
Abstract
Self-healing materials have attracted attention due to their ability to regain their structure and function after damage. In recent years, significant progress has been made in achieving various functions through supramolecular chemistry. This review describes an overview of the strategies used to prepare self-healing and self-restoring materials utilizing reversible and movable crosslinks. Reversible crosslinks, consisting of noncovalent bonds, can reversibly undergo repeated cleavage and reformation. Therefore, self-healing can be achieved by effectively regenerating reversible crosslinks between polymeric chains. Reversible crosslinks exploit many kinds of dynamic covalent bonds and noncovalent bonds, such as hydrogen bonds, metal coordination bonds, ionic interactions, π–π stacking, van der Waals forces, and hydrophobic interactions. Movable crosslinks exhibit self-restoring properties. Self-restoring materials can regain their original shape and mechanical properties after a cycle of loading and unloading external stress. Movable crosslinks consist of polymer chains that penetrate macrocyclic units and have self-restoring properties due to their sliding motion along the polymeric chains. In addition, multiple reversible cross-links produce synergistic effects to simultaneously achieve high toughness and effective self-healing. We believe that self-healing and self-restoring materials will play a substantial role in realizing a sustainable society.
Similar content being viewed by others
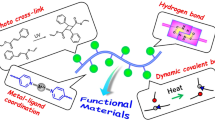

Introduction
It is desirable for polymeric materials, which are mainly derived from petroleum, to have a low environmental impact. To reduce the amount of waste by extending the durable life of the materials, self-healing materials have attracted considerable attention due to their ability to regain their structure and function after damage. To date, several approaches have been applied to successfully fabricate self-healing materials. The simplest approach is repairing fractures by softening the surface with external factors such as heat and light1,2. Much external energy is necessary to heal polymers with high molecular weights due to their low diffusion rate. Another approach is embedding microcapsules/microvasculature-containing repairing agents such as monomers and rehardening catalysts inside the materials3,4. When the materials are damaged, the microcapsules/microvasculature release the monomer to trigger polymerization, which repairs the fracture gap. Even though this method can be used to repair large-volume damage, the disadvantage is that a limited number of repairs can be performed in the same place. Cross-linking reactions by light irradiation of the wound has also been reported5.
In recent years, polymeric materials with reversible crosslinks based on supramolecular chemistry have been developed that exhibit various functions, such as stimuli response6,7,8, toughness9,10, shape memory11,12,13, and self-healing14,15,16,17,18,19,20. Reversible crosslinks consisting of noncovalent bonds can reversibly undergo repeated cleavage and reformation (Fig. 1). Therefore, self-healing can be achieved by the effective regeneration of reversible crosslinks between polymeric chains under mild conditions. This self-healing function helps extend the durable life of materials because significant structural destruction does not occur and repair is enabled. In addition to self-healing, reversible cross-linked materials are typically tough due to the energy dissipation of the sacrificial bonds.

Self-healing and self-restoring materials were constructed by “much amounts of weak bonds”, “synergic effect of strong & weak bonds”, “stress dispersion along nanofiber”, “multiphase of hard and soft domain”, “switching of bond formation”, “switching of ring position”, “restoring assisted by re-association”, and “healing assisted by sliding motion”.
In addition, movable crosslinks show self-restoring properties. “Self-restoration” means that tough elastic materials regain their original shape and mechanical properties after widespread or pinpointed deformation. The driving force of self-restoration is entropic elasticity. General elastic materials are typically not tough because stress concentrates along the fixed covalent cross-links. Movable crosslinks consist of polymer chains penetrating macrocyclic units. Movable cross-linked materials are typically tough due to energy dissipation based on the sliding motion of cross-links along the polymeric chains. The sliding motion is expected to prevent microcracks from cleaving covalent bonds during deformation processes such as stretching. After unloading, movable crosslinks return to their original locations due to their entropic elasticity, thus restoring their original mechanical properties. Similar properties have also been reported in gels with very small amounts of chemical cross-linking points but densely fixed entanglements21,22. Sliding off the fixed entanglements gives high toughness and self-restoring properties, such as movable cross-links.
This review discusses typical strategies for the development of self-healing and self-restoring materials utilizing reversible and movable crosslinks. In addition, materials with multiple reversible crosslinks will be discussed.
Materials with reversible crosslinks
Reversible bonds are key components in the development of self-healing materials. This section focuses on typical self-healing materials with reversible crosslinks such as hydrogen bonds, metal coordination bonds, ionic interactions, host–guest interactions, π–π stacking, dynamic covalent bonding, and van der Waals forces.
Hydrogen bonds
Creating crosslinking polymeric chains via hydrogen bonds is one of the simplest and most effective methods for fabricating self-healing materials. Hydrogen atoms strongly interact with unshared electron pairs on electronegative atoms, such as fluorine, oxygen, and nitrogen. In 1989, supramolecular polymers based on hydrogen bonds were first reported by Kato and Frechet23. In 1997, Meijer et al. prepared a trifurcated polymer terminally modified with 2-ureido-4-pyrimidone, which forms dimers by the strong interactions of four complementary hydrogen bonds (association constant, Ka > 106 M−1) (Table 1)24. The dimers between the terminals of each chain thermodynamically formed a polymeric network. These thermoplastic behaviors can be used in self-healing coating films prepared by hot melting. In 2008, Leibler et al. reported hydrogen-bonded supramolecular materials derived from fatty acids and urea25. Bifunctional and trifunctional fatty acids were condensed with diethylenetriamine and then reacted with urea to produce oligomers with hydrogen bonds. When dodecane was added as a plasticizer, the materials showed rubber-like behavior and self-healing properties at room temperature. To date, most reported self-healing materials have been soft elastic materials. In 2018, Aida et al. reported a hard self-healing material composed of thiourea and ethylene glycol26. They employed relatively low-molecular-weight polymers to achieve excellent segmental motion as well as less ordered hydrogen bonds to simultaneously achieve self-healing properties and a high Young’s modulus (>1 GPa) based on effective bond exchanges of numerous hydrogen bonds.
Metal coordination complexes
One characteristic of metal coordination bonds is that they have a high degree of freedom due to the variety of combinations of metal species and ligands. Metal coordination bonds can be designed for a wide range of binding energies from 100 kJ mol−1 (comparable to hydrogen bonds: 5–200 kJ mol−1) to 300 kJ mol−1 (comparable to covalent bonds (C–C): 300–450 kJ mol−1)17. Lehn et al. reported supramolecular polymers with Zn2+ or Ni2+ coordination to telechelic polydimethylsiloxanes modified with acyl hydrazone-pyridine or acyl hydrazone-quinoline (Table 2)27. The two different supramolecular polymeric films were overlaid at 50 °C for 24 h, yielding metal–ligand exchange-based random copolymer films.
Promotion of self-healing by external stimuli
The self-healing conditions (time and temperature) depend on dissociation and reassociation of the metal coordination complex. To achieve self-healing in a short time and under mild conditions, the promotion of bond exchange by external stimuli has been studied. Rowan et al. reported a supramolecular polymer with Zn2+ coordination to an ethylene–butylene copolymer modified with 2,6-bis(1′-methylbenzimidazolyl)pyridine at both ends28. Upon UV light irradiation, the metal coordination bonds generate heat due to excitation. This accelerates the association and dissociation of the metal coordination bonds, promoting self-healing.
Control of viscoelastic properties
The viscoelastic properties can be controlled by switching between association and dissociation of the complexes, enabling self-healing. Dissociation increases the fluidity of the materials, leading to wound filling. Then, reassociation completes the repair. Waite et al. reported a pH-responsive sol–gel transition material that takes advantage of the pH-dependent change in the coordination number of catechol to Fe3+ 29. The materials crosslinked by a mixture of Fe3+ and terminal catechol-modified four-chain polyethylene glycol are fluids under acidic conditions but form hydrogels at high pH (>8).
Switching kinetics of metal cages
The exchange kinetics of metal coordination complexes in metal–organic cage (MOC) structures depend on the material topology. Then, self-healing can be controlled by introducing a unit whose binding angle changes upon exposure to external stimuli. Johnson et al. reported an organogel in which the MOC structure of the small Pd3L6 ring was connected to polyethylene glycol30. Small Pd3L6 rings showed re-adhesion via fast ligand exchange. The MOC structures changed from a small Pd3L6 ring to Pd24L48 rhombicuboctahedra upon UV irradiation due to a change in the bond angle. Pd24L48 rhombicuboctahedra did not show re-adhesion due to slow ligand exchange. The MOC structures reverse from the Pd24L48 rhombicuboctahedra to the small Pd3L6 ring upon visible light irradiation. Thus, control of reattachment properties was realized by switching the kinetics of ligand exchange.
Ionic interactions
Ionic pairs in polymers form crosslinks based on electrostatic forces and enable self-healing by reforming aggregates. The strength and kinetics of these associations can be controlled by their charges and the size of the aggregation.
Association with dispersed nanosheets
Aida et al. reported a supramolecular hydrogel formed by mixing negatively charged poly(sodium acrylate)-wrapped clay nanosheets with positively charged guanidinium ion end-modified dendrimers (Table 3)31. The supramolecular network structure of the material collapsed and formed a liquid upon application of oscillatory force but recovered to a solid state within a few seconds after the oscillatory force was stopped. The material exhibited thixotropic properties due to the repeated collapse and reformation of the supramolecular network structure. It also showed reattachment properties.
Random distribution of ionic pairs
Gong et al. reported strong hydrogels (polyampholyte hydrogels) using linear amphoteric polyelectrolyte polymers with ionic bonds as crosslinks32. In the material, strong and weak associations were formed depending on the number of ion pairs forming the aggregate: the former as permanent crosslinks and the latter as sacrificial bonds. The presence of these crosslinks with different strengths resulted in the formation of strong, elastic, and tough hydrogels. Furthermore, these hydrogels have been reported to reattach and self-repair by recombination of bonds when placed in water.
Large numbers of weak bonds
In general, self-healing of materials with a high Young’s modulus is difficult because of low segmental motion. To overcome this drawback, Zuo et al. fabricated a self-healing material by introducing many relatively weak bonds and taking advantage of the rapid decrease in Young’s modulus upon heating33. Polydimethylsiloxane cross-linked with Zn-carboxylate interactions was rigid (Young’s modulus: E = 800 MPa) at room temperature. The cross-linked polydimethylsiloxane material was rigid at room temperature (E = 800 MPa) but rapidly became flexible (E = 0.9 MPa) upon heating to 80 °C, showing self-healing properties. Furthermore, the material melted when heated to 120 °C, indicating that it could be used in 3D printing.
Promoting bond exchange
The promotion of bond exchange by external stimuli facilitates self-healing in a short time and under mild conditions. Miwa et al. reported a material in which the self-healing properties were controlled by CO2 gas; this material was synthesized by neutralizing polydimethylsiloxane modified with amide groups and carboxyl groups with sodium hydrate34. Microphase separation of polar ion-associated clusters and nonpolar main chains was observed, resulting in reversible crosslinks. CO2 gas accelerated self-healing by enhancing the mobility of ionic aggregates, although the self-healing ratio in room-temperature air was limited.
Other interactions
π–π stacking
Attractive interactions between π electrons on aromatic moieties result in π–π stacking interactions. In particular, self-healing materials based on donor–acceptor π–π stacking between pyrene units (π electron-rich) and naphthalene-diimide units (π electron poor) have been developed35,36. Blending the naphthalene-diimide-based polyimide and terminally pyrene-modified polymer results in homogeneous films because π–π stacking interactions prevent phase separation. The obtained films were cut into two pieces and then rejoined. Upon exposure to high temperatures, the contact surface disappeared, and the healed film regained its initial Young’s modulus.
Dynamic covalent bonds
General covalent bonds are irreversible, and no bond exchange occurs. However, dynamic covalent bonds exist in a thermodynamic equilibrium state of forming and breaking under specific conditions (temperature, light, pH, and chemical stimuli). Many self-healing materials based on dynamic covalent bonds have been reported, such as Diels-Alder reaction products37 and materials containing hydrazine bonds38, disulfide bonds39, and thiocarbonate bonds40. A vitrimer is a chemically cross-linked network that undergoes a bond exchange reaction upon heating. Even for ester bonds, it is possible to create vitrimers by activating the exchange reaction with a catalyst41.
van der Waals forces
As mentioned above, self-healing materials have been achieved mainly by using functional substituents and additives. Recently, self-healing materials with van der Waals interactions between the polymer chains have also been reported. Random copolymers with a methyl methacrylate/n-butyl acrylate molar ratio of 45/55 to 50/50 have been shown to exhibit self-healing properties42. The van der Waals forces between the segments resulted in a lock-and-key conformation, and this interaction was responsible for the self-healing properties. The tensile test showed that they were almost completely repaired after 14 h. Self-healing materials using crystalline nanodomains of random copolymers as reversible interaction sites have also been reported; copolymers of ethylene and anisyl propylene derivatives exhibited self-healing properties based on the crystalline nanodomains of the ethylene segment43
Host–guest interactions
The host–guest interaction is an interaction in which a macrocyclic molecule (host molecule) incorporates a suitably sized molecule (guest molecule) to form an inclusion complex. Crown ethers44, cucurbit[n]urils (CB[n]s)45, calix[n]arenes46, pillar[n]arenes47, and cyclodextrins (CDs)48 have been employed as host molecules. These inclusion complexes have various association constants depending on the size, shape, and charge state of the guest molecule. Material design based on molecular recognition is a feature of host–guest interactions. Molecular recognition realizes macroscopic self-assembly49 and sol–gel transitions50, inspiring the creation of self-healing materials based on host-guest interactions. This subsection focuses on three kinds of designs for self-healing materials using host–guest interactions.
Mixing between host and guest polymer
In this approach, host molecules or guest molecules are grafted onto the polymer. By mixing the obtained host polymers and guest polymers, inclusion complexes of the guest and host residues of the polymer side chains are formed. As a result, supramolecular materials with reversible crosslinks based on inclusion complexes are obtained. By employing guest molecules that change their association constants in response to external stimuli, stimulus-responsive materials can be fabricated. Redox-responsive hydrogels have been obtained by mixing βCD-modified poly(acrylic acid) and ferrocene (Fc)-modified poly(acrylic acid) (Table 4)51. The high association constants between βCD and Fc (Ka = 1.1 × 103 M−1) resulted in inclusion complexes with reversible crosslinks. The obtained hydrogel was cut into two pieces and then rejoined. After standing for 24 h, the contact surface disappeared, and the gel showed 84% of its initial strength. The addition of an oxidant (sodium hypochlorite) converted the hydrogel into a solution. The oxidized ferrocenium (Fc+) cation exhibited a low affinity for βCD, leading to dissociation of the inclusion complexes. Then, the addition of a reductant (glutathione) reduced Fc+ to Fc and reformed the inclusion complex, yielding a hydrogel again.
In contrast to CD, which disfavors the cationic guest, crown ethers form an inclusion complex with a cationic guest. Unshared electron pairing of oxygen atoms in the crown ether stabilizes the cationic guest. pH-responsive organogels were prepared by mixing dibenzo[24]crown-8 (DB24C8)-modified poly(methyl methacrylate) and bisammonium crosslinkers52. The organogels showed gel-sol and sol-gel transitions upon the addition of a base (triethylamine) and an acid (trifluoroacetic acid). The organogels also showed self-healing properties, and the cracks on the sample disappeared within 4 min.
Pillar[n]arenes act as host molecules for cationic or electron-deficient guest molecules. Redox-responsive organogels were prepared by mixing pillar[6]arene-modified poly(methyl acrylate) and Fc+-modified polystyrene53. The organogels showed gel-sol and sol-gel transitions upon the addition of a reductant (hydrazine) and an oxidant {silver tetrakis[3,5-bis(trifluoromethyl)phenyl]borate}. Since pillar[n]arenes are relatively new macrocyclic molecules, there have been few reports of self-healing materials that use them. However, there are potential host-guest materials that use the unique properties of pillar[n]arenes, which include self-assembly and planar chirality.
When supramolecular elastomers are prepared by drying a solution of host and guest polymers, not only the chemical structure but also processing is important. The elastomer obtained by drying an N-methyl pyrrolidone solution of peracetylated CD-modified poly(ethyl acrylate) and adamantane-modified poly(ethyl acrylate) after ball milling treatment showed higher toughness than the elastomer obtained by normal stirring and casting methods54. Scratches made with a hard brush on the surface of the elastomer disappeared quickly upon heating. The high stress of ball milling detangled the polymer chains, resulting in chains with high mobility. This high mobility facilitated the reformation of inclusion complexes to improve the mechanical properties and self-healing properties.
2-Type host–guest interactions
Another family of macrocyclic molecules is CB[n]s. While CB[5], CB[6], and CB[7] form inclusion complexes with single guest molecules, CB[8] simultaneously captures two guest molecules and even forms 1:1:1 heteroternary complexes with dissimilar guest molecules. Supramolecular materials can be obtained from native CB[8]s and guest polymers and are free from complicated modification of macrocyclic molecules. Mixing CB[8], viologen-modified poly(2-hydroxyethyl acrylate) and naphthalene-modified poly(N,N-dimethyl acrylamide) yielded supramolecular hydrogels55. The thermal reversibility of crosslinks in hydrogels was demonstrated, as the hydrogels showed gel–sol and sol–gel transitions upon heating and cooling. CB[n]s can form inclusions with two identical guest molecules with 1:2 homoternary complexes. After the formation of inclusion complexes of CB[8] and two guest monomers, copolymerization between the inclusion complex and acrylamide yields supramolecular networks56. The obtained hydrogels showed high fracture strain (over 10,000%) and complete healing at room temperature.
Polymerization of inclusion complexes
To achieve good self-healing properties, it is necessary to efficiently form inclusion complexes between the host and guest units in the material. Mixing host and guest polymers is considered to be disadvantageous because inclusion complexes form imperfectly. As the viscosity increases with the formation of the inclusion complex, molecular mobility is impaired before all the host and guest moieties form inclusion complexes. To solve this problem, another method of polymerizing inclusion complexes of host and guest monomers was developed. Insoluble guest monomers can be solubilized via complexation with host monomers. The obtained polymer chain bears both host units and guest units.
After the formation of inclusion complexes of βCD monomers and adamantane monomers, copolymerization between the inclusion complex and acrylamide yielded supramolecular hydrogels (Table 5)57. The obtained hydrogel was cut into two pieces and then rejoined. After standing for 24 h, the contact surface disappeared, and the hydrogel possessed 99% of its initial strength.
The polymerization of inclusion complexes successfully improved the healing efficiencies. However, supramolecular materials with poly(acrylamide) main chains showed no self-healing properties under dry conditions. Rather, humid conditions were still needed to achieve self-healing. To overcome this, poly(methoxy triethyleneglycol acrylate) (polyTEGA) was employed as the main chain polymer with a lower glass transition temperature (Tg of polyTEGA = −50 °C, Tg of polyacrylamide = 165 °C)58. The self-healing xerogels with polyTEGA main chains regained 60% of their initial strength at 100 °C after standing for 24 h. In addition, a high association constant of the guest unit with the βCD unit was found to result in high healing ratios.
By using guest molecules whose absorption wavelengths change between the inclusion and free states, it is possible to create materials that change color in response to stimuli. Phenolphthalein is purple in basic aqueous solution, but the inclusion complex that forms between βCD and phenolphthalein is colorless59. Thus, stimulus-responsive hydrogels with βCD and phenolphthalein inclusion complexes as crosslinks were prepared. The obtained hydrogel was still colorless upon immersion in KH2PO4/NaOH basic buffer (pH = 8). This colorless gel turned purple upon heating to 87 °C because the inclusion complex was dissociated. The gel became colorless again upon cooling due to reassociation. The hydrogel also underwent a color change when competing guest molecules or currents (Joule heating) were applied. Furthermore, self-healing xerogels were obtained by removing the solvent.
Another preparation method of dry self-healing materials is bulk polymerization without a solvent. In this method, the inclusion complex between the host monomer and guest monomer needs to be dissolved in the liquid main chain monomer. However, CD monomers with a large number of hydroxyl groups cannot be dissolved in most general hydrophobic main chain monomers. Then, self-healing elastomers were fabricated using hydrophobic CD monomers with all hydroxyl groups acetylated60. The bulk copolymerization of peracetylated CD monomer, 2-ethyladamantyl acrylate, and ethyl acrylate was carried out. The obtained elastomer was cut into two pieces and then rejoined. The contact surface disappeared, and the gel showed 95% of its initial strength within 4 h at 80 °C. In addition, this elastomer can be recycled. The toluene-swollen sample was ground with a ball mill to form a slurry, and the toluene was removed to obtain a recycled sample with the original properties.
Materials with movable crosslinks
Movable crosslinks consist of a mechanically interlocked architecture in which polymers penetrate macrocyclic molecules. The movable crosslinked materials are tougher upon stress dispersion, which occurs due to the sliding motion of macrocyclic molecules along polymers. Self-restoring properties are also expected because the movable crosslinks return to their original position by entropic elasticity after unloading. This section focuses on a typical approach to obtain movable cross-linked materials.
Polyrotaxanes as starting materials
Polyrotaxanes are macromolecules composed of macrocyclic molecules threaded by axle polymers and that have bulky stoppers at both ends. In 1990, αCDs were threaded onto a PEG chain to yield poly-pseudo-rotaxanes for the first time61. In 1992, a polyrotaxane was prepared by the end-capping reaction of complexes between αCDs and PEG bisamines with 2,4-dinitrofluorobenzene62.
Movable crosslinked materials can be fabricated by using polyrotaxanes as cross-linkers. Ito et al. prepared a “topological gel” by a crosslinking reaction between hydroxyl groups on two αCDs in different polyrotaxanes using cyanuric chloride (Table 6)63. In contrast to covalent crosslinks, which cause stress concentration, movable cross-links act like pulleys to equalize the tension of the polymer chain. This stress dispersion restricts the breakdown of the polymer chains to realize tough materials. This “pulley effect” has been industrially applied to scratch-healing coatings64. The movable crosslinks prevent cracks, and the coating can return to its original shape because of its elasticity. Movable crosslinked materials with polyrotaxanes of pillar[5]arene were also reported by Ogoshi et al.65.
The sliding motion of the movable crosslinks can also be utilized to promote the reformation of reversible crosslinks and improve the self-healing properties. Self-healing materials were prepared by copolymerization between acrylamide and 4-vinylphenylboronic acid in the presence of polyrotaxanes with hydroxypropyl-modified αCDs (Table 7)66. The obtained hydrogel contained dynamic covalent bonds between boronic acid and the diol of αCDs in polyrotaxanes. These hydrogels showed a higher healing ratio and a shorter healing time (~100%, 15 min) than the reference gel with dynamic covalent bonds involving the diol of linear polysaccharides (~20%, 60 min). Efficient healing was realized by movable crosslinks on the polyrotaxanes. Self-healing hydrogels consisting of αCDs in polyrotaxanes crosslinked by an inclusion complex between βCD and adamantane have also been reported67.
Formation of rotaxane/polyrotaxane structures on side chains
Movable cross-linked materials with rotaxanes/polyrotaxanes in the side chains are advantageous because the rotaxanes/polyrotaxanes and the main chain polymer can be designed. Takada et al. prepared a rotaxane crosslinker with methacrylate groups modified on the poly(δ-valerolactone) axle and a crown ether ring in [2]rotaxane, and then copolymerization with the main chain monomer (butyl acrylate or 2-ethylhexyl acrylate) was carried out68. The obtained movable crosslinked elastomer showed higher fracture strain and stress than the reference sample with covalent crosslinks (Table 8).
γCDs can simultaneously capture two PEG chains to form double-stranded inclusion complexes69. This property enables the design of movable crosslinked materials free from complicated modification of γCD. Guest macromonomers were synthesized by PEG terminal modification with a methacrylate group and bulky stopper70. Movable crosslinked microgels were prepared by precipitation copolymerization of the main chain monomer (N-isopropyl methacrylate) and the inclusion complex between the guest macromonomers and γCDs. γCDs in the obtained microgel formed aggregate-based hydrogen bonds at pH = 7 and disaggregated by weakening the hydrogen bonds at pH = 10. As a result, the swelling rate and thus the particle size of the microgels in water changed with pH. In addition, the microgels were thermoresponsive based on the lower critical solution temperature of the poly(N-isopropyl methacrylate) main chain.
Formation of rotaxane/polyrotaxane structures on the main chain
This approach can be further divided into two main methods. The first method is polycondensation that occurs after the formation of a pseudorotaxane/rotaxane structure. Photoresponsive movable crosslinked materials were fabricated using pseudo[2]rotaxane consisting of lysine-modified αCD and diamine-modified azobenzene (Table 9)71. Polycondensation with succinimidyl-modified PEG yielded movable crosslinked hydrogels. Azobenzene (Azo) showed photoinduced isomerization between trans-Azo and cis-Azo. trans-Azo possessed a high affinity for αCD, but cis-Azo possessed low affinity. The dried xerogels quickly bent toward the light source upon UV irradiation (wavelength: 365 nm−1) because of the isomerization to cis-Azo and unthreading from the αCD cavity, leading to the sliding of CD units along the PEG chain.
The self-restoration of movable crosslinked materials can be improved by introducing a high-affinity guest into the axle of [2]rotaxane. Yan et al. fabricated movable crosslinked materials using a [2]rotaxane dibenzo-24-crown-8 ring and dibenzylammonium salt72. The ring and axle were modified with two vinyl groups, and then a thiol-ene click reaction with 3,6-dioxa-1,8-octanediol yielded movable crosslinked materials. The macrocyclic units favor cationic guest units due to their high affinity. Upon application of stress to the materials, the macrocyclic units were pulled away from the cationic guest units. This behavior dissipated the stress to realize high toughness. After the stress was unloaded, host–guest interactions facilitated reassociation, leading to fast deformation recovery.
The second method is the formation of movable crosslinks through copolymerization between the macrocyclic monomer and main chain monomer. Movable crosslinked elastomers were prepared by bulk copolymerization between hydrophobic CD monomers and alkyl acrylate73. The formation of movable crosslinks depends on the cavity diameter of CD and the size of the main chain monomer. In the case of ethyl acrylate (EA) and peracetylated γCD monomers, poly(EA) main chains threaded γCD units and acted as movable crosslinks. In contrast, copolymerization using butyl acrylate and peracetylated βCD monomers yielded only a small number of movable crosslinks. The formation of movable crosslinks depends on the cavity diameter of CD and the size of the main chain monomer. The obtained elastomer showed higher toughness than the poly(EA) with covalent crosslinks. As CD monomers are soluble in liquid acrylamide monomers (N,N-dimethyl acrylamide, 4-acryloylmorpholine, and N,N-dimethylaminopropyl acrylamide), hydrophilic movable crosslinked materials were also prepared in the same manner74. The preparation of polyrotaxanes/poly-pseudo-rotaxanes consisting of poly(alkyl acrylate)s or poly(acrylamide) derivatives as the axles have not been achieved by mixing CD and these polymers in water. Then, these preparation methods enabled polyrotaxanes/poly-pseudo-rotaxanes that cannot be obtained by conventional methods. Using similar preparation methods, movable crosslinked adhesives with high peel strength were also prepared75.
Materials with multiple crosslinks
Recently, material scientists have attempted to develop self-healing materials with multiple reversible bonds. This section describes typical strategies for fabricating high-performance materials using multiple reversible bonds.
The reformation of reversible bonds for self-healing requires mobile polymer chains. Generally, it is difficult to achieve both a high healing ratio and stiffness without external energy. To overcome this challenge, Guan et al. designed self-healing materials with hard-soft multiphase (Table 10)76. Hydrogen bonding brush polymers consist of polystyrene backbones and polyacrylate amide brushes. Transmission electron microscopy images revealed their core-shell morphologies with polystyrene nanospheres (hard phase) dispersed in a polyacrylate amide matrix (soft phase). The hard phase exhibited a high Young’s modulus. The high mobility of polymer chains in the soft phase allowed reassociation of the hydrogen bonds, realizing a high healing ratio (λ/λo ~ 90%) at room temperature.
As shown in Sections “Introduction”–“Materials and movable crosslinks”, the combination of strong and weak ionic bonds resulted in both high toughness and high healing ratio32. This concept has inspired researchers to develop self-healing materials with strong and weak reversible bonds. Zheng et al. prepared poly(cis-1,4-isoprene)-modified amide-triazole groups and then added ZnCl277. Some amide-triazole groups form coordination bonds with Zn ions, and the others form hydrogen bonds between themselves. The obtained elastomers were cut into two pieces and rejoined at 80 °C for 24 h, resulting in the healed sample regaining high toughness (60 and 42.8 MJ m−1 for original and healing samples, respectively). The weak hydrogen bonds first re-formed between the separated surfaces, which re-adhered and helped re-form the strong coordination bonds.
Multivalent effects can be used to design strong and weak bonds. Bao et al. prepared self-healing materials by polycondensation of bis(3-aminopropyl)-terminated poly(dimethylsiloxane), 4,4′-methylenebis(phenyl isocyanate), and isophorone diisocyanate78. The obtained elastomer contained 4,4′-methylenebis(phenylurea) (MPU) units and isophorone bisurea (IU) units as hydrogen bonding motifs. As a strong bond, quadruple hydrogen bonding can form between two MPU units due to their rigid symmetrical structures. In contrast, the IU unit can only form a maximum of dual hydrogen bonds with other IU or MPU units due to steric hindrance.
In contrast to the above two works, complexes have been designed in which strong bonds facilitate the re-formation of weak bonds. Bao et al. reported a self-healing elastomer with poly(dimethylsiloxane) polymer chains cross-linked by 1:2 coordination complexes between Fe3+ and 2,6-pyridinedicarboxamide ligands79. These complexes contain strong Fe3+–Npyridyl bonds (145.0 kcal mol−1), intermediate Fe3+–Namide bonds (82.7 kcal mol−1), and intermediate Fe3+–Oamide bonds (40.7 kcal mol−1). These bonds are stabilized by chelation. However, Fe3+–Namide and Fe3+–Oamide bond cleavage weakens the Fe3+–Npyridyl bonds, leading to effective stress dispersion. After dissociation of the crosslinks, Fe3+–Npyridyl bonds hold them in close proximity and allow rapid bond re-formation. This rapid ligand exchange achieves high rupture strain (>10,000%) and self-healing at even low temperatures (−20 °C).
The introduction of nanofibers is a well-known method of improving the mechanical properties of composite materials. Applied stress on the materials is dispersed along nanofibers to realize higher toughness. Self-healing composite materials were prepared by copolymerization of peracetylated γCD monomers, 12-acrylamido dodecanoic acid, and 2-hydroxyethyl acrylate in the presence of citric acid-modified cellulose (CAC)80. The CAC-based composites realized high toughness (151 MJ m−3) and a high healing ratio (Gf/Gf0 = 84%) at 80 °C based on host–guest interactions and hydrogen bonds between the nanofibers and polymeric matrix.
Conclusion
In recent years, significant progress has been made in achieving various functions based on supramolecular chemistry. This review provides an overview of the strategies used to achieve self-healing and self-restoring materials by using reversible and movable crosslinks. The reversible bonds endow self-healing properties by re-formation. Reversible crosslinks exploit many kinds of dynamic covalent bonds and noncovalent bonds, such as hydrogen bonds, metal coordination bonds, ionic interactions, π–π stacking, van der Waals forces, and host–guest interactions. In particular, highly flexible designs were realized by the molecular recognition of host–guest interactions based on their size, shape and charge. Other kinds of crosslinks, movable crosslinks, show self-restoring properties due to their sliding motion. In addition, the multiple reversible crosslinks produce synergistic effects to achieve high toughness and effective self-healing simultaneously. These self-healing and self-restoring properties are expected to prolong the lifetime of polymeric materials, leading to a reduction in waste and a low environmental impact. We believe that supramolecular materials will play a substantial role in realizing a sustainable society.
References
Prager, S. & Tirrell, M. The healing process at polymer-polymer interfaces. J. Chem. Phys. 75, 5194–5198 (1981).
Kim, Y. H. & Wool, R. P. Polymer-polymer interface chain motions in amorphous polymeric. Macromolecules 16, 1115–1120 (1983).
White, S. R. et al. Autonomic healing of polymer composites. Nature 409, 794–797 (2001).
Toohey, K. S., Sottos, N. R., Lewis, J. A., Moore, J. S. & White, S. R. Self-healing materials with microvascular networks. Nat. Mater. 6, 581–585 (2007).
Weiner, S. & Biomineralization, O. Self-repairing oxetane-substituted. Science 323, 1458–1460 (2009).
Roy, D., Cambre, J. N. & Sumerlin, B. S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 35, 278–301 (2010).
Yan, X., Wang, F., Zheng, B. & Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 41, 6042–6065 (2012).
Harada, A., Takashima, Y. & Nakahata, M. Supramolecular polymeric materials via cyclodextrin-guest interactions. Acc. Chem. Res. 47, 2128–2140 (2014).
Peak, C. W., Wilker, J. J. & Schmidt, G. A review on tough and sticky hydrogels. Colloid Polym. Sci. 291, 2031–2047 (2013).
Zhao, X. Multi-scale multi-mechanism design of tough hydrogels: Building dissipation into stretchy networks. Soft Matter 10, 672–687 (2014).
Habault, D., Zhang, H. & Zhao, Y. Light-triggered self-healing and shape-memory polymers. Chem. Soc. Rev. 42, 7244–7256 (2013).
Zhao, Q., Qi, H. J. & Xie, T. Recent progress in shape memory polymer: New behavior, enabling materials, and mechanistic understanding. Prog. Polym. Sci. 49–50, 79–120 (2015).
Lu, W., Le, X., Zhang, J., Huang, Y. & Chen, T. Supramolecular shape memory hydrogels: a new bridge between stimuli-responsive polymers and supramolecular chemistry. Chem. Soc. Rev. 46, 1284–1294 (2017).
Wojtecki, R. J., Meador, M. A. & Rowan, S. J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 10, 14–27 (2011).
Webber, M. J., Appel, E. A., Meijer, E. W. & Langer, R. Supramolecular biomaterials. Nat. Mater. 15, 13–26 (2015).
Roy, N., Bruchmann, B. & Lehn, J. M. DYNAMERS: dynamic polymers as self-healing materials. Chem. Soc. Rev. 44, 3786–3807 (2015).
Zhao, X. et al. Soft materials by design: unconventional polymer networks give extreme properties. Chem. Rev. 121, 4309–4372 (2021).
Harada, A. Supramolecular Polymer Chemistry. (Wiley-VCH Verlag & Co. KGaA, Berlin, Germany, 2012).
Binder, W. H. Self-Healing Polymers: From Principles to Applications. (Wiley-VCH Verlag & Co. KGaA, Berlin, Germany, 2013).
Burattini, S., Greenland, B. W., Chappell, D., Colquhoun, H. M. & Hayes, W. Healable polymeric materials: a tutorial review. Chem. Soc. Rev. 39, 1973–1985 (2010).
Norioka, C., Inamoto, Y., Hajime, C., Kawamura, A. & Miyata, T. A universal method to easily design tough and stretchable hydrogels. NPG Asia Mater. 13, 34 (2021).
Kim, J., Zhang, G., Shi, M. & Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 216, 212–216 (2021).
Kato, T. & Fréchet, J. M. J. New approach to mesophase stabilization through hydrogen-bonding molecular interactions in binary mixtures. J. Am. Chem. Soc. 111, 8533–8534 (1989).
Sijbesma, R. P. et al. Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding. Science 278, 1601–1604 (1997).
Cordier, P., Tournilhac, F., Soulié-Ziakovic, C. & Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 451, 977–980 (2008).
Yanagisawa, Y., Nan, Y., Okuro, K. & Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 359, 72–76 (2018).
Chow, C.-F., Fujii, S. & Lehn, J.-M. Metallodynamers: neutral dynamic metallosupramolecular polymers displaying transformation of mechanical and optical properties on constitutional exchange. Angew. Chem. 119, 5095–5098 (2007).
Burnworth, M. et al. Optically healable supramolecular polymers. Nature 472, 334–337 (2011).
Holten-Andersen, N. et al. pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc. Natl Acad. Sci. USA 108, 2651–2655 (2011).
Gu, Y. et al. Photoswitching topology in polymer networks with metal–organic cages as crosslinks. Nature 560, 65–69 (2018).
Wang, Q. et al. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463, 339–343 (2010).
Sun, T. L. et al. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 12, 932–937 (2013).
Lai, J. C. et al. A rigid and healable polymer cross-linked by weak but abundant Zn(II)-carboxylate interactions. Nat. Commun. 9, 2725 (2018).
Miwa, Y., Taira, K., Kurachi, J., Udagawa, T. & Kutsumizu, S. A gas-plastic elastomer that quickly self-heals damage with the aid of CO 2 gas. Nat. Commun. 10, 1828 (2019).
Burattini, S. et al. A self-repairing, supramolecular polymer system: Healability as a consequence of donor-acceptor π-π stacking interactions. Chem. Commun. 44, 6717–6719 (2009).
Burattini, S. et al. A healable supramolecular polymer blend based on aromatic π-π stacking and hydrogen-bonding interactions. J. Am. Chem. Soc. 132, 12051–12058 (2010).
Chen, X. et al. A thermally re-mendable cross-linked polymeric material. Science 295, 1698–1702 (2002).
Ono, T., Nobori, T. & Lehn, J. M. Dynamic polymer blends—component recombination between neat dynamic covalent polymers at room temperature. Chem. Commun. 12, 1522–1524 (2005).
Rekondo, A. et al. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 1, 237–240 (2014).
Amamoto, Y., Kamada, J., Otsuka, H., Takahara, A. & Matyjaszewski, K. Repeatable photoinduced self-healing of covalently cross-linked polymers through reshuffling of trithiocarbonate units. Angew. Chem. Int. Ed. 50, 1660–1663 (2011).
Montarnal, D., Capelot, M., Tournilhac, F. & Leibler, L. Silica-like malleable materials from permanent organic networks. Science 334, 965–968 (2011).
Urban, M. W. et al. Key-and-lock commodity self-healing copolymers. Science 362, 220–225 (2018).
Wang, H. et al. Synthesis of self-healing polymers by scandium-catalyzed copolymerization of ethylene and anisylpropylenes. J. Am. Chem. Soc. 141, 3249–3257 (2019).
Pedersen, C. J. Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 89, 7017–7036 (1967).
Freeman, W. A., Mock, W. L. & Shih, N. Y. Cucurbituril. J. Am. Chem. Soc. 103, 7367–7368 (1981).
Gutsche, C. D., Dhawan, B., No, K. H. & Muthukrishnan, R. Calixarenes. 4. The synthesis, characterization, and properties of the calixarenes from p-tert-butylphenol. J. Am. Chem. Soc. 103, 3782–3792 (1981).
Ogoshi, T., Kanai, S., Fujinami, S., Yamagishi, T. & Nakamoto, Y. para-Bridged symmetrical pillar[5]arenes: their Lewis acid catalyzed synthesis and host–guest property. J. Am. Chem. Soc. 130, 5022–5023 (2008).
Komiyama, M. & Bender M. L. Cyclodextrin Chemistry. (Springer-Verlag, Berlin, 1978).
Harada, A., Kobayashi, R., Takashima, Y., Hashidzume, A. & Yamaguchi, H. Macroscopic self-assembly through molecular recognition. Nat. Chem. 3, 34–37 (2011).
Tamesue, S., Takashima, Y., Yamaguchi, H., Shinkai, S. & Harada, A. Photoswitchable supramolecular hydrogels formed by cyclodextrins and azobenzene polymers. Angew. Chem. Int. Ed. 49, 7461–7464 (2010).
Nakahata, M., Takashima, Y., Yamaguchi, H. & Harada, A. Redox-responsive self-healing materials formed from host-guest polymers. Nat. Commun. 2, 511 (2011).
Zhang, M. et al. Self-healing supramolecular gels formed by crown ether based host-guest interactions. Angew. Chem. Int. Ed. 51, 7011–7015 (2012).
Kardelis, V. et al. Supramolecular organogels prepared from pillar[5]arene-functionalized conjugated polymers. Macromolecules 50, 9144–9150 (2017).
Park, J. et al. Extremely rapid self-healable and recyclable supramolecular materials through planetary ball milling and host–guest interactions. Adv. Mater. 32, 2002008 (2020).
Appel, E. A. et al. Supramolecular cross-linked networks via host-guest complexation with cucurbit[8]uril. J. Am. Chem. Soc. 132, 14251–14260 (2010).
Liu, J. et al. Tough supramolecular polymer networks with extreme stretchability and fast room-temperature self-healing. Adv. Mater. 29, 1–7 (2017).
Kakuta, T. et al. Preorganized hydrogel: self-healing properties of supramolecular hydrogels formed by polymerization of host-guest-monomers that contain cyclodextrins and hydrophobic guest groups. Adv. Mater. 25, 2849–2853 (2013).
Takashima, Y. et al. Supramolecular materials cross-linked by host-guest inclusion complexes: the effect of side chain molecules on mechanical properties. Macromolecules 50, 3254–3261 (2017).
Takashima, Y. et al. Multifunctional stimuli-responsive supramolecular materials with stretching, coloring, and self-healing properties functionalized via host-guest interactions. Macromolecules 50, 4144–4150 (2017).
Nomimura, S. et al. Self-healing alkyl acrylate-based supramolecular elastomers cross-linked via host-guest interactions. Macromolecules 52, 2659–2668 (2019).
Harada, A. & Kamachi, M. Complex formation between poly (ethylene glycol) and a-cyclodextrin. Macromolecules 23, 2823–2824 (1990).
Harada, A., Li, J. & Kamachi, M. The molecular necklace: a rotaxane containing many threaded α-cyclodextrins. Nature 356, 325–327 (1992).
Okumura, Y. & Ito, K. The polyrotaxane gel: a topological gel by figure-of-eight cross-links. Adv. Mater. 13, 485–487 (2001).
Noda, Y., Hayashi, Y. & Ito, K. From topological gels to slide-ring materials. J. Appl. Polym. Sci. 131, 1–9 (2014).
Ogoshi, T., Aoki, T., Ueda, S., Tamura, Y. & Yamagishi, T. A. Pillar[5]arene-based nonionic polyrotaxanes and a topological gel prepared from cyclic host liquids. Chem. Commun. 50, 6607–6609 (2014).
Nakahata, M., Mori, S., Takashima, Y., Yamaguchi, H. & Harada, A. Self-healing materials formed by cross-linked polyrotaxanes with reversible bonds. Chemistry 1, 766–775 (2016).
Kobayashi, Y., Zheng, Y., Takashima, Y., Yamaguchi, H. & Harada, A. Physical and adhesion properties of supramolecular hydrogels cross-linked by movable cross-linking molecule and host-guest interactions. Chem. Lett. 47, 1387–1390 (2018).
Sawada, J., Aoki, D., Uchida, S., Otsuka, H. & Takata, T. Synthesis of vinylic macromolecular rotaxane cross-linkers endowing network polymers with toughness. ACS Macro Lett. 4, 598–601 (2015).
Harada, A., Li, J. & Kamachi, M. Double-stranded inclusion complexes of cyclodextrin threaded on poly(ethylene glycol). Nature 370, 1992–1994 (1994).
Kureha, T. et al. Decoupled thermo- and pH-responsive hydrogel microspheres cross-linked by rotaxane networks. Angew. Chem. Int. Ed. 56, 15393–15396 (2017).
Takashima, Y. et al. A photoresponsive polymeric actuator topologically cross-linked by movable units based on a [2]rotaxane. Macromolecules 51, 4688–4693 (2018).
Zhao, D. et al. Mortise-and-tenon joint inspired mechanically interlocked network. Angew. Chem. Int. Ed. 60, 16224–16229 (2021).
Ikura, R. et al. Supramolecular elastomers with movable cross-linkers showing high fracture energy based on stress dispersion. Macromolecules 52, 6953–6962 (2019).
Ikura, R. et al. Preparation of hydrophilic polymeric materials with movable cross-linkers and their mechanical property. Polymer 196, 122465 (2020).
Yi, M. B. et al. Movable cross-linking in adhesives: superior stretching and adhesion properties via a supramolecular sliding effect. ACS Appl. Polym. Mater. 3, 2678–2686 (2021).
Chen, Y., Kushner, A. M., Williams, G. A. & Guan, Z. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 4, 467–472 (2012).
Lin, P., Ma, S., Wang, X. & Zhou, F. Molecularly engineered dual-crosslinked hydrogel with ultrahigh mechanical strength, toughness, and good self-recovery. Adv. Mater. 27, 2054–2059 (2015).
Kang, J. et al. Tough and water-insensitive self-healing elastomer for robust electronic skin. Adv. Mater. 30, 1–8 (2018).
Li, C. H. et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 8, 618–624 (2016).
Sinawang, G. et al. Citric acid-modified cellulose-based tough and self-healable composite formed by two kinds of noncovalent bonding. ACS Appl. Polym. Mater. 2, 2274–2283 (2020).
Acknowledgements
This research was funded by a Grant-in-Aid for Scientific Research (B) (JP18H02035) from JSPS of Japan and Scientific Research on Innovative Area (JP19H05721) from MEXT of Japan, JST-Mirai Program Grant Number JPMJMI18E3, the Ogasawara Foundation for the Promotion of Science & Engineering, the Asahi Glass Foundation, the Kao Foundation for the Promotion of Science & Engineering, and the Yazaki Memorial Foundation for Science. We also want to express our gratitude to the Murata Science Foundation and ENEOS Tonen-general Research/Development Encouragement & Scholarship Foundation.
Author information
Authors and Affiliations
Contributions
Y.T. conceived and directed the review article. R.I., J.P. and Y.T. co-wrote the paper. M.O., H.Y. and A.H. gave valuable suggestions. Y.T. oversaw the project and contributed to the execution of the experiments and interpretation of the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ikura, R., Park, J., Osaki, M. et al. Design of self-healing and self-restoring materials utilizing reversible and movable crosslinks. NPG Asia Mater 14, 10 (2022). https://doi.org/10.1038/s41427-021-00349-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-021-00349-1
This article is cited by
-
Thermal, mechanical, and self-healing properties of polymer networks produced by photo-polymerizing α-cyclodextrin-glycidyl methacrylate adduct and poly(ethylene glycol) methacrylate
Journal of Polymer Research (2024)
-
High-strength, stretchable, and NIR-induced rapid self-healing polyurethane nanocomposites with bio-inspired hybrid crosslinked network
Nano Research (2024)
-
Macromolecular architectures constructed by biscalix[5]arene–[60]fullerene host–guest interactions
Polymer Journal (2023)
-
State of Art Review on Applications and Mechanism of Self-Healing Materials and Structure
Archives of Computational Methods in Engineering (2023)
-
Emerging trends in self-healable nanomaterials for triboelectric nanogenerators: A comprehensive review and roadmap
Frontiers in Energy (2023)
