
Abstract
Machine learning accelerates virtual screening in which material candidates are selected from existing databases, facilitating materials discovery in a broad chemical search space. Machine learning models quickly predict a target property from explanatory material features called descriptors. However, a major bottleneck of the machine learning model is an insufficient amount of training data in materials science, especially data with non-equilibrium properties. Here, we develop an alternative virtual-screening process via ensemble-based machine learning with one handcrafted and two generic descriptors to maximize the inference ability even using a small training dataset. A joint representation with the three descriptors translates the physical and chemical properties of a material as well as its underlying short- and long-range atomic structures to describe a multifaceted perspective of the material. As an application, the ensemble-scope descriptor learning model was trained with only 29 entries in the training dataset, and it selected potential oxygen-ion conductors from 13,384 oxides in the inorganic crystal structure database. The experiments confirmed that we successfully discovered five compounds that have not been reported, to the best of our knowledge, as oxygen-ion conductors.
Similar content being viewed by others

Introduction
Data science is an emerging field in materials science that has given rise to the materials informatics (MI) paradigm due to urgent demands for lean development in clean energy technology1,2. A common strategy for a materials search in MI is virtual screening. Material candidates are selected from existing databases based on high-throughput evaluations of target properties. An ab initio calculation is used as a powerful evaluator of the static properties of materials in a restricted chemical space designed by researchers3,4. However, the number of ab initio evaluations critically drops to a few tens of evaluations when exploring non-equilibrium properties (e.g., ionic conductivity) because the computational cost is too large even with a massive multicore architecture5,6. A combination of ab initio calculations and machine learning is therefore employed to broaden the search space5,7,8,9. The rapid inference of machine learning yields potential candidates from hundreds of thousands of compounds in a database as a first-pass screening. Then, the ab initio calculations precisely examine this subset of the candidates. These virtual screenings have been increasingly employed for materials discoveries in catalysis10,11, organic light-emitting diodes7, thermoelectric compounds12, photovoltaic materials13, and Li-ion cathodes3,14,15.
However, this machine learning-assisted screening approach remains a fundamental challenge since “materials data are not big data”16,17,18. While an image recognition task typically uses many thousands of training images, the number of well-curated materials data of non-equilibrium properties is very limited2. Indeed, we extensively collected training data of oxygen-ion conductors, which are used in solid oxide fuel cells, from research papers and databases such as the Material Project19 and Citrine Informatics (https://citrination.com/data_views/147/matrix_search?from=0; https://citrination.com/datasets/151085/show_search?searchMatchOption=fuzzyMatch). However, available data on the structures and ionic conductivity were found to be quite limited. As a result, we compiled only 29 effective training datasets, as shown in the Supplementary information.
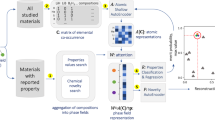
This study proposes a fast virtual screening that maximizes the use of such a small amount of training data in materials science. An evaluator of the present screening is ensemble learning. This scheme is a well-known strategy of machine learning to improve the generalization performance by merging predictions from several weak classifiers20. A prominent technique in our ensemble learning is the descriptor, which is an encoded material feature through a certain protocol, as digital arrays suitable to inputs for the machine learning model21. The next section shows a handcrafted descriptor and two state-of-the-art generic descriptors: the smooth overlap of atomic positions (SOAP)22,23 and the reciprocal 3D voxel space (R3DVS)24. These descriptors are used to highlight the material attributes in the present approach. Here, we call the joint use of these three descriptors an ensemble-scope descriptor that resolves the insufficient data issue. Then, we show the results of the virtual screening for oxygen-ion conductors along with the experimental examinations. Finally, we discuss the prediction model and discovered materials.
Methodology
An outline of the ensemble-scope descriptor is schematically presented in Fig. 1. The three machine learning models were independently trained with the dataset of oxygen-ion conductors shown in the Supplementary information (Table S1).

The present scheme is composed of three independent machine learning models for the oxygen-ion conductivity (σ) as the target property. The first model employs a knowledge-based handcrafted descriptor whose dimensionality is optimally reduced by the PLS model. The second descriptor is SOAP, which calculates a metric between compounds with respect to the atomic geometry and electronic negativity. The SOAP is used for the kernel-ridge regression model. The third descriptor is R3DVS. This descriptor encodes the BVS voxel data, which approximate the oxygen-ion transportation path into a fixed-length array that is suitable for the 3D-CNN model.
Handcrafted descriptor
The handcrafted descriptor is a knowledge-based design policy. Relevant chemical and physical properties are selected by experts to correlate their objective properties. We included the atomic properties, bond valence sum (BVS)25 and porosity properties as elements of the descriptor generated from the crystal structures. For example, the ion number density and polyhedron distortion are calculated from the atomic positions. The porosity is generated using the open-source ZeO++ (http://zeoplusplus.org/) software package. The oxygen-ion pathway is taken into account by using the BVS and BVS potential VBVS, which are defined by the relationship between the nearest ions at each position in a crystal as
where r0 and B are parameters found in ref. 25 (also see http://www.iucr.org/_data_assets/file/0006/81087/bvparm2013_orig.cif) and D0, α, and Rmin are listed in ref. 26. Because the BVS estimates the stable region of ions in a crystal, it has been used as a useful quantity for the prediction of Li-ion conductors27. These features were used to train the partial least squares (PLS) regression model28,29. The number of features was determined by the nonlinear iterative PLS method via leave-one-out cross validation. We omitted the features whose variance importance in projection scores was <0.8. Consequently, 26 features were selected, as shown in the Supplementary information.
As a result of training, the top six features that are correlated with conductivity (the detailed definitions are shown in the Supplementary information) were the atomic density, oxygen density, distortion, log_MM, min_bvs, and min_vbvs. Figure 2a shows that the higher the oxygen-ion conductivity is, the stronger the magnitudes of these features. The atomic and oxygen densities in the unit cell are considered to correlate with the oxygen carrier concentration. The distortion introduced by the elastic strain in a bulk is also known to be an important factor for the oxygen-ion conductivity30.

a A radar chart of the top six correlated features in the handcrafted descriptor. b A projection of SOAP kernel distances on a two-dimensional map and the corresponding atomic structures highlighted with the ionic polyhedra. c The BVS isosurfaces with a bandwidth of 1.85 ≤ BVS ≤ 2.15 for two oxides included in the training data along with their oxygen-ion conductivities σ.
SOAP descriptor
We adopted the alchemically extended SOAP23 as the second descriptor. This feature was generated from the atomic geometries of a unit cell as the input data. This descriptor gives a similarity metric of two local atomic environments with overlapping atomic-neighbor densities and electronegativities, generated to ensure the rotation-symmetry invariances. The SOAP descriptor was used as a kernel in the kernel ridge regression. The SOAP parameters, which are the cutoff radius (rcut), the smoothing parameter for atomic density (ρ) and the width of the Gaussian function to evaluate the similarity of the electronegativities (δ), were set to rcut = 5.0 Å, ρ = 0.5 Å, and δ = 1. These parameters were determined according to relevant studies23,24. We set the oxygen sites as the origins of the atomic environments to evaluate SOAP, embodying the ionic-polyhedral chains made of cations and oxygen ions. The regularization parameter of the ridge regression model was 0.5.
Figure 2b shows a two-dimensional representation of the SOAP distances between the training data of conductors by multidimensional scaling31, which is implemented in scikit-learn (https://scikit-learn.org/). We can see clusters that correspond to the classes of solid structures. Therefore, the SOAP descriptor represents the structural feature that characterizes the ionic-polyhedral networks that they have. For example, the perovskite structure forms a network of corner-sharing \({\mathrm{O}}_6^{2 - }\) octahedra. It is well known that the flexibility in the deformation of these network units appears to be a key for the ion conduction mechanism because the migration of the oxygen carrier occurs by hopping along the ionic polyhedral network32.
R3DVS descriptor
As a complementary descriptor to the SOAP, which can incorporate the short-range atomic structure, we used the R3DVS descriptor for any field quantities distributed over a solid unit cell (e.g., electronic density, local potential)24. We chose the BVS voxel data as the field feature. The R3DVS converts the BVS to a voxel in reciprocal space with fixed array lengths. These formatted voxel data are suitable to be imported into three-dimensional convolutional neural networks (3D-CNNs)33. To accentuate the region with +2 BVS values, where the oxygen ion (O2−) is expected to be stable, the raw BVS data \(f^{0}\left({\mathbf{r}} \right)\) are transformed to f(r) by
The R3DVS parameters were δL* = 0.4 Å and L* = 12.8 Å, where δL* and L* are the recaptured real-space resolution and the cutoff radius24, respectively. These parameters render the BVS data into 323 voxels that are formatted in the R3DVS descriptor. The architecture of the 3D-CNNs is shown in the Supplementary information.
Figure 2c shows that the BVS in YBi3O6, which is a prominent oxygen conductor, distributes more broadly than that of the lower-oxygen conductor Ba2Zr2O6. Broadening the BVS over a unit cell in real space may be relevant to high ion transportation since the BVS estimates the stable region of an ion in a crystal. The 3D-CNNs are expected to detect this long-range correlation of the BVS distribution to the conductivity, leveraging its object-pattern recognition.
Validations
The average of the three predictions of the oxygen-ion conductivities was used as the prediction value of ensemble-scope descriptor learning. Table 1 shows the leave-one-out cross validations of these machine learning models. The three descriptors provide non-small prediction errors due to the sparsity of the training data. Ensemble-scope descriptor learning possesses good regression accuracy similar to the R3DVS descriptor. In particular, this approach shows the best classification accuracy to judge whether the conductivity is greater than the target conductivity of yttria-stabilized zirconia (YSZ), which is popularly used for solid oxide fuel cells. Given the regression and classification measures, we decided to employ ensemble-scope descriptor learning as a navigator of this material search.
Experimental conditions
We synthesized compounds that were selected from the virtual screening by using the conventional solid-state reaction method. The raw materials were K2CO3, MgO, CaCO3, SrCO3, BaCO3, Y2O3, Nb2O5, Ta2O5, B2O3, Al2O3, Ga2O3, GeO2, MnO, Fe2O3, CuO, RuO2, La2O3, Pr2O3, Nd2O3, Eu2O3, Er2O3, Dy2O3, Ho2O3, and Bi2O3 in powder form with purities above 3N, as were provided by Kojundo Chemical Laboratory Co., Ltd., Saitama, Japan. The doping elements were selected by using the concept of the Hume-Rothery rules, in which an element with an ionic radius similar to the host element is expected to be doped34. Accordingly, we chose dopants such as Ca2+ for Eu3+, La3+ for Ca2+, La3+ for Ba2+, and Ca2+ for Dy3+, as listed in Table S2. The raw materials for each selected compound were weighed in the nominal ratio and mixed via ball milling using a 250 ml pot with 80 ml volume of the ZrO2 balls (ϕ = 5 mm) and 80 ml of ethanol. The pot was rotated to mix and pulverize the raw materials for 24 h. The mixed raw materials were then collected from the slurry via evaporation. The obtained powder was calcined in air at 900−1100 °C for 10 h. The calcined powder was then sufficiently pulverized using a mortar. The fine powder was compacted to a disc via die compaction without any lubricants. The green compacts were heated at a rate of 6 °C/min and sintered at 900−1600 °C under O2 gas flow for 1 h. The surface of the sintered bodies was polished using abrasive papers to remove any altered layers.
The sintered bodies were characterized as follows. The densities were determined from the dimensions and weight of the discs. X-ray diffraction (XRD) patterns were obtained to check the crystal structures. The specimens were densely sintered with a relative density higher than 89%. After the Pt electrodes were deposited on the surface of the specimen by a sputtering method, the conductivities were measured at 700 °C via the AC impedance method using IM 3536 (Hioki E.E. Corporation, Nagano, Japan) with a 100 mV signal from 50 Hz to 5 MHz. The measurement atmosphere was either in air or under a N2 gas flow (PO2: 10 ppm, dew point: below −70 °C). The specimens were kept at the measurement temperature for 15 min before the conductivity was measured. The transport number of oxygen ions was measured by an oxygen concentration cell method. The oxygen-rich side was under O2 gas flow (PO2: 1 atm), and the oxygen-poor side was under N2 gas flow (PO2: 10 ppm). The electromotive force due to the difference in O2 pressures was corrected using a YSZ sintered body, which was 231.2 mV at 700 °C.
Results and discussion
Discovery of oxygen-ion conductors by virtual screening
By using ensemble-scope learning trained by 29 entries of the training dataset, we performed the virtual screening of 13,384 oxides recorded in the inorganic crystal structure database (ICSD) (https://icsd.fiz-karlsruhe.de), where we excluded oxides including hydrogen atoms because most hydroxides are unstable at high temperatures, such as 700 °C. We then chose 18 oxides with a predicted conductivity higher than 10−4 S/cm and synthesis accessibility for our experimental facility (details of the selection procedures are described in Sec. 4 of the Supplementary information). Eight oxides, EuGe2KO6, Ca3Fe2Ge3O12, BaCu2Ge2O7, and DyAlO3, and four bismuth oxides were found as oxygen-ion conductors. Their crystal structures are shown in Fig. 3a. Among them, SrTa2Bi2O9, SrNb2Bi2O9, and BaNb2Bi2O9 have been reported as oxygen-ion conductors in previous works35,36; however, we did not include them as training data since the transport numbers at 700 °C were not known. We thus present these bismuth oxides as those recommended by virtual screening with our ensemble-scope learning trained by the 29 entries. The other compounds did not show ionic conductivities (Table S2 of Supplementary information), which may indicate further experimental optimization, such as the densification and proper selection of dopants. Nevertheless, our prediction led to a surprisingly high rate of discovery, as discussed later. Figure 3b summarizes the experimental and prediction conductivities. The experimental values are listed in Table S3 in the Supplementary information. These materials seem to be selected from a wide range of chemical and structural space, and such a discovery is supposed to be difficult via only a conventional knowledge-based interpolation based on the crystal space group and chemical formula of the already-known high-performance materials. In fact, EuGe2KO6 contains an alkali-metal element, and Ca3Fe2Ge3O12 exhibits the garnet structure; they are apparently very different from the oxygen-ion conductors in our training set. Some of the oxides in Fig. 3b have a larger deviation than the MAE or RMSE in Table 1, which may encourage the further experimental optimization of doping, defects, and microstructures.

a Graphics of the unit cells of the host materials of oxygen-ion conductors. b Experimental and prediction values for the oxygen-ion conductivities σ at 700 °C.
Here, we note that transport properties are very affected by the defects and the doping of inorganic materials. The oxygen conductors typically need dopants to open a channel for the oxygen-ion path. However, the ICSD dataset includes mostly pure crystals without identifying subtle defects and doping. To reconcile this common issue of virtual screening for solids, we make the following assumptions. First, the experimental data reported in publications are regarded as optimum through various synthesis processes in terms of defects and doping in each study. Second, while the presence of defects and doping is important in the sense of the density of the transport carriers, the transport coefficient of one carrier itself should be determined mainly by the intrinsic chemical and crystal structures of the base materials recorded in the ICSD. Finally, after training to correlate a stoichiometric crystal structure with the experimentally reported conductivity, our machine learning model predicts an attainable value via experimental optimization. We examined whether our machine learning model could predict an optimal value tuned by doping from a perfect crystal structure. The crystal structures of the doped materials were prepared as follows. The lattice constants of the doped materials were determined by XRD analysis. The sites of the doping atom and vacancy or interstitial oxygen were randomly introduced into a supercell of the non-doped crystal structure registered in the ICSD. The size of the supercell was determined in such a way to approximate the ratio of the doping/defect components observed in the experiments. Then, the atomic positions were optimized by fixing the lattice constants to the experimental values by first-principles calculations using the Vienna Ab initio Simulation Package (VASP)37,38. Table 2 shows that stoichiometric and experimentally optimized structures provide similar predictions, which supports our assumption.
We compare these discoveries with the reported oxygen-ion conductors in a dataset of 241 conductors downloaded from the Citrine-Informatics website (https://citrination.com/data_views/147/matrix_search?from=0), in which the dataset is provided as a result of Advanced Research Projects Agency-Energy IONICS program (https://citrination.com/datasets/151085/show_search?searchMatchOption=fuzzyMatch). Because the Citrine database records the pre-exponential factors and activation energies of the oxygen-ion conductors, we estimated the conductivity at 700 °C using these quantities by the Arrhenius plot. We sorted the conductivities by compound types defined in the dataset. Figure 4 shows that the oxygen-ion conductivities of the present oxides do not reach the level of YSZ. The four bismuth oxides are classified in the “bismuth oxide” family, which is the highest proportion in the dataset. The remainder of the four discoveries are unclassified in the types of reported oxygen-ion conductors, indicating new directions of the materials design, such as potential sources for oxygen-ion conductors. Regarding the number of discoveries, we find that 241 materials have been reported as oxygen-ion conductors in the Citrine dataset (https://citrination.com/data_views/147/matrix_search?from=0) over the past 41 years since 1975, meaning that six materials per year are newly registered on average. This finding is indeed comparable to the amount of our discovery, i.e., five compounds (EuGe2KO6, Ca3Fe2Ge3O12, BaCu2Ge2O7, DyAlO3, and CaTa2Bi2O9). In addition, as shown in Fig. S3 in the Supplementary information, we examined all the experimentally verified candidates that were predicted to have conductivities higher than 10−4 S/cm. The number of oxides that exceeds 10−4 S/cm in the experiments is 7 in the 18 candidates. This result implies that the present screening process is very efficient for this search task.

The reported data are prepared from the Citrine Informatics dataset (https://citrination.com/data_views/147/matrix_search?from=0), which was downloaded in 2017. The labels for the compound types are defined in the dataset. The horizontal line indicates the conductivity of YSZ, which is popularly used for solid oxide fuel cells. The quantity σ indicates the oxygen-ion conductivity at 700 °C.
Structural similarity
The ensemble-scope descriptor should recognize the three-dimensional atomic information through the SOAP and BVS descriptors. Figure 5a shows the atomic geometries of the conductors together with those of the training data entries closest to the experimentally verified materials in terms of the SOAP distance. In particular, focusing on the ionic polyhedra, we can see that the features of the polyhedral network, such as the shape and connections of the vertices and edges, are similar to each other. Furthermore, the BVS of the discovered oxides shown in Fig. 5b is also distributed broadly in their unit cell. This finding implies that oxygen ions are likely to be transported as a conductance carrier in the bulk. Such multifaceted perspectives have facilitated the discovery of those novel classes beyond human perception in the vast chemical space and provide physical and chemical interpretations behind the predictions. In addition, these observations may lead to the conclusion that the ensemble-scope descriptor still searches in the vicinity of the training data and thus indicates a certain limitation in the present predictions but a promising scope for a new area of discovery with the enrichment of the training data.

a Atomic coordinates highlighted on the oxygen-corner polyhedral structures of the screened and nearest training data of oxides measured in the SOAP distance. b BVS isosurfaces with a bandwidth of 1.85 ≤ BVS ≤ 2.15.
Conclusion
We have shown the concept of ensemble-scope descriptor learning that provides a pivotal advance in virtual screening. This concept demonstrates an efficient search capability using only a few tens of training datasets. This result is encouraging because the common perception is that many hundreds of datasets are necessary for material-search informatics. The generic descriptors used here, SOAP and R3DVS, can be flexibly applied to explore any kind of functional material because they only require atomic coordinates. In addition, the knowledge-based handcrafted descriptor can be employed to determine a correlation with the target property in a compact way. This powerful, flexible scheme, which effectively utilizes a small dataset for large-scale searches, may provide new opportunities for researchers to apply material-search informatics in their respective research fields.
References
Tabor, D. P. et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat. Rev. Mater. 3, 5 (2018).
Luna, P. D. et al. Use machine learning to find energy materials. Nature 552, 23 (2017).
Hautier, G. et al. Phosphates as lithium-ion battery cathodes: an evaluation based on highthroughput ab initio calculations. Chem. Mater. 23, 3495–3508 (2011).
Hayashi, H. et al. Discovery of a novel Sn(II)‐based oxide β‐SnMoO4 for daylight‐driven photocatalysis. Adv. Sci. 4, 1600246 (2017).
Sendek, A. D. et al. Machine learning-assisted discovery of many new solid Li-ion conducting materials. Chem. Mater. 31, 342–352 (2019).
Kajita, S., Kinjo, N. & Nishi, T. Autonomous Molecular design by monte-carlo tree search and rapid evaluations using molecular dynamics simulations. Commun. Phys. in press.
Gomez-Bombarelli, R. et al. Design of efficient molecular organic light-emitting diodes by a highthroughput virtual screening and experimental approach. Nat. Mater. 15, 1120 (2016).
Narayan, A. et al. Computational and experimental investigation for new transition metal selenides and sulfides: the importance of experimental verification for stability. Phys. Rev. B 94, 045105 (2016).
Lee, J., Ohba, N. & Asahi, R. Discovery of zirconium dioxides for the design of better oxygen-ion conductors using efficient algorithms beyond data mining. RSC Adv. 8, 25534–25545 (2018).
Studt, F. et al. CO hydrogenation to methanol on Cu-Ni catalysts: theory and experiment. J. Catal. 293, 51–61 (2012).
Ohba, N., Yojoya, T., Kajita, S. & Takechi, K. Search for high-capacity oxygen storage materials by materials informatics. RSC Adv. 9, 41811 (2019).
Seko, A. et al. Prediction of low-thermal-conductivity compounds with first-principles anharmonic lattice-dynamics calculations and Bayesian optimization. Phys. Rev. Lett. 115, 205901 (2015).
Yu, L., Kokenyesi, R. S., Keszler, D. A. & Zunger, A. Inverse design of high absorption thin‐film photovoltaic materials. Adv. Energy Mater. 3, 43–48 (2013).
Nishijima, M. et al. Accelerated discovery of cathode materials with prolonged cycle life for lithiumion battery. Nat. Comm. 5, 4553 (2014).
Chen, H. et al. Carbonophosphates: a new family of cathode materials for Li-ion batteries identified computationally. Chem. Mater. 24, 2009–2016 (2012).
McBride, M., Persson, N., Reichmanis, E. & Grover, M. A. Solving materials’ small data problem with dynamic experimental databases. Processes 6, 79 (2018).
Broderick, S. & Rajan, K. Informatics derived materials databases for multifunctional properties. Sci. Technol. Adv. Mater. 16, 013501 (2015).
Balachandran, P. V., Xue, D., Theiler, J., Hogden, J. & Lookman, T. Adaptive strategies for materials design using uncertainties. Sci. Rep. 6, 19669 (2016).
Jain, A., Persson, K. A. & Ceder, G. Research Update: The materials genome initiative: Data sharing and the impact of collaborative ab initio databases. APL Mater. 4, 053102 (2016).
Murphy, K. P. Machine Learning—A Probabilistic Perspective (The MIT Press, Cambridge, 2012).
Jain, A., Hautier, G., Ong, S. P. & Persson, K. New opportunities for materials informatics: resources and data mining techniques for uncovering hidden relationships. J. Mater. Res. 31, 977–994 (2016).
Bartok, A. P., Kondor, R. & Csanyi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013).
De, S., Bartok, A. P., Csanyi, G. & Ceriotti, M. Comparing molecules and solids across structural and alchemical space. Phys. Chem. Chem. Phys. 18, 13754–13769 (2016).
Kajita, S., Ohba, N., Jinnouchi, R. & Asahi, R. A universal 3D voxel descriptor for solid-state material informatics with deep convolutional neural networks. Sci. Rep. 7, 16991 (2017).
Brown, I. D. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 12, 109 (2009).
Adams, S. & Rao, R. P. High power lithium ion battery materials by computational design. Phys. Status Solidi (A) 208, 1746 (2011).
Avdeev, M., Sale, M., Adams, S. & Rao, R. P. Screening of the alkali-metal ion containing materials from the Inorganic Crystal Structure Database (ICSD) for high ionic conductivity pathways using the bond valence method. Solid State Ion. 225, 43 (2012).
Wold, S., Sjostrom, M. & Eriksson, L. PLS-regression: a basic tool of chemometrics. Chemom. Intell. Lab. Syst. 58, 109 (2001).
Chong, I. G. & Jun, C. H. Performance of some variable selection methods when multicolliniarity is present. Chemom. Intell. Lab. Syst. 78, 103 (2005).
Kilner, J. A. & Brook, R. J. A study of oxygen ion conductivity in-doped non-stoichiometric oxides. Solid State Ion. 6, 237–252 (1982).
Ingwer, B. & Groenen, P. Modern multidimensional scaling: theory and applications. J. Educ. Meas. 40, 277–280 (2003).
Malavasi, L., Fisher, C. A. J. & Islam, M. S. Oxide-ion and proton conducting electrolyte materials for clean energy applications: structural and mechanistic features. Chem. Soc. Rev. 39, 4370–4387 (2010).
Socher, R., Huval, B., Bath, B., Manning, C. D. & Ng, A. Y. Convolutional-recursive deep learning for 3d object classification. Adv. Neural Inf. Process. Syst. 656–664 (2012).
Kingery, W. D., Bowen, H. K. & Uhlmann, D. R. Introduction to Ceramics 2nd edn (Wiley-Interscience Publication, New York, 1976).
Thomas, J. K., Anderson, M. E., Krause, W. E. & Loye, H. Z. Oxygen ion conductivity in a new class of layered bismuth oxide compounds. Mater. Res. Soc. Symp. Proc. 293, 295–300 (1993).
Palanduz, A. C. & Smyth, D. M. The similar defect chemistry of highly doped SrBi2Ta2O9 and SrBi2Nb2O9. J. Electroceram. 14, 123–132 (2005).
Kresse, G. & Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Acknowledgements
We thank Y. Masuoka, Dr. R. Jinnouchi, and M. Nagaya for sample evaluations, the implementation of the SOAP kernel, and the data collection, respectively.
Author information
Authors and Affiliations
Contributions
S.K. performed the 3D-CNNs with R3DVS. S.K. and N.O. performed the SOAP-kernel regression. N.O., A.S., and R.A. developed the handcrafted descriptor. S.K. integrated all the descriptors into ensemble scope descriptor learning. S.T. synthesized and measured the oxygen-ion conductors. R.A. designed this research project. All the authors contributed to the writing of the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kajita, S., Ohba, N., Suzumura, A. et al. Discovery of superionic conductors by ensemble-scope descriptor. NPG Asia Mater 12, 31 (2020). https://doi.org/10.1038/s41427-020-0211-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-020-0211-1
This article is cited by
-
Small data machine learning in materials science
npj Computational Materials (2023)
-
Training data augmentation using generative models with statistical guarantees for materials informatics
Soft Computing (2022)
-
Autonomous molecular design by Monte-Carlo tree search and rapid evaluations using molecular dynamics simulations
Communications Physics (2020)
