of the molecular orientation and conformational changes of biomolecules, and the adhesion, extension, apoptosis, proliferation, and differentiation of living cells [6]. Although a considerable number of complicated cascading molecular processes lead to the above responses, the contact of biomolecules and cells with materials is the starting point of the processes. Therefore, understanding the interactions of biomaterials with biomolecules and cells is critical to discussing the mechanisms that underlie their responses [7].
In general, interactions between objects in water are described by the well-known DLVO theory, which was established by Derjaguin, Landau, Verwey, and Overbeek [8, 9]. In this theory, the interactions are described by an electric double layer and van der Waals interactions. However, with the DLVO theory alone, it is difficult to explain protein and cell resistance. Vogler et al. [10] pointed out this issue and discussed the role of water-mediated force. They analyzed previous reports on surface force measurements and found that the water-mediated force correlates well with macroscopic water wettability (water contact angle). Unfortunately, the authors analyzed only the results of quartz surfaces whose macroscopic wettability was controlled by hydrophobic silanes. Because both the quartz and modified surfaces adsorbed proteins, it was difficult to discuss the correlation between the water-mediated force and protein adsorption.
The macroscopic water wettability also cannot explicate the mechanism underlying the high-blood compatibility (avoiding the activation of blood cells) of poly(ethylene glycol) (PEG), poly(2-methoxyethyl acrylate) (PMEA), and poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC), which have been widely used for bioinert (avoiding the adsorption and adhesion of biomolecules and cells) coatings [11,12,13,14,15,16]. Their compatibility critically depends on the method of surface coating (spin coat, grafting, the formation of a polymer brush, etc.), as different methods result in different surface structures and molecular densities. Therefore, the clarification of the mechanism requires interfacial analysis using well-defined platforms regarding structure, the ordering of molecules, and physicochemical properties.
Self-assembled monolayers in biology
Self-assembled monolayers (SAMs) (Fig. 1) provide versatile platforms for various surface and interfacial studies such as tribology, adhesion, adsorption, and corrosion, since we can modify the physicochemical properties (wettability, surface charges, polarity, etc.) of solid surfaces by employing molecules with different terminal groups [17]. By choosing the combinations of anchoring groups of molecules that constitute the SAMs and substrates forming chemical bonds with the anchoring groups, we can fabricate highly ordered and densely packed monolayers on various solid substrates such as noble metals, semiconductors, and oxides.

Schematic illustration of self-assembled monolayers (SAMs)
In the 1990s, the research group of Prof. George Whitesides initiated the use of SAMs as model organic surfaces to investigate the interactions of proteins with organic materials [18, 19]. Later, they expanded their studies to cell adhesion testing [20,21,22,23]. The most important finding they reported is that protein adsorption and cell adhesion can be easily controlled by changing the terminal groups of the molecules. This finding is currently widely used in the design of biosensors and cell assays. In these applications, the formation of bioinert (protein- and cell-resistant) surfaces is essential, in addition to the promotion of protein adsorption and cell adhesion.
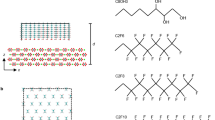
Thus far, many kinds of bioinert SAMs have been reported (Fig. 2) [24]. Roughly, the bioinert SAMs are categorized into two types: nonionic and zwitterionic. The nonionic SAMs consisting of poly(ethylene glycol) (PEG)- and oligo(ethylene glycol) (OEG)-terminated alkanethiols on gold are the most widely used bioinert SAMs. Later, saccharide-functionalized monolayers were reported to exhibit both a specific affinity to the target protein and bioinert properties [25, 26]. Among SAMs of the zwitterionic type, two types have been particularly studied, namely, betaine- and phosphorylcholine (PC)-terminated alkanethiol SAMs [27, 28]. SAMs consisting of mixed negatively and positively charged molecules [29] exhibited excellent bioinertness.

Chemical structures of thiols to form the following bioinert SAMs: oligo(ethylene glycol)-terminated (OEG), tri(methylamine)-terminated (TMA), sulfonic acid-terminated (SA), sulfobetaine-terminated (SB), and phosphocholine-terminated (PC) SAMs. Note that TMA and SA molecules form MC-SAM
History of research on bioinert SAMs
An old-fashioned method of rendering solid surfaces bioinert is coating the substrate with hydrophilic polymers. The polymer chains on the substrate are fully hydrated, and their configuration has a high degree of freedom. In this situation, the approach of biomolecules to the surface is energetically unfavorable from the stand points of enthalpy and entropy, as the process would require the dehydration of polymers and a decrease in the degree of freedom of the polymer chains [30]. This idea is referred to as ‘‘steric repulsion’’, which is often used to explain the bioinertness of surfaces covered with hydrophilic polymers.
In contrast to the above case of polymers, the molecules constituting SAMs are densely packed (typically 3 to 5 molecules/nm2) [31,32,33,34]. Therefore, steric repulsion is not expected to contribute to the bioinertness of the surface in the case of SAMs. Therefore, the mystery underlying the bioinertness of the above SAMs has attracted many researchers [24]. There are a considerable number of models to explain the underlying mechanism, e.g., electrostatic repulsion [35,36,37,38] or a physical barrier of water in an anomalous state [39,40,41,42].
A hint to the answer to this question is the strong dependence of the bioinertness on the packing density of the molecules in the SAMs. Systematic modifications of the OEG-terminals of alkanethiols and assays of protein adsorption by Herrwerth et al. indicated this dependence. In their work, the OEG-SAMs with lower packing density exhibited protein resistance, while OEG-SAMs with higher packing density did not, and a clear threshold was observed between the two categories (4.0 molecules/nm2).
Here, we introduce two examples showing that even for the same molecule, bioinertness shows a dependence on the molecular packing density. One is the report by Jiang et al. [43]. They controlled the molecular packing density of EG4-OH thiol on gold substrates by changing the ratio of water and ethanol dissolving the thiol, and they found that only monolayers with thiol molecules at a density between 55 and 75% of the highest packing density exhibited protein resistance. These findings are in good agreement with the report by Herrwerth et al. [44].
The dependence of the bioinertness of the methoxy-tri(ethylene glycol) terminated (EG3-OMe) alkanethiol SAMs on its packing density was reported by Harder et al. [45]. They reported that a loosely packed Au-supported SAM exhibited strong resistance to the adsorption of fibrinogen, whereas a densely packed Ag-supported SAM allowed adsorption. Grunze et al. attempted to explain the origin of this difference in protein resistance by the difference in the molecular conformation of the OEG chains and the electric field they generated [46,47,48,49]. Later, neutron reflectivity measurements and computer simulation using the grand canonical Monte Carlo method revealed that the interfacial behavior of water is completely different between Au- and Ag-supported EG3-OMe SAMs [49, 50]. That is, the water molecules penetrate into the OEG region of the Au-supported SAM. On the other hand, the Ag-supported SAM does not allow the penetration of water because of its high-molecular packing density. Based on their results, interfacial water was found to be involved in the origin of bioinertness.
Jiang et al. reported on the water-induced interaction between a lysozyme molecule and OEG-SAMs or other bioinert SAMs by using molecular dynamics simulation [39,40,41, 51, 52]. They found that repulsive interaction operated between the lysozyme molecule and the SAMs within a range of 2 nm and that the water molecules responsible for the repulsion have much slower dynamics in terms of the diffusion coefficient. Similar behavior was observed for other types of bioinert SAMs.
The first experimental result showing repulsive force induced by OEG-SAMs was reported by Kim et al. [42]. They employed their original interface force microscopy to measure the force operating between OEG-SAMs directly and found that the repulsive force obeys the Stokes-Einstein law, indicating that the viscosity of the interfacial water is different from that of bulk water.
Later, Hayashi et al. [53] performed a systematic analysis of the force operating between SAMs and found a clear correlation between the strength of the water-induced repulsive force and protein and cell resistance. These results showed for the first time that the interfacial water plays the role of a physical barrier to prevent the approach of proteins and cells to the bioinert SAMs [53,54,55].
As introduced above, it has become clear that the interfacial water is the main player in determining the bioinertness of SAMs. However, the detailed physicochemical properties of the interfacial water in the vicinity of bioinert and bioadhering SAMs have not been revealed yet. In the following chapter, we review the recent progress and prospects in this field.
Interfacial behavior of water near bioinert SAMs
Bioinert SAMs composed of synthetic molecules
As discussed above, understanding the interfacial behavior of molecules and ions is essential for understanding the underlying mechanism. We previously performed surface force analysis to measure the interaction induced by bioinert SAMs as well as analyses of protein adsorption (Fig. 3a) and cell adhesion. In these works, we fabricated identical SAMs on gold substrates and gold-coated silica colloids attached to the cantilever of an atomic force microscope (AFM) (the so-called colloid probe) (Fig. 3b). The measured force–distance curves are presented in (Fig. 3c. An attractive force due to hydrophobic and van der Waals interaction was observed for methyl-terminated SAMs. The hydrophilic sulfonic acid (SA)- and tri(methyl) amine (TMA)-terminated SAMs exhibited only van der Waals attraction. In contrast to the above bioadhering SAMs, bioinert OEG-, MC-, and SB-SAMs exhibited similar water-induced repulsion. The important finding here is that the water-induced repulsive force was always observed for bioinert SAMs, without any exception.

a Protein adsorption onto SAMs measured by a quartz crystal microbalance (QCM). C8 denotes hydrophobic n-octanethiol. b Schematic illustration of surface force measurements using AFM, and c force–distance curves measured for bioadhering and bioinert SAMs
Surface-enhanced infrared absorption spectroscopy (SEIRAS) is a surface-sensitive vibrational analysis technique that can explore hydrogen-bonding states of water in the vicinity of SAMs. We recently found that the OH-stretching band of the water near the SAMs correlates well with the bioinertness of the SAMs [54]. This result supports the findings obtained by surface force analysis. The interaction of water with SAMs modulates the hydrogen-bonding state of the interfacial water and affects the interactions with proteins and cells.
Biomolecule-based SAMs
Biomolecules (e.g., proteins, peptides, polysaccharide, RNA, and DNA) are key players in maintaining homeostasis in living things. Their chain-reaction of molecular recognitions and chemical reactions allow us to maintain our homeostatic balance. These biomolecules possess two complementary functions, i.e., the function of binding their target molecules and the function of rejecting nontargets.
Inspired by the selectivity and specificity of the molecular processes conducted by biomolecules, several works on bioinert SAMs consisting of biomolecules have been reported. White et al. [56] analyzed the population of each amino acid on the surfaces of human proteins (1162 in number) and found that glutamic acid (E) and lysine (K) have the highest and second-highest population, respectively. Based on this finding, they designed a peptide consisting of zwitterionic pairs of anionic and cationic residues: EKEKEKE-PPPP-C (bioinert, spacer, and anchoring groups, respectively) [57]. This SAM exhibited excellent cell resistance. Moreover, this SAM can control cell adhesion by docking a cell-adhering RGD (ligand-binding to integrin) moiety at the end of the peptide, allowing us to endow peptide-based SAMs with both bioinertness and specificity to cells by designing the sequence of amino acids.
Our question is the physical origin of the bioinert property of the peptide-based SAMs. We performed surface force analysis to measure the force operating near peptide-fouled SAMs or SAMs with bioinert terminal groups [ERERER (denoted as ER) and EKEKEK (EK), respectively] ((Fig. 4). Although both peptide surfaces exhibited water-induced repulsion, the bioinert SAMs exhibited long-range repulsion, whereas the fouled SAMs induced only short-range repulsion. These results indicate that the zwitterionic pair of amino acids induces the formation of a physical barrier of water, and the mechanical stability (thickness and rigidity) of a SAM depends on the combination of cationic and anionic amino acids. The details of the adsorption state of the amino acids, surface force, and bioinert properties are described elsewhere [58].

a Chemical structures of the peptides constituting EK and ER SAMs. b Force-distance curves measured on approach by the colloidal probe method. The same peptide SAMs are formed on both the probe and the substrate
Next, we move to the topic of DNA-based SAMs. Maeda et al. discovered that the colloidal stability of gold nanoparticles (denoted hereafter as AuNPs) covered with DNA molecules critically depends on the complementarity of the terminal base pairs [59,60,61]. That is, AuNPs covered with fully complementary DNA (cDNA) aggregate at high ion concentration (0.25 M of NaCl), whereas AuNPs capped with DNA molecules with a single mismatch at the terminal (mDNA) exhibit excellent colloidal stability (monodispersity) at a concentration of 1 M of NaCl. The distinct difference in the colloidal stability depending on the complementarity of just one terminal base pair cannot be explained by the DLVO theory.
(Figure 5 displays force–distance profiles observed for the cDNA-SAMs (filled circles) and mDNA-SAMs (open squares) in phosphate buffer (10 mM) containing NaCl (250 mM). As shown, we observed attraction between the cDNA-SAMs. On the other hand, repulsive force always operated between mDNA-SAMs, which showed good agreement with the colloidal stability of AuNPs covered with these SAMs. The origin of the attraction between cDNA-SAMs is a mixture of van der Waals and hydrophobic attraction. This specific attraction between terminal base pairs is in general referred to as ‘‘blunt-end’’ stacking, which is frequently used to build self-assembled nanostructures of DNA molecules (so-called DNA origami) [62].

a Chemical structures of cDNA and mDNA molecules. b Force–distance curves measured on approach by the colloidal probe method. The same DNA SAMs are formed on both the probe and substrate
A prominent feature here is the extremely long-range repulsion between mDNA-SAMs. The decay length of the repulsion is approximately 3 nm, which is much longer than the Debye length (decay length of electrostatic double layer force) of the solution (approximately 0.5 nm). In our recent paper, we concluded that the repulsion is induced by the interfacial water. The working distances of the water-mediated repulsion induced by the peptide- and DNA-based SAMs are longer than those of SAMs of alkanethiols. Unfortunately, we cannot clearly distinguish the steric repulsion induced by flexible hydrated peptide or DNA chains from the water-mediated repulsion. To answer this question, we require further systematic force measurements for peptide and DNA molecules with different molecular lengths.
To summarize this discussion, both synthetic and biological molecules can modify the states of interfacial water, and the interfacial water plays an important role in protein adsorption and cell adhesion. The flexibility of the molecular design of SAMs, together with their mechanical and chemical stability in various biological assays, expands their potential for further applications in biology. However, the mechanism underlying the formation of a physical barrier of water requires further investigation. In the next section, we introduce several recent signs of progress in the analysis of interfacial water.
Future perspectives and summary
The importance of interfacial water in the responses of biomolecules and cells to artificial materials has been shown. A similar phenomenon is also found for other molecular systems, such as lipid molecules [63, 64]. As is generally accepted, interfacial water has been found to play important roles in fundamental interfacial phenomena such as friction, corrosion, and catalysis [65, 66]. In these phenomena, only one or two hydration layers are considered to be involved. However, we have found that both synthetic molecule- and biomolecule-based SAMs repel proteins and cells via a physical barrier of interfacial water with a thickness of several nm, which is incredibly large. X-ray and neutron diffraction and recent computer simulations suggested the long-range ordering (structural correlation longer than several nm) of water molecules [67,68,69]. Recently, Harada et al. reported the distinct states of water confined in polymer brushes by X-ray absorption and emission spectroscopy. We expect new analytical techniques to reveal the details of the interfacial water that are responsible for biocompatibility in future.
It should not be forgotten that protein molecules (and probably other biomolecules) are covered by a hydration shell with a thickness >1 nm [70, 71]. Therefore, artificial materials “communicate” with biomolecules via their interfacial water molecules, and the response of the biomolecules can be determined. Therefore, we believe that it is time to move on to analyses of the interfacial properties of water at such heterogeneous interfaces to understand the ‘‘real’’ biomolecular processes in living systems.
References
Gupta A, Patel VK, Kant R, Bhattacharya S. Surface modification strategies for fabrication of nano-biodevices: A critical review. Rev Adhes Adhes. 2016;4:166–91.
Tiwari, A Nordin, AN. Advanced biomaterials and biodevices. Wiley; 2014.
Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Mediation of biomaterial-cell interactions by adsorbed proteins: A review. Tiss Eng. 2005;11:1–18.
Onuki Y, Bhardwaj U, Papadimitrakopoulos F, Burgess DJ. A review of the biocompatibility of implantable devices: current challenges to overcome foreign body response. J Diabetes Sci Technol. 2008;2:1003–15.
Teo AJT, Mishra A, Park I, Kim YJ, Park WT, Yoon YJ. Polymeric biomaterials for medical implants and devices. ACS Biomater Sci Eng. 2016;2:454–72.
Li Y, Xiao Y, Liu C. The horizon of materiobiology: A perspective on material-guided cell behaviors and tissue engineering. Chem Rev. 2017;117:4376–421.
Williams, D. Essential Biomaterials Science. Cambridge University Press; 2014.
Israelachvili, J. Intermolecular and Surface Forces. Academic Press; 1992.
Leckband D, Israelachvili J. Intermolecular forces in biology. Q Rev Biophys. 2001;34:105–267.
Vogler EA. Structure and reactivity of water at biomaterial surfaces. Adv Colloid Interface Sci. 1998;74:69.
Ishihara K. Bioinspired phospholipid polymer biomaterials for making high performance artificial organs. Sci Technol Adv Mater. 2000;1:131–8.
Tanaka M, Mochizuki A. Effect of water structure on blood compatibility-thermal analysis of water in poly(meth)acrylate. J Biomed Mater Res A. 2004;68A:684–95.
Tanaka M, Motomura T, Ishii N, Shimura K, Onishi M, Mochizuki A, Hatakeyama T. Cold crystallization of water in hydrated poly(2-methoxyethyl acrylate) (PMEA). Polym Int. 2000;49:1709–13.
Tanaka M, Motomura T, Kawada M, Anzai T, Kasori Y, Shiroya T, Shimura K, Onishi M, Mochizuki A. Blood compatible aspects of poly(2-methoxyethylacrylate) (PMEA)-relationship between protein adsorption and platelet adhesion on PMEA surface. Biomaterials. 2000;21:1471–81.
Tanaka M, Hayashi T, Morita S. The roles of water molecules at the biointerface of medical polymers. Polym J. 2013;45:701–10.
Hayashi T, Tanaka M, Yamamoto S, Shimomura M, Hara M. Direct observation of interaction between proteins and blood-compatible polymer surfaces. Biointerphases. 2007;2:119–25.
Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem Rev. 2005;105:1103–69.
Prime KL, Whitesides GM. Self-assembled organic monolayers-model systems for studying adsorption of proteins at surfaces. Science. 1991;252:1164–7.
Prime KL, Whitesides GM. Adsorption of proteins onto surfaces containing end-attached oligo(ethylene oxide)-a model system using self-assembled monolayers. J Am Chem Soc. 1993;115:10714–21.
Mrksich M, Whitesides GM. Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cells. Annu Rev Biophys Biomol Struct. 1996;25:55–78.
Mrksich M, Dike LE, Tien J, Ingber DE, Whitesides GM. Using microcontact printing to pattern the attachment of mammalian cells to self-assembled monolayers of alkanethiolates on transparent films of gold and silver. Exp Cell Res. 1997;235:305–13.
Mrksich M, Chen CS, Xia Y, Dike LE, Ingber DE, Whitesides GM. Controlling cell attachment on contoured surfaces with self-assembled monolayers of alkanethiolates on gold. Proc Natl Acad Sci USA. 1996;93:10775–8.
Mrksich M. Tailored substrates for studies of attached cell culture. Cell Mol Life Sci. 1998;54:653–62.
Hayashi T, Hara M. Nonfouling self-assembled monolayers: mechanisms underlying protein and cell resistance. Curr Phys Chem. 2011;1:90–8.
Yonzon CR, Jeoung E, Zou S, Schatz GC, Mrksich M, Van Duyne RP. A comparative analysis of localized and propagating surface plasmon resonance sensors: The binding of concanavalin a to a monosaccharide functionalized self-assembled monolayer. J Am Chem Soc. 2004;126:12669–76.
Revell DJ, Knight JR, Blyth DJ, Haines AH, Russell DA. Self-assembled carbohydrate monolayers: Formation and surface selective molecular recognition. Langmuir. 1998;14:4517–24.
Ostuni E, Chapman RG, Liang MN, Meluleni G, Pier G, Ingber DE, Whitesides GM. Self-assembled monolayers that resist the adsorption of proteins and the adhesion of bacterial and mammalian cells. Langmuir. 2001;17:6336–43.
Chen SF, Zheng J, Li LY, Jiang SY. Strong resistance of phosphorylcholine self-assembled monolayers to protein adsorption: Insights into nonfouling properties of zwitterionic materials. J Am Chem Soc. 2005;127:14473–8.
Chen SF, Yu FC, Yu QM, He Y, Jiang SY. Strong resistance of a thin crystalline layer of balanced charged groups to protein adsorption. Langmuir. 2006;22:8186–91.
Jeon SI, Lee JH, Andrade JD, Degennes PG. Protein surface interactions in the presence of polyethylene oxide .1. Simplified theory. J Colloid Interf Sci. 1991;142:149–58.
Kodama C, Hayashi T, Nozoye H. Decomposition of alkanethiols adsorbed on Au (111) at low temperature. Appl Surf Sci. 2001;169–70:264–7.
Hayashi T, Morikawa Y, Nozoye H. Adsorption state of dimethyl disulfide on Au(111): Evidence for adsorption as thiolate at the bridge site. J Chem Phys. 2001;114:7615–21.
Hayashi T, Kodama C, Nozoye H. Structural evolution of dibutyldisulfide adsorbed on Au(111). Appl Surf Sci. 2001;169-170:100–3.
Hayashi T, Fricke A, Katsura K, Kodama C, Nozoye H. Adsorption state of diethyldisulfide on Au(111) studied with a combined system of HREELS and STM. Surf Sci. 1999;427-428:393–7.
Chan YHM, Schweiss R, Werner C, Grunze M. Electrokinetic characterization of oligo- and poly(ethylene glycol)-terminated self-assembled monolayers on gold and glass surfaces. Langmuir. 2003;19:7380–5.
Dicke C, Hahner G. Interaction between a hydrophobic probe and tri(ethylene glycol)-containing self-assembled monolayers on gold studied with force spectroscopy in aqueous electrolyte solution. J Phys Chem B. 2002;106:4450–6.
Feldman K, Hahner G, Spencer ND, Harder P, Grunze M. Probing resistance to protein adsorption of oligo(ethylene glycol)-terminated self-assembled monolayers by scanning force microscopy. J Am Chem Soc. 1999;121:10134–41.
Kreuzer HJ, Wang RL, Grunze M. Hydroxide ion adsorption on self-assembled monolayers. J Am Chem Soc. 2003;125:8384–9.
He Y, Chang Y, Hower JC, Zheng J, Chen SF, Jiang S. Origin of repulsive force and structure/dynamics of interfacial water in OEG-protein interactions: a molecular simulation study. Phys Chem Chem Phys. 2008;10:5539–44.
Zheng J, Li LY, Chen SF, Jiang SY. Molecular simulation study of water interactions with oligo (ethylene glycol)-terminated alkanethiol self-assembled monolayers. Langmuir. 2004;20:8931–8.
Zheng J, Li LY, Tsao HK, Sheng YJ, Chen SF, Jiang SY. Strong repulsive forces between protein and oligo (ethylene glycol) self-assembled monolayers: A molecular simulation study. Biophys J. 2005;89:158–66.
Kim HI, Kushmerick JG, Houston JE, Bunker BC. Viscous “interphase” water adjacent to oligo(ethylene glycol)-terminated monolayers. Langmuir. 2003;19:9271–5.
Li LY, Chen SF, Zheng J, Ratner BD, Jiang SY. Protein adsorption on oligo(ethylene glycol)-terminated alkanethiolate self-assembled monolayers: The molecular basis for nonfouling behavior. J Phys Chem B. 2005;109:2934–41.
Herrwerth S, Eck W, Reinhardt S, Grunze M. Factors that determine the protein resistance of oligoether self-assembled monolayers--internal hydrophilicity, terminal hydrophilicity, and lateral packing density. J Am Chem Soc. 2003;125:9359–66.
Harder P, Grunze M, Dahint R, Whitesides GM, Laibinis PE. Molecular conformation in oligo(ethylene glycol)-terminated self-assembled monolayers on gold and silver surfaces determines their ability to resist protein adsorption. J Phys Chem B. 1998;102:426–36.
Pertsin AJ, Grunze M. Computer simulation of water near the surface of oligo(ethylene glycol)-terminated alkanethiol self-assembled monolayers. Langmuir. 2000;16:8829–41.
Pertsin AJ, Grunze M, Garbuzova IA. Low-energy configurations of methoxy triethylene glycol terminated alkanethiol self-assembled monolayers and their relevance to protein adsorption. J Phys Chem B. 1998;102:4918–26.
Pertsin AJ, Grunze M, Kreuzer HJ, Wang RLC. The effect of electrostatic fields on an oligo(ethylene glycol) terminated alkanethiol self-assembled monolayer. Phys Chem Chem Phys. 2000;2:1729–33.
Pertsin AJ, Hayashi T, Grunze M. Grand canonical Monte Carlo Simulations of the hydration interaction between oligo(ethylene glycol)-terminated alkanethiol self-assembled monolayers. J Phys Chem B. 2002;106:12274–81.
Schwendel D, Hayashi T, Dahint R, Pertsin A, Grunze M, Steitz R, Schreiber F. Interaction of water with self-assembled monolayers: Neutron reflectivity measurements of the water density in the interface region. Langmuir. 2003;19:2284–93.
He Y, Hower J, Chen SF, Bernards MT, Chang Y, Jiang SY. Molecular simulation studies of protein interactions with zwitterionic phosphorylcholine self-assembled monolayers in the presence of water. Langmuir. 2008;24:10358–64.
Hower JC, He Y, Jiang SY. A molecular simulation study of methylated and hydroxyl sugar-based self-assembled monolayers: Surface hydration and resistance to protein adsorption. J Chem Phys. 2008;129:215101.
Hayashi T, Tanaka Y, Koide Y, Tanaka M, Hara M. Mechanism underlying bioinertness of self-assembled monolayers of oligo(ethyleneglycol)-terminated alkanethiols on gold: protein adsorption, platelet adhesion, and surface forces. Phys Chem Chem Phys: PCCP. 2012;14:10196–206.
Sekine T, Asatyas S, Sato C, Morita S, Tanaka M, Hayashi T. Surface force and vibrational spectroscopic analyses of interfacial water molecules in the vicinity of methoxy-tri(ethylene glycol)-terminated monolayers: mechanisms underlying the effect of lateral packing density on bioinertness. J Biomater Sci Polym Ed. 2017;28:1231–43.
Sekine T, Tanaka Y, Sato C, Tanaka M, Hayashi T. Evaluation of factors to determine platelet compatibility by using self-assembled monolayers with a chemical gradient. Langmuir. 2015;31:7100–5.
White AD, Nowinski AK, Huang W, Keefe AJ, Sun F, Jiang S. Decoding nonspecific interactions from nature. Chem Sci. 2012;3:3488–94.
Nowinski AK, Sun F, White AD, Keefe AJ, Jiang S. Sequence, structure, and function of peptide self-assembled monolayers. J Am Chem Soc. 2012;134:6000–5.
Chang, R & Hayashi, T. (in preparation).
Isoda K, Kanayama N, Fujita M, Takarada T, Maeda M. DNA terminal mismatch-induced stabilization of polymer micelles from RAFT-generated poly(N-isopropylacrylamide)-DNA block copolymers. Chem Asian J. 2013;8:3079–84.
Sato K, Hosokawa K, Maeda M. Rapid aggregation of gold nanoparticles induced by non-cross-linking DNA hybridization. J Am Chem Soc. 2003;125:8102–3.
Wang G, Akiyama Y, Shiraishi S, Kanayama N, Takarada T, Maeda M. Cross-linking versus non-cross-linking aggregation of gold nanoparticles induced by DNA hybridization: A comparison of the rapidity of solution color change. Bioconjug Chem. 2017;28:270–7.
Rothemund PWK. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297.
Jackman JA, Spackova B, Linardy E, Kim MC, Yoon BK, Homola J, Cho NJ. Nanoplasmonic ruler to measure lipid vesicle deformation. Chem Commun (Camb). 2016;52:76–9.
Jackman JA, Zan GH, Zhao Z, Cho NJ. Contribution of the hydration force to vesicle adhesion on titanium oxide. Langmuir. 2014;30:5368–72.
Sakuma H, Otsuki K, Kurihara K. Viscosity and lubricity of aqueous NaCl solution confined between mica surfaces studied by shear resonance measurement. Phys Rev Lett. 2006;96:046104.
Ledezma-Yanez, I, Wallace, WDZ, Sebastian-Pascual, P, Climent, V, Feliu, JM, Koper, MTM. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat Energy. 2017;2:17031.
Pluharova E, Laage D, Jungwirth P. Size and origins of long-range orientational water correlations in dilute aqueous salt solutions. J Phys Chem Lett. 2017;8:2031–5.
Tokushima T, Harada Y, Takahashi O, Senba Y, Ohashi H, Pettersson LGM, Nilsson A, Shin S. High resolution X-ray emission spectroscopy of liquid water: The observation of two structural motifs. Chem Phys Lett. 2008;460:387–400.
Wikfeldt KT, Leetmaa M, Ljungberg MP, Nilsson A, Pettersson LG. On the range of water structure models compatible with X-ray and neutron diffraction data. J Phys Chem B. 2009;113:6246–55.
Petsev DN, Vekilov PG. Evidence for non-DLVO hydration interactions in solutions of the protein apoferritin. Phys Rev Lett. 2000;84:1339–42.
Svergun DI, Richard S, Koch MHJ, Sayers Z, Kuprin S, Zaccai G. Protein hydration in solution: Experimental observation by x-ray and neutron scattering. Proc Natl Acad Sci USA. 1998;95:2267–72.
Acknowledgements
We acknowledge members of Prof. Teiji Tsuruta’s forum for many discussions on the mechanisms of biocompatibility over the years. A part of this work was performed under the Cooperative Research Program of “NJRC Mater. & Dev.” This work is also supported by JST-PRESTO and MEXT KAKENHI (17K20095 and 15KK0184)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Chang, R., Asatyas, S., Lkhamsuren, G. et al. Water near bioinert self-assembled monolayers. Polym J 50, 563–571 (2018). https://doi.org/10.1038/s41428-018-0075-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0075-1
