
Abstract
Currently, wound infection is an important health problem for the public. Wound infection can not only hinder healing but it can also lead to serious complications. Injectable wound dressings with biocompatible and antibacterial properties can promote wound healing during skin infections and reduce antibiotic use. Here, we used glycidyl methacrylate (GMA) to modify ε-polylysine (ε-PL) and γ-poly(glutamic acid) (γ-PGA) to produce ε-polylysine-glycidyl methacrylate (ε-PL-GMA) and γ-poly(glutamic acid)-glycidyl methacrylate (γ-PGA-GMA). Subsequently, ε-PL-GMA- and γ-PGA-GMA-based hydrogels were developed through photopolymerization using visible light. The hydrogels were injectable, could rapidly gelatinize, were biocompatible, and showed a wide spectrum of antibacterial activity. The hydrogels also promoted wound healing. The results show that these hydrogels inhibit bacterial infection and shorten the wound healing time of skin defects in Staphylococcus aureus models. This demonstrates that the hydrogels hold potential for clinical antimicrobial and wound healing therapy.
Similar content being viewed by others
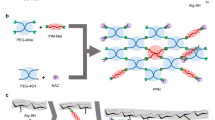


Introduction
Skin forms a unique physical barrier that prevents the invasion of microorganisms1,2. When the skin is damaged by physical and chemical factors, such as trauma, burns, or surgical incisions, its integrity is destroyed, and pathogenic bacterial infections can occur3,4. Surgical infections are common during surgical disease, accounting for 1/3–1/2 of all surgical cases, among which Staphylococcus aureus is the most common infection source5,6. Bacteria invade lymphangion and blood, causing fatal sepsis and severe metastatic infections, such as arthritis, osteomyelitis, and pneumonia. Staphylococcus aureus also produces a variety of toxins and enzymes that slow wound closure and reduce immune function7,8. Poorly hydrated wounds are also difficult to repair9. Due to the aging population and increasing costs of medical care, the burden of health care-related infections in developing countries is high. However, antibiotics are still used in the clinical treatment of infection, the frequent use of which promotes resistance. Wound dressings often lack biocompatibility and fail to stimulate the skin, leading to further health care-related infections10,11. The development of hydrogels with moisturizing, biocompatible, and antimicrobial activity as a wound dressing can overcome these issues and promote wound healing12,13.
Hydrogels are popular biomedical polymers that have been widely applied in the fields of drug delivery, implantation, and tissue engineering14,15,16. Hydrogels are composed of three-dimensional molecular networks that contain a large amount of water17,18. Hydrogels provide high biocompatibility and a moist healing environment for skin wounds that promotes tissue repair and regeneration19,20. Chitosan has inherent antimicrobial activity and contains a large number of amino groups that can be chemically modified and crosslinked to produce antimicrobial hydrogels21,22. Antimicrobial hydrogels also possess antibacterial activity through the loading of antibacterial substances (Ag+, antibiotics) and sterilization through photothermal effects23,24,25. However, hydrogels show limited biodegradation, gel formation, bacterial resistance, and a small therapeutic window, limiting their potential for practical use26.
Epsilon-poly-l-lysine (ε-PL) is a water-soluble, biocompatible polypeptide. ε-PL can be broken down into lysine, forming one of the eight essential amino acids for human health27. ε-PL is a nutritional antibacterial agent that is safer than food preservatives, including potassium sorbate and sodium benzoate28. ε-PL has broad-spectrum antibacterial activity against Gram-positive and Gram-negative bacteria and can kill microorganisms in a manner comparable to other antimicrobial peptides (AMPs). ε-PL causes physiological damage to cells and reduces microbial resistance by the adsorption through microbial membranes29. ε-PL can be used in wound dressings to effectively treat bacterial infections30,31. γ-Poly(glutamic acid) (γ-PGA) is a poly-amino acid produced by microbial fermentation that consists of a high-molecular-weight polymer, with glutamic acid units that form peptide bonds through α-amino and γ-carboxyl groups. γ-PGA has excellent water solubility and biocompatibility due to its similarity to the extracellular matrix (ECM). Due to its high biocompatibility and capacity for tissue repair, γ-PGA is used in bone regeneration and skin repair. Similar to natural ECM, hydrogels based on polysaccharides and peptides are more suitable for clinical use and to improve wound healing32,33.
In this study, we used GMA to prepare ε-PL-GMA and γ-PGA-GMA, which were mixed and added to the visible light initiator lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate (LAP)34. Both were photopolymerized under visible light to obtain a biodegradable, broad-spectrum antimicrobial hydrogel. Visible light was used to prevent ultraviolet rays from injection into the tissue around the wound35. These hydrogels had the advantage of an injectable and fast in situ gel. The hydrogels showed high antimicrobial activity, biodegradability, and adhesion to S. aureus and Escherichia coli. The hydrogel was used to treat bacterial (S. aureus) infections in Sprague–Dawley rats from models of subcutaneous infection (Fig. 1). The hydrogels inhibited bacterial infection and promoted wound healing. These data demonstrate that the injectable visible light-polymerized antimicrobial hydrogel is an ideal candidate for the repair of skin infections.

a γ-PGA-ε-PL hydrogel was synthesized by γ-PGAGMA and ε-PL-GMA. b Build the infection model and treat it with γ-PGA-ε-PL hydrogel.
Materials and methods
Materials
γ-PGA (Mn: 2000 kDa), ε-polylysine (Mn: 2–5 kDa), and GMA (purity 97%, containing 100 p.p.m. MEHQ stabilizer) were obtained from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). Tetrabutylammonium bromide (TBAB) and LAP were obtained from Sigma-Aldrich (USA).
Synthesis of γ-PGA-GMA and ε-polylysine-GMA
γ-PGA was dissolved in deionized (DI) water at a concentration of 5% (wt/vol) at 60 °C as previously described. GMA and TBAB were then added36. The mixture was adjusted to pH 5.0 and stirred vigorously for 6 h. The reaction was dialyzed in 8–12 kDa dialysis bags for 3 days. The solution was lyophilized to obtain purified γ-PGA-GMA.
ε-PL was dissolved in DI water at a concentration of 3% (wt/vol) at 60 °C. GMA and TBAB were then added. The mixture was adjusted to pH 5.0 and stirred vigorously for 8 h. The reaction mixture was dialyzed in 1000 Da dialysis bags for 4 days. The solution was then lyophilized to obtain purified ε-PL-GMA.
Preparation of the γ-PGA-ε-PL hydrogel
γ-PGA-GMA (0.5 g) was dissolved in 5 mL of PBS (0.01 M, pH 7.4). Next, 0.5 g of ε-PL-GMA was dissolved in 5 mL of PBS (0.01 M, pH 7.4). After mixing, LAP was added at a concentration of 0.05 wt% (relative to the volume of the solution). The hydrogel was obtained after irradiation with a visible light source (maximum emission wavelength 405 nm, 60 mW/cm2, China) at room temperature. Four γ-PGA-ε-PL hydrogel systems were prepared by varying the ratios of the components (shown in Fig. 2).

a Synthesis of γ-PGA-GMA. b Synthesis of ε-PL-GMA.
Characterization
1H-nuclear magnetic resonance (1H-NMR) spectra of γ-PGA-GMA and ε-PL-GMA were measured using a 400 MHz 1H-NMR (Bruker Inc., Germany) with D2O as the solvent.
The characterization of γ-PGA-GMA and ε-PL-GMA were performed in the range of 400–4000 cm−1 by using Fourier transform infrared spectroscopy (FT-IR, Bruker Daltonics, Bremen, Germany).
The morphologies of the freeze-dried hydrogels were observed using scanning electron microscopy (SEM; JSM-7500F; JEOL, Japan). The surface of the freeze-dried hydrogels was coated with gold prior to observations.
The gelation time was recorded through tube inversion. The mixture (2 mL) was added to a glass bottle at room temperature and irradiated with a visible light source. The gelation time was determined by inhibiting the flow of the mixture.
The swelling ratio (SR) of the hydrogels was measured as previously described37. Each freeze-dried hydrogel was of an identical shape and size, and all freeze-dried hydrogels (weighted as W0) were soaked in phosphate-buffered saline (PBS) at 37 °C. After incubation, the hydrogels were dried with filter paper and weighed as WS. The SR was calculated using the following equation:
Rheology test of the γ-PGA-ε-PL hydrogel
The rheological properties of the hydrogels were assessed on a rotational HAAKE Rheostress 6000 rheometer (Thermo Scientific, USA) with a cone plate of 20 mm and an upper 4° as previously reported38. The oscillatory rheology of the hydrogels was measured under linear viscoelastic conditions. Dynamic frequency scanning at a fixed strain and dynamic strain scanning at a fixed frequency were performed to measure the storage modulus G′ and loss modulus G″, respectively. The hydrogels with the different components were formed into disks at a radius of 10 mm and a height of 1 mm.
In vitro antibacterial properties of the γ-PGA-ε-PL hydrogels
The antibacterial activity of the γ-PGA-ε-PL hydrogels was evaluated using E. coli and S. aureus as reported previously37. First, the bacteria were incubated overnight in Mueller-Hinton Broth (MHB) at 37 °C with shaking. The bacteria were then diluted in MHB to a concentration of 106 colony-forming unit (CFU)/mL. Next, 200 μL of the γ-PGA-GMA and ε-PL-GMA mixture were added to the wells of a 48-well microplate and mixed. Subsequently, 100 μL of the bacterial solution was seeded onto the hydrogel surface in the 48-well plates. The plates were incubated at 37 °C for 2 h, and 1 mL of PBS was added to each well to resuspend the viable bacteria. Suspensions (100 μL) from each well were plated in Luria−Bertani agar (LB agar). LB plates were incubated at 37 °C for 24 h, and the number of bacterial colonies was recorded. Experiments were performed three times in each group. Data were expressed as kill % as follows:
For the morphology assessments, 100 μL of the bacterial suspensions were sprayed onto γ-PGA-ε-PL hydrogels and incubated at 37 °C for 2 h. Next, 100 μL of the bacterial dispersions were sprayed onto γ-PGA-ε-PL hydrogels at time 0 h. The hydrogels were immediately fixed with 5 mL of 2.5% glutaraldehyde for 4 h and dehydrated in a graded ethanol series (20–100%). Morphological changes in the microorganisms were observed using SEM (JSM-7500F; JEOL, Japan).
Biocompatibility test
The in vitro cytotoxicity of γ-PGA-ε-PL hydrogels was quantitatively determined using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assays. Hydrogels were immersed in Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum for 8 h at 37 °C. Hydrogels were then removed to obtain the extracts. NIH 3T3 cells at a concentration of 3×103 cells per well were added into 96-well culture plates and incubated overnight. Each well was treated with extracts, except for the control wells. Following culture for an additional 24, 48, or 72 h, cell viability was evaluated via MTT assay. Optical density values at 490 nm were measured on a microplate reader (iMark; Bio-Rad, Hercules, CA, USA).
The incompatibility of γ-PGA-ε-PL hydrogels was assessed through fluorescence microscopy observations of NIH 3T3 cells. γ-PGA-ε-PL hydrogels were seeded into 24-well plates and ultraviolet (UV) sterilized for 1 h. NIH 3T3 cells (2 × 104 cells per well) were seeded on the surface of the UV-sterilized hydrogels. After culturing for 24, 48, or 72 h, cell proliferation assays were performed using Calcein-AM/PI Double Staining Kits. Images were obtained with a fluorescence microscope (Nikon TS 100; Japan). Control cells were seeded into wells lacking hydrogels.
In vivo assay of the antimicrobial activity and wound healing properties
Infection models of full-thickness skin defects were established in female SD rats using reported methods38. Each rat was anesthetized with isoflurane. A full-thickness round wound with a diameter of 10 mm was introduced into the middle of the back. Subsequently, 100 µL of inoculum containing 1011 CFU/mL S. aureus was applied evenly to the wound. After 12 h, the wound showed pus. Hydrogel precursors were then added (200 μL) and injected onto the wound surface and gelled using visible light. Wounds not treated with the hydrogels were used as a control group. Wounds treated with 200 μL of mupirocin ointment were compared to determine the healing effects of the experimental group. Wounds were covered with sterilized gauze and sutured with 4-0 silk sutures. On days 4, 8, 12, and 16, wounds were imaged with a digital camera. Images were taken on the meter scale for distance calibrations and standardization. Wound closure was calculated using the following formula: (area of original wound − area of actual wound)/area of original wound × 100%.
Rats were euthanized on days 4, 8, 12, and 16 postoperation. Skin tissues were fixed in 4% phosphoric acid buffer paraformaldehyde and buried in paraffin. A total of 10-μm-thick continuous sections were cut from paraffin-embedded blocks and stained with hematoxylin and eosin (H&E) and Masson’s stain. Dyed sections were imaged with a DXM 1200F microscope (Nikon H600L; Germany).
Immunofluorescent staining
Immunofluorescence was used to assess the expression of interleukin‐6 (IL-6) and transforming growth factor-β (TGF-β) in the wound tissues to evaluate the inflammatory response14,30. Briefly, sections were rehydrated and incubated with primary antibodies for IL-6 and TGF-β (Abcam, USA). After rinsing, sections were incubated with fluorescent secondary antibodies (Abcam, USA). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich, USA). Data were examined with a fluorescent microscope (Nikon TS 100; Japan).
Results
Synthesis of the hydrogels
γ-PGA-GMA and ε-PL-GMA were prepared with GMA. The solutions of γ-PGA-GMA and ε-PL-GMA were mixed with the LAP initiator and then photopolymerized under visible light to obtain the γ-PGA-ε-PL hydrogel.
Characterization
1H-NMR was used to confirm that γ-PGA-GMA and ε-PL-GMA were successfully modified by GMA (Fig. 3a, b). The spectrum of γ-PGA showed 1H-NMR signals at 2.4 (a-CH2), 1.9 and 2.1 (b-CH2), and 4.4 p.p.m. (c-CH). Additional signals at 5.40 and 5.64 p.p.m. were attributed to the two hydrogens of the double bond of the methacrylamide group. The additional signals at 1.8 p.p.m. (f-CH3) were attributed to the methyl protons. The degree of substitution of the methacryloyl groups (DS, molar ratio of methacryloyl groups per glutamyl unit) was determined by 1H-NMR spectroscopy. The degree of substitution of γ-PGA-GMA at a molar ratio of 2:1 was 20.29%:

a, b 1H-NMR spectra of γ-PGA-GMA and ε-PL-GMA. c, d FT-IR spectra of γ-PGA-GMA and ε-PL-GMA. e Morphologies of the hydrogels. f The components and gelation times of the hydrogels. g Swelling ratio of the hydrogels.
The spectrum of ε-PL showed additional 1H-NMR signals at 3.2 (a-CH2), 3.9 (b-CH), 1.5 (c-CH2), 1.3 (d-CH2), and 1.9 p.p.m. (e-CH2). The additional signals at 5.40 and 5.64 p.p.m. were attributed to the two hydrogens on the double bond of the methacrylamide group. The degree of substitution of ε-PL-GMA at a molar ratio of 2:1 was 26.58%:
FT-IR spectra confirmed that the methylacryloyl chloride was grafted onto γ-PGA and ε-PL (Fig. 3c, d). The appearance of a peak at 1689 cm−1 revealed the presence of C = O groups in the conjugated system. The peak at 1643 cm−1 showed the presence of C = C groups in γ-PGA-GMA. The peak at 1666 cm−1 showed the presence of C = O groups in the conjugated system. The peak at 1644 cm−1 showed the presence of C = C groups in ε-PL-GMA.
The multiporous framework of the hydrogels was investigated by SEM (Fig. 3e). The pore sizes of the γ-PGA-ε-PL-1, γ-PGA-ε-PL-2, γ-PGA-ε-PL-3, and γ-PGA-ε-PL-4 hydrogels were between 80 and 120 μm. This indicated that γ-PGA-ε-PL hydrogels had sufficient pore sizes to exchange nutrients and enable the cells to propagate and migrate.
The gelation time of the hydrogels decreased from 10 to 6 s with increasing proportion of ε-PL-GMA (Fig. 3f). This was due to the increase in cross-linking density of the hydrogel. All hydrogels showed characteristically fast gelation and could be used as injectable hydrogels.
γ-PGA-ε-PL-2, γ-PGA-ε-PL-3, and γ-PGA-ε-PL-4 hydrogels had a lower swelling percentage than the γ-PGA-ε-PL-1 hydrogels (Fig. 3g). This was attributed to the hydrogel cross-linking density. As the proportion of ε-PL-GMA increased, the increase in cross-linking enhanced the network structure of the hydrogels. This resulted in a loss of water absorption.
Rheology test of the γ-PGA-ε-PL hydrogel
To obtain the mechanical properties of hydrogels, rheological analysis was performed at different frequencies and strains (Fig. 4). The storage modulus (G′) was found to be greater than the loss modulus (G″). This was a remarkable feature of the gel. The G′ did not significantly change according to strain. This indicated that the hydrogel was homogeneous and that the cross-linking was relatively stable. With increasing frequency, the G′ of the γ-PGA-ε-PL-1 hydrogel and γ-PGA-ε-PL-2 hydrogel significantly changed. As the G′ of γ-PGA-ε-PL-3 hydrogels and γ-PGA-ε-PL-4 hydrogels did not significantly differ, this indicated that increasing the proportion of ε-PL-GMA could increase the cross-linking density of the hydrogels and improve the stability and mechanical strength. The γ-PGA-ε-PL-4 hydrogels showed the most favorable mechanical properties. However, hydrogels above this ratio did not mix or precipitate.

a, b Storage modulus (G’) and loss modulus (G’’) values are investigated through dynamic strain sweep. c, d Storage modulus (G’) and loss modulus (G’’) values are investigated through dynamic frequency sweep.
Antibacterial activities of the γ-PGA-ε-PL hydrogel
To treat skin infections, wound dressings must display antibacterial activity. The antibacterial activity of the γ-PGA-ε-PL hydrogels was therefore evaluated using two common bacterial wound infections (Fig. 5a, b). Compared with hydrogels without ε-PL-GMA, hydrogels containing ε-PL-GMA showed high levels of activity against S. aureus and E. coli. γ-PGA-ε-PL-4 hydrogels reduced the bacterial levels to lower than 103 CFU/mL, and ≥99% of S. aureus and E. coli were killed. These results showed that the γ-PGA-ε-PL hydrogels showed high levels of antibacterial activity.

a Images of surviving bacterial clones on agar plates. b Evaluation of the resistance of hydrogels to E. coli and S. aureus. c SEM images of S. aureus seeded on the γ-PGA-ε-PL hydrogel.
To observe the morphology of the bacteria after contact with the hydrogels, S. aureus was seeded onto γ-PGA-ε-PL-4 hydrogels for 2 h and imaged using SEM (Fig. 5c). Staphylococcus aureus cells were seeded onto γ-PGA-ε-PL-4 hydrogels for 0 h as a control. The morphology of S. aureus after contact with γ-PGA-ε-PL-4 hydrogels changed compared with the control. The membranes of S. aureus were also damaged. These data suggest that γ-PGA-ε-PL hydrogels possess killing activity against S. aureus through cell membrane disruption. This mechanism of action is consistent with the known effects of ε-PL on microorganisms29.
Cytotoxicity of the γ-PGA-ε-PL hydrogel
As a biomaterial for wound healing, the biocompatibility of γ-PGA-ε-PL hydrogels is important. Due to the high antibacterial activity of γ-PGA-ε-PL-4 hydrogels, we measured only the cytotoxicity of γ-PGA-ε-PL-4 hydrogels. As shown in Fig. 6a, compared with the control groups, the hydrogels showed no obvious cytotoxicity, with over 90% of the cells remaining viable. Similarly, live/dead staining (Fig. 6b) showed that the hydrogels caused no obvious damage to 3T3 cells. After 3 days, the cells adhered to the hydrogel and showed a spindle shape. These data suggest that the hydrogels display a high level of biocompatibility for biomedical applications.

a Cell viability. b Live/dead staining of 3T3 cells in contact with the hydrogel surface on the third day. Scale bar: 200 μm.
Wound closure
As shown in Fig. 7a, the γ-PGA-ε-PL-4 hydrogel group showed improved wound healing compared with the untreated and antibiotic (mupirocin ointment) groups. On day 4, the γ-PGA-ε-PL-4 hydrogels showed wounds of similar sizes, which were smaller than those of the untreated and mupirocin ointment groups. On day 8, compared with the untreated and mupirocin ointment groups, the wounds of the γ-PGA-ε-PL-4 hydrogel group were reduced. On day 12, more than half of the wounds healed in the mupirocin ointment group. Interestingly, wounds treated with the γ-PGA-ε-PL-4 hydrogel exhibited complete closure. On day 16, the hydrogel group had fully healed, while the untreated and mupirocin ointment groups retained open wounds.

a Digital photos of wound contraction at the given time. b, c Images of wound tissue after H&E and Masson staining. The red arrows show epidermal tissue, and the imaginary line indicates the inflammatory response. The black arrows show blood vessels and hair follicles. The scale bar is 200 µm.
As shown through H&E and Masson staining (Fig. 7b, c), inflammatory cells were produced in all groups on day 4. On day 8, the number of inflammatory cells decreased in the γ-PGA-ε-PL-4 hydrogel group and mupirocin ointment group. Epithelial tissue formation at the wound site was also evident in the γ-PGA-ε-PL-4 hydrogel and mupirocin ointment groups. On day 12, the wounds treated with γ-PGA-ε-PL-4 hydrogels were covered by epidermal tissue, and new tissue, such as hair follicles, were generated. In the mupirocin ointment and control groups, part of the epidermis was formed, and some of the wound was covered by the epidermis. On day 16, the corneum in the γ-PGA-ε-PL-4 hydrogel group continued to form, and blood vessels continued to be repaired. These results indicated that γ-PGA-ε-PL-4 hydrogels inhibit bacteria, promote wound healing, and hold potential for wound dressings.
Immunofluorescent staining
The therapeutic effects of the hydrogels on wound infections were evaluated through immunofluorescence analysis. The secretion of two typical proinflammatory cytokines, IL‐6 and TGF-β, at the wound site was investigated39,40. As shown in Fig. 8, the untreated groups showed high expression of IL-6 and TGF-β, indicating a severe inflammatory response. Interestingly, the levels of IL-6 and TGF-β were lower in the hydrogel group. This suggested that the hydrogels could effectively control inflammation and promote wound healing, most likely due to the antibacterial effects of ε-PL-GMA coupled to its high biocompatibility and ECM-like structure.

The white arrows show the location and amount of IL‐6 and TNF‐α expression. The scale bar is 100 µm.
Conclusions
In this study, we designed an injectable antibacterial hydrogel that was photopolymerized by visible light for the treatment of skin infections. The hydrogel consists of γ-PGA-GMA and ε-PL-GMA. The gel time, mechanical strength, and swelling rate of the hydrogels could be modified through adjustment of the γ-PGA-GMA and ε-PL-GMA ratio. The hydrogels showed characteristics of injectable and rapid gels and were easy to use. Importantly, the hydrogels demonstrated high levels of antibacterial activity and biocompatibility. In in vivo infection models, the hydrogels reduced inflammation, promoted wound healing, and shortened the healing time. This highlights these hydrogels as promising candidates for anti-infection and wound healing.
References
Clark, R. A. F., Ghosh, K. & Tonnesen, M. G. Tissue engineering for cutaneous wounds. J. Invest. Dermatol. 127, 1018–1029 (2007).
Kamel, R. A., Ong, J. F., Eriksson, E., Junker, J. P. E. & Caterson, E. J. Tissue engineering of skin. J. Am. Coll. Surg. 217, 533–555 (2013).
Sorg, H., Tilkorn, D. J., Hager, S., Hauser, J. & Mirastschijski, U. Skin wound healing: an update on the current knowledge and concepts. Eur. Surg. Res. 58, 81–94 (2017).
Sen, C. K. et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Rep. Regen. 17, 763–771 (2009).
de Lissovoy, G. et al. Surgical site infection: Incidence and impact on hospital utilization and treatment costs. Am. J. Infect. Control 37, 387–397 (2009).
Allegranzi, B. et al. Burden of endemic health-care-associated infection in developing countries: systematic review and meta-analysis. Lancet 377, 228–241 (2011).
Li, W., Nie, A., Li, Q., Xie, N. & Ling, Y. Establishment of rapid diagnostic method for the identification of Staphylococcus aureus in bacterial conjunctivitis. Mater. Express 9, 484–491 (2019).
Percival, S. L. et al. A review of the scientific evidence for biofilms in wounds. Wound Rep. Regen. 20, 647–657 (2012).
Stevens, D. L. et al. Executive summary: practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 59, 147–159 (2014).
Pallin, D. J., Camargo, C. A. Jr. & Schuur, J. D. Skin infections and antibiotic stewardship: analysis of emergency department prescribing practices, 2007–2010. West J. Emerg. Med. 15, 282–289 (2014).
Han, G. & Ceilley, R. Chronic wound healing: a review of current management and treatments. Adv. Ther. 34, 599–610 (2017).
Gurtner, G. C., Werner, S., Barrandon, Y. & Longaker, M. T. Wound repair and regeneration. Nature 453, 314–321 (2008).
Groeber, F., Holeiter, M., Hampel, M., Hinderer, S. & Schenke-Layland, K. Skin tissue engineering—in vivo and in vitro applications. Adv. Drug Deliv. Rev. 63, 352–366 (2011).
Liang, Y., Zhao, X., Hu, T., Han, Y. & Guo, B. Mussel-inspired, antibacterial, conductive, antioxidant, injectable composite hydrogel wound dressing to promote the regeneration of infected skin. J. Colloid Interface Sci. 556, 514–528 (2019).
Mohan, N., Mohanan, P. V., Sabareeswaran, A. & Nair, P. Chitosan-hyaluronic acid hydrogel for cartilage repair. Int. J. Biol. Macromol. 104, 1936–1945 (2017).
Chen, X. et al. Light-induced hydrogel based on tumor-targeting mesoporous silica nanoparticles as a theranostic platform for sustained cancer treatment. ACS Appl Mater. Interfaces 8, 15857–15863 (2016).
Wang, Q., Hou, R., Cheng, Y. & Fu, J. Super-tough double-network hydrogels reinforced by covalently compositing with silica-nanoparticles. Soft Matter 8, 6048–6056 (2012).
Niu, Y., Yang, T., Ke, R. & Wang, C. Preparation and characterization of pH-responsive sodium alginate/humic acid/konjac hydrogel for l-ascorbic acid controlled release. Mater. Express 9, 563–569 (2019).
Bannasch, H. et al. Skin tissue engineering. Clin. Plast. Surg. 30, 573–579 (2003).
Li, X. et al. Nanofiber-hydrogel composite-mediated angiogenesis for soft tissue reconstruction. Sci. Transl. Med. 11, eaau6210 (2019).
Verma, J., Kanoujia, J., Parashar, P., Tripathi, C. B. & Saraf, S. A. Wound healing applications of sericin/chitosan-capped silver nanoparticles incorporated hydrogel. Drug Deliv. Transl. Res. 7, 77–88 (2017).
Dong, R., Zhao, X., Guo, B. & Ma, P. X. Self-healing conductive injectable hydrogels with antibacterial activity as cell delivery carrier for cardiac cell therapy. ACS Appl. Mater. Interfaces 8, 17138–17150 (2016).
Chen, M. et al. Fabrication of self-healing hydrogels with surface functionalized microcapsules from stellate mesoporous silica. Polym. Chem. 10, 503–511 (2019).
He, M., Wang, Q., Zhang, J., Zhao, W. & Zhao, C. Substrate-independent Ag-nanoparticle-loaded hydrogel coating with regenerable bactericidal and thermoresponsive antibacterial properties. ACS Appl. Mater. Interfaces 9, 44782–44791 (2017).
Zhu, Q. et al. Enhanced healing activity of burn wound infection by a dextran-HA hydrogel enriched with sanguinarine. Biomater. Sci. 6, 2472–2486 (2018).
Tiwari, A., Grailer, J. J., Pilla, S., Steeber, D. A. & Gong, S. Biodegradable hydrogels based on novel photopolymerizable guar gum-methacrylate macromonomers for in situ fabrication of tissue engineering scaffolds. Acta Biomater. 5, 3441–3452 (2009).
Wang, R. et al. Antimicrobial and biocompatible ε-polylysine-γ-poly(glutamic acid)-based hydrogel system for wound healing. J. Bioact. Compat. Polym. 31, 242–259 (2016).
Hyldgaard, M. et al. The antimicrobial mechanism of action of epsilon-poly-l-lysine. Appl. Environ. Microbiol. 80, 7758–7770 (2014).
Zhou, C. et al. A photopolymerized antimicrobial hydrogel coating derived from epsilon-poly-l-lysine. Biomaterials 32, 2704–2712 (2011).
Wang, R. et al. A biomimetic mussel-inspired ε-poly-l-lysine hydrogel with robust tissue-anchor and anti-infection capacity. Adv. Funct. Mater. 27, 1604894 (2017).
Zhou, L. et al. Injectable self-healing antibacterial bioactive polypeptide-based hybrid nanosystems for efficiently treating multidrug resistant infection, skin-tumor therapy, and enhancing wound healing. Adv. Funct. Mater. 29, 1806883 (2019).
Shi, L. et al. A novel poly(γ-glutamic acid)/silk-sericin hydrogel for wound dressing: synthesis, characterization and biological evaluation. Mater. Sci. Eng. C 48, 533–540 (2015).
Yang, R. et al. Bioinspired poly (γ-glutamic acid) hydrogels for enhanced chondrogenesis of bone marrow-derived mesenchymal stem cells. Int. J. Biol. Macromol. 142, 332–344 (2019).
Fairbanks, B. D., Schwartz, M. P., Bowman, C. N. & Anseth, K. S. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials 30, 6702–6707 (2009).
Hu, J. et al. Visible light crosslinkable chitosan hydrogels for tissue engineering. Acta Biomater. 8, 1730–1738 (2012).
Yang, Q., Song, F., Zou, X. & Liao, L. Preparation and mineralization of a biocompatible double network hydrogel. J. Biomater. Sci. Polym. Ed. 28, 431–443 (2017).
Chen, H. et al. An injectable self-healing hydrogel with adhesive and antibacterial properties effectively promotes wound healing. Carbohydr. Polym. 201, 522–531 (2018).
Tian, R. et al. Fabrication of self-healing hydrogels with on-demand antimicrobial activity and sustained biomolecule release for infected skin regeneration. ACS Appl. Mater. Interfaces 10, 17018–17027 (2018).
Lichtman, M. K., Otero-Vinas, M. & Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Rep. Regen. 24, 215–222 (2016).
Swartzlander, M. D. et al. Immunomodulation by mesenchymal stem cells combats the foreign body response to cell-laden synthetic hydrogels. Biomaterials 41, 79–88 (2015).
Acknowledgements
This work was financially supported by the National Natural Science Fund for Distinguished Young Scholars (NSFC31525009), the National Natural Science Foundation of China (NSFC31930067, 31771096), the National Key Research and Development Program of China (No. 2017YFC1103502), and the 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD18002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, A., He, X., Li, L. et al. An injectable photopolymerized hydrogel with antimicrobial and biocompatible properties for infected skin regeneration. NPG Asia Mater 12, 25 (2020). https://doi.org/10.1038/s41427-020-0206-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-020-0206-y
This article is cited by
-
Bioactive polymers for cosmetics: unleashing the potential of Chromohalobacter canadensis 28
Macromolecular Research (2024)
-
Synthesis, optimization, and multifunctional evaluation of amla-based novel biodegradable hydrogel
Polymer Bulletin (2024)
-
Advances in preparation and application of antibacterial hydrogels
Journal of Nanobiotechnology (2023)
-
Therapeutic Potential of Antimicrobial Peptides for Wound Healing
International Journal of Peptide Research and Therapeutics (2022)
-
Development of novel mannitol blended PVA hydrogel membrane and its anticancer and antimicrobial drug delivery potential for wound dressing applications
Journal of Sol-Gel Science and Technology (2022)
