
Abstract
The NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is a multiprotein complex involved in the release of mature interleukin-1β and triggering of pyroptosis, which is of paramount importance in a variety of physiological and pathological conditions. Over the past decade, considerable advances have been made in elucidating the molecular mechanisms underlying the priming/licensing (Signal 1) and assembly (Signal 2) involved in NLRP3 inflammasome activation. Recently, a number of studies have indicated that the priming/licensing step is regulated by complicated mechanisms at both the transcriptional and posttranslational levels. In this review, we discuss the current understanding of the mechanistic details of NLRP3 inflammasome activation with a particular emphasis on protein-protein interactions, posttranslational modifications, and spatiotemporal regulation of the NLRP3 inflammasome machinery. We also present a detailed summary of multiple positive and/or negative regulatory pathways providing upstream signals that culminate in NLRP3 inflammasome complex assembly. A better understanding of the molecular mechanisms underlying NLRP3 inflammasome activation will provide opportunities for the development of methods for the prevention and treatment of NLRP3 inflammasome-related diseases.
Similar content being viewed by others
Introduction
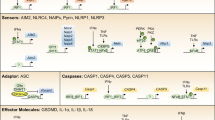
Inflammasomes are cytoplasmic high-molecular-weight protein platforms of caspase-1 activation in response to microbial invasion and damage signals.1,2 Inflammasomes consist of the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family, the adapter apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and the effector protease caspase-1. The formation of these protein complexes results in the activation of caspase-1, which is involved in the maturation of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 into biologically active forms, and cleavage of gasdermin D (GSDMD) to promote pyroptotic cell death (pyroptosis).3,4,5,6
Among inflammasomes, the NOD-, leucine-rich repeat (LRR)-, and pyrin domain (PYD)-containing protein 3 (NLRP3) inflammasome has been studied extensively and was found to be activated by a wide spectrum of stimuli. It is generally accepted that NLRP3 inflammasome activation is regulated through a two-step process, with priming at the transcriptional and posttranslational levels (Signal 1) and assembly by multiple pathways in response to a variety of exogenous pathogen-derived or endogenous danger molecules (Signal 2). Recently, there has been a renaissance in our understanding of the posttranslational modification (PTM) and protein-protein interactions of NLRP3 inflammasome components that license cells for full activation of inflammasome assembly.7,8,9,10 The breadth of our current understanding extends to the regulation of the priming that is involved in NLRP3 inflammasome complex assembly, including accumulating evidence indicating a number of molecular mechanisms underlying the positive or negative regulation of NLRP3 inflammasome activation. Indeed, inflammasome and IL-1β activity are important for host defense against numerous bacterial, viral, and fungal infections. However, excessive or altered regulation of NLRP3 inflammasome activity is related to the pathogenesis of a wide variety of inflammatory, autoimmune, and degenerative diseases.11,12 The pleiotropic roles of the NLRP3 inflammasome have been reviewed extensively elsewhere in terms of physiological responses and in the context of a variety of human diseases.13,14,15,16 In addition, the mechanisms of noncanonical and one-step NLRP3 inflammasome activation are beyond the scope of this review.
Here, we summarize the current understanding of the molecular details involved in the priming/licensing step of NLRP3 inflammasome activation. We then cover the protein–protein interactions and spatiotemporal regulation of the NLRP3 inflammasome machinery. Finally, we discuss the various positive/negative regulatory mechanisms that orchestrate optimal regulation of the NLRP3 inflammasome.
Overview of NLRP3 inflammasome activation
NLRP3 is an NLR that contains an N-terminal PYD, a central NAIP, CIITA, HET-E, and TP1 (NACHT) or NOD that hydrolyzes adenosine triphosphate (ATP) into adenosine diphosphate (ADP), and a C-terminal LRR domain. During inflammasome assembly, NLRP3 interacts with the N-terminus of the adapter protein ASC via PYD–PYD interactions; the C-terminus of ASC has a caspase recruitment domain (CARD) that can bind to procaspase-1 via CARD–CARD interactions to promote caspase dimerization and activation. Due to its prion-like properties, ASC forms large fibrillar aggregates known as “specks”.11,12,17,18,19,20
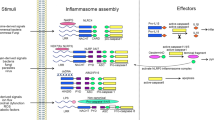
NLRP3 inflammasome activation generally requires two steps, i.e., priming (Signal 1) and protein complex assembly (Signal 2) (Fig. 1). The priming process is triggered by pattern recognition receptor signaling, e.g., Toll-like receptor (TLR) 4 activation or tumor necrosis factor (TNF) signaling, which subsequently leads to the transcriptional activation of NLRP3, pro-IL-1β, and pro-IL-18 via nuclear factor-κB (NF-κB)-dependent pathways.11,12 However, emerging data suggest that the priming step of NLRP3 inflammasome activation is complicated, involving transcriptional and posttranslational mechanisms, and requires numerous protein binding partners.11,12 The activation signal (Signal 2) is induced by various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), including extracellular ATP, pore-forming toxins, RNA viruses, and particulate matter. For Signal 2 activation, numerous molecular or cellular events, including mitochondrial dysfunction and reactive oxygen species (ROS) generation, ion flux (K+/Cl− efflux, and Ca2+ influx), and lysosomal damage, are involved in the activation of NLRP3 inflammasome assembly.11,12,17,18,19,20 Both Signal 1 and 2 are triggered by microbial or sterile inflammatory stimuli, although microbial signals are different than sterile signals in terms of their kinetics and magnitude.21

Overview of NLRP3 inflammasome priming and activation. NLRP3 inflammasome activation involves two steps, i.e., Signal 1 (priming) and Signal 2 (protein complex assembly). Signal 1 is triggered by pattern recognition receptor signaling or cytokines, leading to the transcriptional activation of NLRP3 inflammasome components. Licensing of the NLRP3 protein is important for the priming step of the NLRP3 inflammasome. The activation signal (Signal 2) is induced by various pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Interleukin 1β (IL-1β)/IL-1R1, lipopolysaccharide (LPS)/Toll-like receptor 4 (TLR4), tumor necrosis factor (TNF)/TNF receptor (TNFR), sphingosine 1-phosphate (S1P)/S1P receptor 2 (S1PR2), adenosine diphosphate (ADP)/P2Y12, α-synuclein/CD36, and bromodomain-containing protein 4 (BRD4) inhibitor JQ1 each activate NF-κB and then upregulate the transcription level of the component required for NLRP3 inflammasome formation. Caspase-8 and fas-associated protein with death domain (FADD) are upstream regulators of NF-κB signaling that activate both the transcriptional priming and PTM of NLRP3 inflammasome pathway components. Upon TLR3 stimulation, FADD/caspase-8 scaffolding is involved in the PTM associated with Signal 1 in the intermediate pathway or activates receptor-interacting serine/threonine-protein kinases 3 (RIPK3)/mixed lineage kinase domain like pseudokinase (MLKL) function required for both Signal 1 and Signal 2 in the late pathway. Extracellular Ca2+ can activate the NLRP3 inflammasome through calcium-sensing receptor (CaSR), and CaSR triggers the phospholipase C (PLC)/inositol-1,4,5-trisphosphate (InsP3) pathway to induce intracellular Ca2+ release from the endoplasmic reticulum (ER). Ca2+ flux by transient receptor potential melastatin 2 (TRPM2) or apolipoprotein C3 (ApoC3) is mediated by reactive oxygen species (ROS) to activate NLRP3. It is currently recognized that TXNIP binds to NLRP3. ADP/P2Y1 induces Ca2+ movement, and various DAMPs/PAMPs trigger K+ efflux through pannexin-1 to activate NLRP3. In addition, P2X7 receptor (P2X7R) and tandem pore domains in weak inward rectifying K+ channel 2 (TWIK2) act as K+ efflux channels and are required for NLRP3 inflammasome activation. Testosterone, imiquimod, CLO97, K+ efflux, and α-synuclein generate mitochondrial ROS (mtROS), which activate the NLRP3 inflammasome. Severe fever with thrombocytopenia syndrome virus (SFTSV) infection triggers BCL2 antagonist/killer 1 (BAK)/BCL2-associated X (BAX) signaling and leads to oxidized mitochondrial DNA (ox-mtDNA). Furthermore, cardiolipin can directly bind to NLRP3 and activate NLRP3 inflammasome assembly. Particulates and crystals, nicotine, lysophosphatidylcholine (LPC), and Leu-Leu-O-methyl ester (LLME) induce lysosomal damage, and damaged lysosomes release cathepsin B. In addition, damaged lysosomes induce K+ efflux, which causes Cl- efflux through chloride intracellular channels (CLICs). The complement system activates NLRP3 by forming a membrane attack complex (MAC). In endothelial cells, immunoglobulin M (IgM)-mediated MAC induces NF-κB-inducing kinase (NIK) stabilization and causes NLRP3 inflammasome activation. C5a-C5aR2 signaling also activates the NLRP3 inflammasome through protein kinase R (PKR) in macrophages. Moreover, pore-forming toxins and ATP induce K+ efflux and activate NLRP3 inflammasome. During Mycobacterium tuberculosis (Mtb) infection, plasma membrane damage causes K+ efflux and NLRP3 activation. In addition, ADP also induces K+ efflux through P2Y12. During RNA virus infection, mitofusin 2 (MFN2) and mitochondrial antiviral signaling (MAVS) protein directly bind to and activate NLRP3. Cathepsin B also directly binds to NLRP3 in the ER. Z-DNA binding protein 1 (ZBP1) regulates NLRP3 activation in response to influenza A virus infection. Orange arrows indicate direct binding with NLRP3
In addition, NLRP3 inflammasome activation often leads to proinflammatory programmed cell death known as pyroptosis. The inflammasome complex leads to autoproteolytic activation of caspase-1, which subsequently triggers the cleavage of the proinflammatory cytokines IL-1β and IL-18 and the pyroptotic substrate GSDMD.22,23 Upon GSDMD cleavage, the N-terminus of GSDMD (N-GSDMD) oligomerizes and forms plasma membrane pores that mediate cell death and the secretion of the mature forms of IL-1β and IL-18.22,23 Recently, the Z-DNA binding protein 1 (ZBP1)-NLRP3 inflammasome, which is specifically activated by viral RNA products or endogenous nucleic acid ligands, was shown to promote a mixed form of cell death, i.e., pyroptosis, apoptosis, necroptosis (PANoptosis) and PANoptosome assembly.24,25 Another recent study revealed fundamental differences in GSDMD trafficking and cleavage between macrophages and neutrophils.26 In macrophages, N-GSDMD oligomerizes in the plasma membrane to form pores that promote NLRP3 inflammasome-associated pyroptosis.27 In contrast, N-GSDMD does not localize to the plasma membrane or mediate pyroptosis in NLRP3-activated neutrophils, although it is required for IL-1β secretion. Moreover, N-GSDMD associates with azurophilic granules to release neutrophil elastase into the cytosol and promotes secondary cleavage of GSDMD to form an alternatively cleaved form of N-GSDMD.26 We speculate that different NLRP3 inflammasome-activating stimuli in each cell type may determine the fate of cells through distinct patterns of GSDMD trafficking and cleavage. The regulatory mechanisms underlying pyroptosis and PANoptosis pathways are beyond the scope of this review. For information on the regulation of cell death during NLRP3 inflammasome activation, readers are directed to recent extensive and focused reviews in this area.22,23,24,25,28
Signal 1: priming and licensing
It is generally thought that the priming process of NLRP3 inflammasome activation involves the transcriptional induction of NLRP3, as well as pro-IL-1β and pro-IL-18. However, a growing body of evidence suggests that the priming step involves more than transcriptional activation of NLRP3 to license its rapid activation toward Signal 2. Although the mechanisms of priming and licensing are not yet clear, the licensing of the NLRP3 protein is now generally accepted to be required for sufficient induction of functional NLRP3 by PTM regulation and the protein–protein interactions that enable the efficient assembly of inflammasome complexes. Investigations into these areas are rapidly expanding and have recently been extensively reviewed.11,12
While pro-IL-18 is constitutively expressed in monocytes and epithelial cells, it can also be induced by lipopolysaccharide (LPS) (TLR4), CpG oligonucleotides (TLR9), and the Sendai virus.11,12,29 TLR signals, such as TLR4/LPS, are the best known stimuli for transcriptional activation of pro-IL-1β, pro-IL-18, and NLRP3.11,12,29 Cytokines not involved in the inflammasome, such as TNF-α or type I interferon (IFN), enhance the priming process of inflammasome activation by influencing the transcription of inflammasome components.11,29,30 The mechanisms through which non-inflammasome proinflammatory cytokines contribute to priming/licensing remain to be characterized.
NF-κB signaling is essential for the transcriptional activation of priming responses to TLR and cytokine stimulation.11,29,30,31 Sphingosine 1-phosphate (S1P)/S1P receptor (S1PR) signaling is involved in the upregulation of NLRP3 priming through elevation of the gene expression of NLRP3 inflammasome components.32 Recently, the inhibition of bromodomain-containing protein 4 (BRD4), in the bromodomain and extraterminal domain (BET) family member of epigenetic readers, was reported to activate NF-κB signaling and enhance NLRP3 expression at the transcriptional level.33 Upon TLR/IL-1R signaling, TRAF6 is involved in the priming step of NLRP3 inflammasome activation through both transcriptional and nontranscriptional regulation of NLRP3.34 In addition, the TLR downstream adapter MyD88 and the IL-1 receptor-associated kinases IRAK-1 and IRAK-4 play crucial roles in the rapid activation of NLRP3 priming, presumably through PTM.35,36,37 These events lead to acute activation of caspase-1, regardless of new protein synthesis, thus suggesting that PTMs are crucial for the priming and licensing of the NLRP3 inflammasome.11,29,30,31
In addition, several reports show the critical role of fas-associated protein with death domain (FADD) and caspase-8 during the priming process of the NLRP3 inflammasome.38,39 FADD-caspase-8 plays an essential function in both canonical and noncanonical NLRP3 inflammasome activation through NF-κB–dependent transcription of pro-IL-1β and posttranslational activation of the NLRP3 inflammasome.38 Furthermore, FADD/caspase-8 scaffolding induces receptor-interacting serine/threonine-protein kinase (RIPK) 3/mixed lineage kinase domain-like pseudokinase (MLKL) activation required for both Signal 1 and 2 upon TLR3 stimulation.39 Together, these diverse intracellular signaling molecules, most are TLR-dependent, can prime the NLRP3 inflammasome at the transcriptional and posttranslational levels. Further work is needed to determine the precise mechanism by which different signaling molecules/pathways cooperate and cross talk during the transcriptional and posttranslational regulation of the priming/licensing process of canonical and noncanonical activation of the NLRP3 inflammasome.
In the next section, we discuss recent advances in our knowledge of NLRP3 interactions with molecular partners as well as several types of PTMs for NLRP3 priming/licensing, and Signal 2 is subsequently described.
PTMs of NLRP3 and other components of the inflammasome complex
Ubiquitination
Ubiquitination and deubiquitination of NLRP3 and other inflammasome components are essential for the assembly of the inflammasome complex (Fig. 2).7 Ubiquitination of NLRP3 by several E3 ligases is generally thought to abrogate inflammasome activation. Autophagic degradation of the NLRP3 inflammasome is mediated through K63 polyubiquitination of NLRP3 and subsequent interaction with the autophagic adapter p62.40 The E3 ubiquitin ligases RNF125 and Cbl-b are essential for targeting NLRP3 for K63- and K48-linked ubiquitination, respectively, ultimately leading to proteasome-mediated degradation.41

Posttranslational modifications (PTMs) in NLRP3 inflammasome components. The NLRP3 inflammasome is regulated by various PTMs, including ubiquitination/deubiquitination, phosphorylation/dephosphorylation, acetylation/deacetylation, SUMOylation, and nitrosylation, in different domains of NLRP3 (PYD, NACHT, and LRR). Activators and inhibitors of the NLRP3 inflammasome are represented by red and blue ovals, and phosphorylation sites of NLRP3 components from human and mouse species are indicated with (h) and (m), respectively.
The E3 ligase TRIM31 binds and ubiquitinates NLRP3 for protein polyubiquitination and proteasomal degradation.42 In addition, dopamine-mediated inhibition of NLRP3 inflammasome activation is mediated through the E3 ligase MARCH7-mediated ubiquitination and degradation of the NLRP3 protein.43 Cullin1, the key component of the Skp1-Cullin1-F-box E3 ligase, interacts with NLRP3 and promotes the K63‐linked ubiquitination of NLRP3, in which K689 acts as a significant ubiquitin acceptor site in NLRP3.44 This ubiquitination of NLRP3 does not lead to its degradation but is crucial for the prevention of NLRP3 activation.44
Furthermore, Ariadne homolog 2 (ARIH2), the E3 ligase for binding and ubiquitinating NLRP3 at K48 and K63, is a negative regulator of NLRP3 priming activity in macrophages.45 However, another study showed that the E3 ubiquitin ligase Pellino2 is essential for NLRP3 ubiquitination during the priming step, thereby further promoting activation of the NLRP3 inflammasome.46 The precise mechanisms through which multiple E3 ligases and different sites in NLRP3 cooperatively or separately control NLRP3 inflammasome licensing remain to be fully determined.
Deubiquitination
Deubiquitination of NLRP3, another key process for the licensing of NLRP3 inflammasome activation, depends on TLR4 and mitochondrial ROS (mtROS) generation.11,12,47 Priming signals triggered through TLR4 or TLR2 stimulation leads to the induction of Abraxas brother protein 1 (ABRO1), a subunit of the BRCA2-containing complex subunit 3 (BRCC3, human BRCC36) deubiquitinase complex, thereby deubiquitinating the LRR domain of NLRP3 upon inflammasome activation.47,48,49 Notably, vitamin D receptor (VDR) appears to be a negative regulator of NLRP3 oligomerization and activation by blocking BRCC3-mediated deubiquitination of NLRP3.50 In the responses to nigericin and calcium pyrophosphate dihydrate (CPPD) crystals, both ubiquitin-specific peptidase (USP) 7 and USP47 function as essential deubiquitinating enzymes (DUBs) for the removal of ubiquitin from NLRP3 and inhibition of ASC speck formation. Both USP7 and USP47 have functional redundancy in deubiquitinating NLRP3, although ubiquitin linked to NLRP3 at K48 and K63 is not removed by USP7/USP47 upon inflammasome activation.51 Further studies to identify the precise targets for deubiquitination by USP7/USP47 in the context of licensing NLRP3 inflammasome activation are warranted.
Under conditions of cytosolic DNA stimulation and herpes simplex virus type 1 (HSV-1) infection, stimulator of interferon genes (STING) promotes NLRP3 inflammasome activation through recruitment and interaction with NLRP3 via attenuation of K48- and K63-linked polyubiquitination.52 Given that aberrant activation of the cGAS-cyclic GMP-AMP (cGAMP)-STING pathway leads to inflammation, senescence, and cancer,53,54 it is difficult to clarify the potential detrimental effects of the cGAS-STING pathway on NLRP3 licensing for inflammasome assembly. However, studies are beginning to identify the substrate-targeting mechanisms by which E1/E2/E3 ligases and DUBs regulate activation of the NLRP3 inflammasome. Given that current studies encompass only small numbers of E3s and DUBs in the regulation of NLRP3 priming, future structural and biochemical studies are warranted to reveal the functions and mechanisms of other currently uncharacterized ubiquitin ligases/DUBs in terms of NLRP3 licensing.
Phosphorylation and dephosphorylation
Accumulating evidence suggests that the control of phosphorylation/dephosphorylation of inflammasome components is required for the priming/licensing of NLRP3 inflammasome activation (Fig. 2). In the early phase of priming, c-Jun N-terminal kinase 1 (JNK1)-dependent phosphorylation of NLRP3 at human Ser198 (mouse Ser194) is critical for NLRP3 deubiquitination and self-association, which drive inflammasome activation.55 In addition, the NLRP3 inflammasome is phosphorylated at human Ser295 (mouse Ser293), and the role of this phosphorylation in NLRP3 activation is controversial. During priming, NLRP3 is phosphorylated at human Ser295 (mouse Ser293) by protein kinase D (PKD), an effector of diacylglycerol (DAG), at the Golgi apparatus, which is adjacent to mitochondria-associated ER membranes (MAMs), where NLRP3 and ASC assemble to form the inflammasome complex.56 However, another study showed that Ser295 phosphorylation by protein kinase A (PKA) has an inhibitory effect through suppression of the ATPase activity of the NLRP3 NACHT domain, which is critical for NLRP3 oligomerization.57 The molecular details of NLRP3 Ser295 phosphorylation are poorly understood. Further studies are required to explore the mechanisms underlying the dual functions involving the same phosphorylation site.
Interestingly, Bruton’s tyrosine kinase (Btk) may play dual opposite roles in the priming phase of NLRP3 inflammasome activation. A recent report showed that Btk promotes NLRP3 inflammasome activation through phosphorylation of ASC at Tyr144 and physical interaction with NLRP3 and ASC, thereby contributing to postischemic inflammation after stroke.58 However, another study reported that Btk interacts with NLRP3 during priming and functions as a physiological inhibitor of NLRP3 phosphorylation and oligomerization.59 The inhibitory function of Btk is mediated through the maintenance of NLRP3 phosphorylation at human Ser5 (mouse Ser3),59 which is in the PYD interaction interface.9 NLRP3 Ser5 phosphorylation is critical for suppression of NLRP3 inflammasome activation through interference with charge–charge interactions between PYD domains.9 Mechanistically, Btk suppresses protein phosphatase 2 A (PP2A), which dephosphorylates Ser5 of the PYD in NLRP3, thus blocking aberrant activation of the NLRP3 inflammasome and the related inflammation.9,59 These data may explain the observation that Btk-deficient macrophages or monocytes from patients with X-linked agammaglobulinemia (XLA) with Btk mutation have dysregulated NLRP3 inflammasome activity.59
Another transmembrane tyrosine kinase, EphA2, physically interacts with NLRP3 and induces its phosphorylation at Tyr132, thus inhibiting NLRP3 inflammasome assembly in murine airway epithelial cells during reovirus infection.60 In addition, EphA2-mediated NLRP3 phosphorylation is crucial for amelioration of pathological asthmatic exacerbation in a mouse model of asthma.60 The enhanced tyrosine phosphorylation of NLRP3 at Tyr861 negatively regulates inflammasome activation through activation of autophagy for NLRP3 degradation.61 Protein tyrosine phosphatase nonreceptor 22 (PTPN22) targets and dephosphorylates NLRP3 at tyrosine residue Tyr861, thereby activating the NLRP3 inflammasome and IL-1 secretion.61,62 Furthermore, phosphatase and tensin homolog (PTEN) in myeloid cells interacts with and dephosphorylates NLRP3 at Tyr32, thereby promoting assembly of the NLRP3 inflammasome.63 Given that PTEN-NLRP3 functions in enhancing chemotherapy sensitivity and antitumor responses,63 myeloid-specific NLRP3 regulation of phosphorylation may be associated with chemotherapeutic responsiveness in the tumor immune microenvironment.63 As apparent from these studies, NLRP3 activation by phosphorylation/dephosphorylation is regulated in a multilayered manner. Future studies are warranted to clarify how multiple tyrosine kinases and phosphatases orchestrate the fine-tuning of NLRP3 inflammasome activation and their functional consequences in a variety of human diseases.
Other PTMs: acetylation/deacetylation, SUMOylation, and nitrosylation
Several types of PTMs, including acetylation/deacetylation, SUMOylation, and nitrosylation, are also involved in the regulation of NLRP3 inflammasome activation (Fig. 2). Previous studies showed that nitric oxide (NO) and S-nitrosylation of NLRP3 inhibit inflammasome assembly and IL-1 production during mycobacterial infection and LPS stimulation.64,65,66 However, whether nitrosylation is required for NLRP3 priming or feedback regulation after NLRP3 inflammasome activation remains to be fully characterized. In addition, NLRP3 SUMOylation plays either a positive or negative role in NLRP3 inflammasome activation depending on the context. NLRP3 SUMOylation by the small ubiquitin-like modifier (SUMO) E3 ligase MAPL (MUL1) restrains activation of the NLRP3 inflammasome,67 suggesting that SUMO conjugation of NLRP3 at multiple sites is a fundamental negative regulator of innate immune signaling. However, another study showed that SUMOylation of NLRP3 at K204 by SUMO1 facilitates ASC oligomerization and NLRP3 inflammasome activation.68 Additional studies are needed to understand how multiple PTM pathways are selected and coordinated for the priming/licensing of NLRP3 and its oligomerization.
A recent study showed that SIRT2-mediated deacetylation of NLRP3 ameliorates NLRP3 inflammasome activation, thus contributing to protection against aging-associated inflammation and insulin resistance.69 However, it is unclear whether multiple Lys residues of NLRP3 are acetylated or deacetylated under basal conditions and which upstream signals regulate the acetylation of NLRP3 at certain phases of inflammasome activation. In addition, future studies should investigate whether a variety of PTMs play synergistic or redundant roles in NLRP3 priming/licensing. During NLRP3 inflammasome activation, various types of PTMs, including phosphorylation, ubiquitination, and SUMOylation, might be activated sequentially or simultaneously in a context-specific manner. Whether different types of PTMs are activated in an interlinked, overlapping, or independent manner remains a major theme to be explored in terms of the NLRP3 inflammasome licensing step.
NLRP3 interactions with molecular partners
NLRP3 and NEK7 interaction
The mitotic serine and threonine kinase NEK7, a member of the mammalian never in mitosis A (NIMA)-related kinase (NEK) protein family, is a key interacting partner of NLRP3, leading to NLRP3 oligomerization along with ASC speck formation and maturation of IL-1β and IL-18 in response to NLRP3 inflammasome activating signals involving K+ efflux and ROS.70,71,72 NEK7 is also transcriptionally activated by RELA through direct targeting and activation of NLRP3 promoter activity.73 There are two major isoforms of human NLRP3 produced by alternative splicing, i.e., the full-length variant and a variant that lacks exon 5 and cannot interact with NEK7, resulting in the attenuation of NLRP3 inflammasome activation.74 A recent structural modeling study using cryoelectron microscopy highlighted the molecular mechanism of NEK7–NLRP3 interactions—NEK7 was shown to bridge adjacent NLRP3 subunits and facilitate NLRP3 inflammasome oligomerization.10
Whether NEK7 is absolutely required for NLRP3 oligomerization and further facilitation of inflammasome assembly remains an unknown. A recent preprint suggested NEK7-independent but TGF-β-activated kinase-1 (TAK1)-dependent PTM regulation of NLRP3 priming.75 Further understanding of NEK7-dependent and NEK7-independent priming pathways and how they work together or separately will provide more precise insights into the molecular mechanisms involved in licensing NLRP3 inflammasome activation.
NLRP3 interactions with other proteins
Studies showed have shown that thioredoxin-interacting protein (TXNIP) binds to NLRP3 in a redox-dependent manner and plays a critical role in the activation of the NLRP3 inflammasome.76,77,78 In addition, mitochondrial antiviral signaling (MAVS) protein plays a key role in the optimal activity of the NLRP3 inflammasome through binding with and recruitment of NLRP3 in response to RNA viruses and synthetic RNA polyinosinic–polycytidylic acid.79,80,81 During infection with RNA viruses, including influenza virus and encephalomyocarditis virus, the outer mitochondrial membrane protein mitofusin 2 (MFN2) physically interacts with NLRP3 and further induces the secretion of IL-1β through NLRP3 inflammasome activation.82
During cellular stress responses, stress granules are critical to cell survival. A recent study showed that the stress granule protein DEAD-box helicase 3 X-linked (DDX3X) interacts with NLRP3 to promote inflammasome activation. The assembly of stress granules and NLRP3 inflammasome pathways compete for DDX3X in innate immune responses and the determination of cell fate under stress conditions.83 In NLRP3 inflammasome activation, the stress granule protein DDX3X interacts with NLRP3, leading to the formation of pyroptotic ASC specks.83 The rheostat-like function of DDX3X between activation of the NLRP3 inflammasome and stress granule assembly may contribute to the determination of live-or-die cell fate decisions in response to stressors.83
More recent studies have shown that microtubule affinity regulating kinase 4 (MARK4) physically interacts with NLRP3 and drives its localization to the microtubule-organizing center, contributing to the formation of the NLRP3 inflammasome complex.84 It was also reported that E74-like ETS transcription factor 3 (ELF3) increases MARK4 expression upon high glucose-triggered NLRP3 inflammasome activation in vascular endothelial cells.85 In addition, there are several other molecular partners (ZBP1, caspase-8/FADD, etc.) that interact with NLRP3 in the context of pyroptosis or priming signaling. These molecules have been discussed in the relevant section in this review. Further studies are warranted to clarify which protein partners are recruited to NLRP3 to activate the inflammasome complex further and how each can be regulated in the respective context.
Spatiotemporal activation of the NLRP3 inflammasome complex
MAMs and the microtubule-organizing center (MTOC)
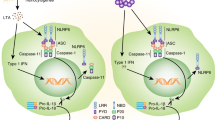
NLRP3 resides in the endoplasmic reticulum (ER) and cytosol, and upon activation by diverse stimuli, NLRP3 in the ER localizes adjacent to ASCs in mitochondria (Fig. 3).86,87 Indeed, the NLRP3 inflammasome complex can be assembled at highly specialized contact sites between the ER and mitochondria known as MAMs.86 Mitochondrial ASC apposition to ER NLRP3 is mediated through acetylated α-tubulin via dynein-dependent mitochondrial transport to the ER.87 The localization of NLRP3 to MAMs/mitochondria may contribute to the immediate recognition of and response to mitochondrial damage, mitochondrial DNA (mtDNA) translocation, and cardiolipin.86

Spatiotemporal regulation of the NLRP3 inflammasome complex. NLRP3, which is present in the endoplasmic reticulum (ER), comes in close proximity to apoptosis-associated speck-like protein containing a CARD (ASC) in mitochondria upon induction by various stimuli. It is known that NLRP3 and ASC are assembled in mitochondria-associated ER membranes (MAMs). NLRP3 binds to microtubule affinity regulating kinase 4 (MARK4) and is translocated to microtubule-organizing center (MTOC). When NLRP3 reaches the MTOC, NIMA related kinase 7 (NEK7) binds with NLRP3, and the inflammasome is assembled. In addition, NLRP3 is recruited to the dispersed trans-Golgi network (dTGN) through phosphatidylinositol-4-phosphate (PIP4) by diverse stimuli. In addition, translocation of the SREBP cleavage-activating protein (SCAP)-sterol regulatory element-binding protein 2 (SREBP2) complex from the ER to the Golgi is important for the activation of NLRP3. Diacylglycerol (DAG) in the Golgi recruits protein kinase D (PKD), which is involved in the assembly and activation of NLRP3
Several recent studies have revealed the molecular mechanisms by which microtubules provide the optimal sites for the activation of the NLRP3 inflammasome. The binding between MARK4 and NLRP3 results in the translocation of NLRP3 into the MTOC, where inflammasome speck formation and assembly are activated.84 Importantly, the localization of NLRP3 to the MTOC leads to its interaction with the centrosome-localized mitotic kinase NEK7 to facilitate NLRP3 inflammasome assembly.10 Furthermore, a recent study showed that the dynein adapter histone deacetylase 6 (HDAC6) is critical for microtubule transport, inflammasome assembly, and autophagosomal degradation of aggresomes at the MTOC, the centrosome.88 Studies are beginning to reveal where when, and how the NLRP3 inflammasome complex is assembled, depending on the context. A deeper understanding of the mechanisms underlying the proximity to several subcellular compartments will contribute to the identification of potential therapeutic targets for NLRP3-related disorders.
Trans-Golgi disassembly
Recent studies have highlighted that the Golgi apparatus and its lipid mediators play essential roles in the aggregation of NLRP3 and the activation of NLRP3 inflammasome assembly.88,89,90 Imaging and biochemical analyses showed that NLRP3 exposed to certain stimuli induces the disassembly of the trans-Golgi network (TGN) into the dispersed TGN (dTGN) and that NLRP3 is recruited to the dTGN via the conserved polybasic region of NLRP3. Indeed, the phospholipid phosphatidylinositol-4-phosphate (PIP4) is exposed on dTGN and recruits and interacts with NLRP3, thus resulting in the formation of multiple NLRP3 puncta and caspase-1 activation. Notably, K+ efflux-independent stimuli (imiquimod) the high activation of NLRP3-dTGN, which leads to aggregation and activation of the NLRP3 inflammasome.89 In addition, NLRP3 inflammasome activation is dependent on the ER-to-Golgi translocation of sterol regulatory element-binding protein (SREBP) 2 and SREBP cleavage-activating protein (SCAP), which form a ternary complex with NLRP3.90 How then does NLRP3 inflammasome assembly occur at the intracellular level in both MAMs and dTGN? Another recent study reconciled this issue by demonstrating that NLRP3 inflammasome stimuli induce the localization of MAMs adjacent to Golgi membranes.56 This interorganelle communication depends on the recruitment of PKD to the sites of DAG at the Golgi, thereby facilitating NLRP3 oligomerization and assembly of the active inflammasome.56
Taken together, these data suggest that the spatial interrelations among the ER-mitochondria-Golgi apparatus are closely related to NLRP3 inflammasome activation (Fig. 3). Given that K+ efflux-dependent and K+ efflux-independent stimuli converge for Golgi disassembly,89 future studies should consider simultaneous measurement of NLRP3–ASC aggregation in different organelles and develop inclusive approaches that address not only signaling and cytokine production but also organellar contacts and tethering to exert crucial control over spatiotemporal activation of the NLRP3 inflammasome. Moreover, recent findings suggest that ASC specks can be secreted or found ex vivo.91,92 Further studies are needed to clarify how spatiotemporal coordination among intracellular organelles regulates the ASC secretion pathway and to decipher the complex interrelationships between ASC and other inflammasome components that affect the distinct physiological and pathological roles of secreted ASC in terms of NLRP3 inflammasome activation.
Signal 2: activation of the NLRP3 inflammasome
A variety of stimuli that perturb intracellular ion homeostasis, i.e., K+ efflux, intracellular Ca2+ flux, and Cl− efflux, can activate the assembly of the NLRP3 inflammasome complex and release mature IL-1β. Other pathways, including mitochondrial dysfunction, lysosomal destabilization, and metabolic alteration pathways, also contribute to NLRP3 inflammasome activation. Recent studies have indicated that several other pathways, including the complement, protein kinase R (PKR), purine receptor signaling, necroptosis, and ZBP1 pathways, are required for Signal 2 of NLRP3 inflammasome activation. In this section, we discuss recent advances in the understanding of the pathways and mechanisms by which Signal 2 triggers activation of the NLRP3 inflammasome complex.
Potassium efflux
K+ efflux has emerged as a common step in the activation of the NLRP3 inflammasome induced by multiple NLRP3 agonists, including nigericin, a well-known K+/H+ ionophore, and extracellular ATP.72,93,94,95 The channel-forming glycoprotein pannexin-1 hemichannels are known to be involved in inflammasome activation through membrane permeability and ATP release during apoptosis.96,97 It was thought that ATP gating of the P2X7 receptor (P2X7R), an ion channel in the purinergic receptor family, promotes IL-1β maturation via K+ efflux.95,98 A recent study showed that tandem pore domains in weak inward rectifying K+ channel 2 (TWIK2) mediate K+ efflux in cooperation with P2X7R-mediated influx of Ca2+ and Na+, leading to ATP-mediated activation of the NLRP3 inflammasome in macrophages.99 Notably, neither nigericin- nor imiquimod-induced NLRP3 inflammasome activation was regulated in TWIK2-deficient macrophages.99
Indeed, numerous signals, including the complement cascade component membrane attack complex (MAC)100,101 and particulate matter signals,95 converge for the induction of K+ efflux to promote NLRP3 inflammasome activation. In addition, Mycobacterium tuberculosis (Mtb) infection results in ESX-1-mediated plasma membrane damage responses that cause K+ efflux, leading to the activation of caspase-1/NLRP3/GSDMD-mediated pyroptosis in human monocytes and macrophages.102 Phospholipids, platelet-activating factor (PAF), and PAF-like lipids can activate the canonical NLRP3 inflammasome through mechanisms involving K+ efflux and Ca2+ influx in a manner independent of the PAF receptor.103 Although K+ efflux is required for the NEK7-NLRP3 association,72 the mechanisms underlying K+ efflux-mediated NLRP3 inflammasome activation are not fully understood. In this regard, further studies are warranted to elucidate how K+ efflux triggered by multiple signals activates the assembly of the NLRP3 inflammasome complex.
Calcium signaling and calcium-sensing receptor (CaSR)
Intracellular Ca2+ flux plays an essential role in the assembly and activation of the NLRP3 inflammasome induced by multiple stimuli.104,105 Studies showed that CaSR signaling promotes NLRP3 inflammasome assembly through intracellular Ca2+ flux and leads to a decrease in the cellular level of cyclic AMP (cAMP), which is an inhibitory signal for inflammasome activation.105 Recent studies have shown that platelets are able to boost NLRP3 inflammasome activation by triggering CaSRs in human macrophages, suggesting the importance of Ca2+ signaling in the activation of the inflammasome linked to cell–cell interactions.106 Indeed, intracellular Ca2+ mobilization seems to be involves in coordinated action with several other signaling pathways to activate the NLRP3 inflammasome complex. Several studies have shown a cooperative relationship between K+ efflux and Ca2+ flux, contributing to the idea of a greater influence on mtROS generation.99,107 However, other studies have shown that K+ efflux-mediated NLRP3 inflammasome signaling is not associated with cytosolic Ca2+ flux.108,109 In addition, Ca2+ flux-mediated calpain activation is required for caspase-1 activation, whereas K+ efflux inhibits calpain.109 Therefore, the cross talk between Ca2+ flux and K+ efflux pathway components is complex, regulated in a context-dependent manner, and remains to be fully elucidated.
Recent studies have revealed relationships of Ca2+ flux and oxidative stress in the activation of the NLRP3 inflammasome. Particulate matter-mediated oxidative stress can trigger activation of the NLRP3 inflammasome through intracellular Ca2+ mobilization.110 In this case, transient receptor potential melastatin 2 (TRPM2), a calcium-permeable cation channel, mediates ROS-associated NLRP3 inflammasome activation.110,111 Apolipoprotein C3 (ApoC3)-triggered alternative NLRP3 inflammasome activation involves intracellular Ca2+ flux and the production of ROS in human monocytes.112
Ca2+ flux-triggered NLRP3 inflammasome activation is closely associated with the pathogenesis of several human autoimmune diseases. Extracellular ADP, a danger signal, is extensively released from injured colonic tissue in inflammatory bowel disease. ADP/P2Y1 receptor signaling activates the NLRP3 inflammasome through intracellular Ca2+ mobilization, thereby aggravating intestinal inflammation.113 In addition, the increased extracellular Ca2+ and phosphate induced by the formation of fetuin-A-based calciprotein particles triggers NLRP3 inflammasome activation through CaSR-mediated signaling, leading to pathological inflammation in inflammatory arthritis.114 Taken together, these data suggest that Ca2+ flux-induced signaling depends on another molecule/pathway to integrate sufficient signals for NLRP3 inflammasome activation. It will be important to explore further the molecular mechanisms by which signals selectively and cooperatively impact the ability of Ca2+ flux to activate the NLRP3 inflammasome.
Chloride efflux
The decreased extracellular Cl− level, which often acts in cooperation with other signals for NLRP3 activation, promotes activation of caspase-1, leading to mature IL-1β secretion.12,115 During inflammasome activation, the chloride intracellular channel (CLIC) proteins CLIC1 and CLIC4 are translocated to the plasma membrane where they mediate Cl− efflux.116 In addition, CLICs function as proximal and upstream signals for priming by synthesizing IL-1β and as downstream signals of the K+ efflux–mtROS axis for NLRP3 inflammasome activation.115,116 However, another report indicated that K+ and Cl− efflux is required for the oligomerization of NLRP3 and ASC, respectively,117 suggesting that both K+ and Cl− efflux pathways function separately in the activation of the NLRP3 inflammasome.
Cystic fibrosis is caused by genetic mutations of cystic fibrosis transmembrane conductance regulator (CFTR), which is an ion channel involved in the transport of chloride and bicarbonate with hyperabsorption of sodium due to a dysregulated epithelial sodium channel (ENaC).118,119 Although the mechanisms underlying excessive inflammation in patients with cystic fibrosis remain a matter of some debate,118 it is suggested that ENaC-mediated Na+ influx, accompanied by defective Cl− efflux, may contribute to exaggerated inflammatory responses and NLRP3 inflammasome activation in this disease.120 Given the role of Cl− efflux in NLRP3 inflammasome induction, a recent study revealed a new mechanism of action of the FDA-approved drug ticagrelor, which is used for the treatment of coronary artery disease.121 Ticagrelor functions by attenuating the oligomerization of ASCs by blocking Cl− efflux via the degradation of CLICs and inhibition of their translocation to the plasma membrane.121 However, another recent study showed that myocardial protection by ticagrelor is mediated through its antiplatelet properties and not an additive effect involving the inhibition of the NLRP3 inflammasome.122 Understanding the mechanisms by which Cl− efflux controls NLRP3 inflammasome activation may facilitate the discovery of novel agents or drugs suitable for repurposing to increase clinical benefit for patients with one of a variety of NLRP3-related diseases.
Mitochondrial dysfunction, oxidative stress, mtDNA, and mitochondrial dynamics
Beyond their role in energy metabolism, mitochondria are emerging as central organelles in the activation of the NLRP3 inflammasome. Mitochondria can play multifaceted roles by serving as docking sites for assembly of the NLRP3 inflammasome, release of danger signals, generation of mtROS, etc.12,123,124,125 Persistent damage and dysfunction of mitochondria, often induced by a wide range of danger signals, are key upstream processes for activation of the NLRP3 inflammasome.1,12,126,127,128 Mitochondrial dysfunction provides the key activation mechanism for the NLRP3 inflammasome complex through excessive generation of mtROS, cytosolic translocation of mtDNA, or relocation of mitochondria to the proximity of NLRP3 by the induction of α-tubulin acetylation.12,17,128,129,130 Increased mitochondrial stress often lead to detrimental consequences that contribute to the pathogenesis of metabolic diseases.128,130 Although the supposition remains controversial, mitochondrial dysfunction has been suggested to be closely linked with other signaling pathways, including K+ efflux or Cl− efflux pathways, for activating the NLRP3 inflammasome.12,17,116,128,130
Several small molecules that target mitochondria lead to the production of mtROS to further activate the NLRP3 inflammasome complex. For example, imiquimod, a small-molecule ligand activates TLR7, and the related compound, CL097, activates the NLRP3 inflammasome through the production of mtROS, but K+ efflux is not involved.131 In addition, oxidation of phosphatidylcholine upon cellular stress and damage activates the NLRP3 inflammasome in macrophages through mtROS downstream of intracellular Ca2+ signaling.132 A recent study of neuroinflammation in a model of Parkinson’s disease showed that the Fyn kinase induces PKCδ-dependent NF-κB-p65 activation and inflammasome priming. This activation and priming facilitate α-synuclein uptake by microglia, contributing to the generation of mtROS and leading to exaggerated neuroinflammation and progression of Parkinson’s disease.133 Human respiratory syncytial virus (RSV) infection triggers macrophage cell lysis through NLRP3 inflammasome-mediated pyroptosis through ROS production134 Moreover, supraphysiological testosterone levels trigger vascular dysfunction through induction of mtROS generation, enhancing NLRP3 inflammasome activation and leading to increased cardiovascular risk.135 In summary, multiple danger or microbial signals are involved in triggering mtROS generation to further activate the NLRP3 inflammasome.
Indeed, NLRP3 signaling activators result in mitochondrial destabilization and the release of mitochondria-derived molecules, such as mtDNA and cardiolipin, to further activate the NLRP3 inflammasome complex.124,130,136 Circulating mitochondrial DAMPs, including formyl peptides and mtDNA, can be produced upon cellular injury, and they induce systemic inflammation.137 More recently, it was shown that mtDNA synthesis following TLR signaling can lead to the formation of oxidized mtDNA fragments that lead to inflammasome activation, indicating that these fragments are critical components of the NLRP3 machinery.138,139 Severe fever with thrombocytopenia syndrome (SFTS) virus (SFTSV) infection triggers the activation of BCL2 antagonist/killer 1 (BAK)/BCL2-associated X (BAX) signaling, leading to mitochondrial dysfunction and release of oxidized mtDNA that activates the NLRP3 inflammasome.140 Furthermore, the mitochondrial phospholipid cardiolipin appears to be a signaling platform for autophagy, apoptosis, and inflammasome activation.141 Cardiolipin binds directly to the LRR domain of NLRP3 and provides an activating signal for NLRP3 inflammasome complex assembly and activation.123,125 A deeper, context-dependent understanding of the roles of mtROS and mtDNA and the interaction of NLRP3 components with mitochondrial molecules is required to appreciate inflammasome formation and understand the pathophysiological effects of the inflammasome.
Mitochondrial dynamic proteins may play key roles in inflammasome activation, which is related to chronic inflammation in type 2 diabetes mellitus (T2DM).142 Under conditions of nutrient excess, the expression of inflammasome-related genes and inflammatory responses are increased in cybrid cells harboring mitochondrial haplogroup B4, which is the type 2 diabetes-associated haplogroup in the Chinese population. Notably, inflammasome-related inflammatory responses are attenuated by inhibition of Drp1 and overexpression of fusion proteins, suggesting that inflammasome activation is regulated by components involved in mitochondrial dynamics.142 However, as mentioned above, MFN2 interacts with NLRP3 and activates the inflammasome during RNA virus infection.82 Thus, the issues of mitochondrial dynamics and inflammasome activation remain to be addressed before we can gain a deeper understanding of the diverse effects of each component in mitochondrial dynamics on inflammasome regulation.
Lysosomal disruption
Studies showed that particulate matter, including uric acid and cholesterol crystals, alum, silica, and asbestos, are canonical stimulators of NLRP3 inflammasome activation through induction of lysosomal damage and rupture, thereby releasing multiple cathepsins into the cytoplasm.143,144 Recent studies have shown that carbon-based nanomaterials, such as multiwalled carbon nanotubes, can activate the NLRP3 inflammasome through lysosomal destabilization and release of cathepsin B.145 Nicotine also induces lysosomal membrane permeability in endothelial cells and triggers the lysosomal release of cathepsin B, thus enhancing NLRP3 inflammasome activation.146 Recent studies have suggested a more generalized function of cathepsin B in the activation of the NLRP3 inflammasome through a direct interaction with NLRP3 at the ER upon stimulation with multiple types of NLRP3 activators, including ATP and nigericin, as well as particulate matter.147
Although the interwoven molecular pathways are not well understood, lysosomal damage and rupture may require another signal for full activation of the NLRP3 inflammasome. For example, Leu-Leu-O-methyl ester (LLME), a soluble lysosomotropic agent, induces NLRP3 inflammasome activation through the combined effects of lysosome membrane permeabilization and increased K+ efflux.148 In addition, lysophosphatidylcholine (LPC), a major lipid component in the plasma membrane, activates foam cell formation and triggers NLRP3 inflammasome activation in human endothelial cells and monocytes upon lysosomal damage and K+ efflux.149 Recent studies have also shown that plasma membrane damage is a key upstream event for lysosomal damage-associated NLRP3 inflammasome activation.102 During Candida albicans infection, the expansion of phagosomes through lysosome recruitment is needed to prevent NLRP3 inflammasome activation and host cell death.150 However, phagosomal rupture and/or lysosomal damage triggers NLRP3 inflammasome activation at least partly through plasma membrane damage.102,150 Future studies should examine the detailed molecular mechanisms underlying the cross talk between molecules involved in plasma damage and lysosomal destabilization-associated NLRP3 inflammasome activation and pyroptosis.
Complement system and PKR pathway
There is accumulating evidence indicating that a variety of elements in innate immune responses are essential activators of the NLRP3 inflammasome. For example, the complement system is essentially involved in the activation of inflammasome pathways in the context of autoimmune and inflammatory responses. As mentioned above, the complement cascade component MAC can trigger NLRP3 inflammasome activation and pathological inflammation.100,101 Ischemia-reperfusion injury (IRI) results in immunoglobulin M (IgM)-dependent complement system activation that induces NLRP3 inflammasome assembly in endothelial cells.151 The internalization of MAC in IFN-γ-primed human endothelial cells causes NLRP3 translocation into endosomes and leads to endosomal NF-κB-inducing kinase (NIK)-dependent inflammasome assembly, resulting in complement-associated pathologies.152 Several studies have revealed that the C5a/C5aR pathway promotes activation of the NLRP3 inflammasome through amplification of dsRNA-dependent PKR expression in macrophages, suggesting that PKR is an important NLRP3-activating factor.153 In addition, the C5a/C5aR2 axis-dependent induction of HMGB1 contributes to pathological damage and renal inflammation through upregulation of NLRP3 inflammasome activation in macrophages.154 Taken together, these studies suggest a molecular link is established between the complement system and the NLRP3 inflammasome in a multilayered and complex way to potentiate inflammatory pathology in a variety of NLRP3-associated disorders. Further studies are needed to determine the precise mechanisms underlying the interrelationship between complement system components, PKR, and inflammasome activation.
Purine receptor signaling
Adenosine and ATP receptors are involved in a variety of metabolic and degenerative diseases through inflammasome activation.155 P2X7R, a distinct ligand-gated ion channel, is recognized as a strong activating signal for NLRP3 inflammasome assembly and secretion of IL-1β. The P2X7R-cathepsin pathway contributes to pathological inflammation in a variety of autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and inflammatory bowel disease (IBD).98 P2Y14 receptor (P2Y14R) participates in the induction of caspase-1-mediated pyroptosis through inhibition of adenylyl cyclase and suppression of cAMP/NLRP3 signaling, thereby contributing to exacerbation of inflammation in acute gouty arthritis and pyroptosis-related diseases.156 In addition, extracellular ADP triggers NF-κB signaling and NLRP3 inflammasome activation to enhance microglial inflammation through the P2Y12 receptor, a metabotropic P2YR expressed in microglia.157 Collectively, these findings warrant a more comprehensive assessment based on purine receptor signaling-mediated inflammasome modulation to explore their clinical therapeutic efficacy in various NLRP3-associated diseases.
Necroptotic signaling and ZBP1
Emerging data suggest a close relation between the NLRP3 inflammasome and RIPK1/3-mediated necroptosis pathways. Necroptotic signaling mediated by RIPK1, RIPK3, and MLKL activates the NLRP3 inflammasome to enhance IL-1β, suggesting that this cell death pathway is closely associated with NLRP3 inflammasome activation and the pathogenesis of heritable autoinflammatory diseases.158,159,160 RIPK1 kinase activity is generally related to PANoptosis (pyroptosis, apoptosis, and necroptosis). In TAK1-deficient macrophages, autocrine TNF signaling, without TLR priming, induces spontaneous RIPK1-dependent NLRP3 inflammasome activation and cell death.161 Orning et al. also reported RIPK1- and caspase-8–dependent cleavage of GSDMD, resulting in pyroptosis and the release of IL-1β and IL-18 by inhibiting TAK1–IκB kinase signaling with the Yersinia effector protein YopJ.162 Interestingly, TLR priming that mimics pathogen-mediated priming triggers RIPK1 kinase activity-independent PANoptosis and activation of the NLRP3 inflammasome in the absence of TAK1.163 Moreover, TAK1 inactivation leads to myeloid proliferation and severe systemic inflammation through the RIPK3-caspase-8 signaling axis in vivo.163 These results support the supposition that TLR priming during TAK1 deficiency bypasses the RIPK1 requirement, but not RIPK3 and caspase-8, which are needed to induce pyroptotic cell death and inflammation in macrophages. We are only beginning to understand the complex regulatory mechanisms between innate immunity, inflammatory cell death, and NLRP3 inflammasome activation. Further studies are needed to identify the potential factors and mechanisms to explain how the sum of these pathways determines the pathophysiological consequences during numerous inflammatory and infectious diseases.
Recent studies have indicated the impact of regulators of necroptosis on NLRP3 activation-related pathologies. For example, sirtuin 3, a major deacetylase involved in mitochondrial homeostasis, is required to control the expression of necroptosis-related RIPK1, RIPK3, and NLRP3, as well as to prevent mitochondrial injury and mtROS, thereby exerting a protective effect in diabetic cardiomyopathy.164 In addition, the RIPK3 inhibitor dabrafenib was shown to be beneficial for amelioration of renal fibrosis, the pathogenesis of which is associated with RIPK3-regulated NLRP3 inflammasome activation.165
As mentioned above, ZBP1 represents the key mediator of NLRP3 inflammasome-related cell death. During influenza virus infection, an innate immune sensor and the interferon-inducible protein ZBP1 can sense Z-RNA and trigger cell death through PANoptosis (pyroptosis, apoptosis, and necroptosis) through the multiprotein PANoptosome complex via formation of the ZBP1-NLRP3 inflammasome.24,166,167,168 The ZBP1 Zα2 domain is crucial for influenza A virus (IAV)-induced PANoptosis, NLRP3 inflammasome activation, and perinatal lethality, which are associated with hyperinflammation and epithelial damage.166,169 In addition, caspase-6 is required for ZBP1-mediated inflammasome activation by facilitating the binding of RIPK3 to ZBP1.170 Furthermore, IFN regulatory factor (IRF)1 is a transcriptional regulator of ZBP1 and promotes activation of the NLRP3 inflammasome and induces cell death during IAV infection.171 Further studies to elucidate the cellular and molecular mechanisms underlying ZBP1-mediated NLRP3 inflammasome activation and PANoptosis may enable the identification of new therapeutic agents useful for the termination of severe viral infections and the design of novel vaccines.
Dual regulatory mechanisms controlling the NLRP3 inflammasome
Immunometabolism (positive regulation)
The metabolic reprogramming of immune cells plays a critical role in the regulation of inflammatory responses and NLRP3 inflammasome activation (Table 1).20,172 Enhanced glycolysis coupled with increased succinate levels increases IL-1β expression by stabilizing HIF-1α in macrophages.173 Glycolysis-related activation of mitochondrial respiration and an increase in mtROS levels contribute to activation of the NLRP3 inflammasome and IL-1β secretion.174 In addition, pyruvate kinase isozyme M2 (PKM2)-mediated aerobic glycolysis drives inflammasome activation through phosphorylation of eukaryotic translation initiation factor 2-alpha kinase 2 (EIF2AK2)/PKR in macrophages175 In postburn responses with abnormal scar formation (keloid), NLRP3 inflammasome activation is correlated with glucose transporter 1 (GLUT1) expression and glycolysis. The inhibition of aberrant glucose metabolism attenuates NLRP3 inflammasome activation, suggesting that Warburg-like metabolism is closely associated with NLRP3-mediated inflammation in postburn responses.176
However, a recent study showed that during bacterial infection, N-acetylglucosamine, a sugar subunit of bacterial cell wall peptidoglycan, can inhibit and drive the relocalization of the glycolytic enzyme hexokinase from mitochondria into the cytosol. This localization leads to NLRP3 inflammasome activation that is independent of K+ efflux or pyroptosis.177 Inhibition of hexokinase, glycolytic inhibitors, and hexokinase relocalization appear to be sufficient to induce inflammasome activation.177 It remains to be determined how hexokinase in the cytosol triggers NLRP3 inflammasome assembly and activation. Although the mechanisms are not clear, competition of C. albicans with host cells for the use of glucose triggers activation of the NLRP3 inflammasome under conditions of glucose starvation caused by increased bacterial load.178 Accumulating data support future directions for study, such as investigating how immunometabolic regulation in the context of host–pathogen interactions shapes the collective outcome of infectious diseases.
As danger signals, cholesterol crystals trigger NLRP3 inflammasome activation, and dysregulated lipid metabolism plays a critical role in inflammasome-related diseases.179 The cholesterol trafficking pathway, the lysosomal efflux of cholesterol through Niemann-Pick C1 (NPC1), is tightly associated with immune responses, particularly NLRP3 inflammasome activation.180 In addition, the interaction of the cholesterol homeostatic regulator SCAP-SREBP2 with NLRP3 to form a complex that is translocated to the Golgi apparatus leads to the activation of the NLRP3 inflammasome in macrophages.90 Indeed, several lipids, including PIP4, contribute to NLRP3 aggregation and activation of the inflammasome complex.89,179 These data suggest that metabolic enzymes and metabolite changes directly activate the NLRP3 inflammasome complex. It is likely that data on the detailed molecular mechanisms underlying the regulation of immunometabolism will continue to accumulate in the context of inflammasome activation.
Immunometabolism and immune reprogramming (negative regulation)
Depending on which metabolites or metabolic pathways are predominant in individual immune cells in response to external cues, immunometabolic remodeling mechanisms can act as checkpoints to inhibit NLRP3 inflammasome activation.20 The important immunometabolite itaconate attenuates LPS-induced IL-1β secretion by impairing glycolytic flux by targeting the glycolytic enzymes fructose-bisphosphate aldolase A and GAPDH to enhance anti-inflammatory responses.181,182 In addition, itaconate has a negative regulatory role in the activation of the NLRP3 inflammasome complex by blocking the interaction between NLRP3 and NEK7 and preventing the induction of dicarboxypropylated C548 on NLRP3.183
A recent ex vivo study showed that treatment of macrophages obtained from T2DM patients with the sodium-glucose cotransporter 2 (SGLT2) inhibitor empagliflozin significantly inhibited IL-1β secretion, which was accompanied by increased levels of serum β-hydroxybutyrate (BHB).184 Given the inhibitory functions of BHB on NLRP3 inflammasome activity,185 these data suggest that NLRP3 inflammasome activation is regulated by ketone bodies. The BHB-induced inhibition of NLRP3 inflammasome activation seems to be mediated through prevention of K+ efflux and reduction of ASC oligomerization and speck formation.185 Another recent study showed that BHB induces increased expression of the forkhead box O (FOXO) 1 transcription factor and its target gene, heme oxygenase 1, an antioxidative enzyme, thereby providing a protective function against liver injury.186 However, the detailed molecular mechanisms by which BHB regulates NLRP3 inflammasome activation remain to be characterized.
In addition, the Src kinase-Cbl pathway has a negative regulatory role in the activation of the NLRP3 inflammasome,187 at least partly through the inhibition of GLUT1 expression and decreased glycolytic capacity.174 Given that Cbl-b is required for the ubiquitination and proteasomal degradation of NLRP341 and decreases the phosphorylated Pyk2 level,187 Cbl may function in multiple ways in the negative regulation of the NLRP3 inflammasome.
β-Glucan-induced immune reprogramming, which is critical for innate immune memory, suppresses ASC oligomerization and speck formation activation in human macrophages to attenuate the NLRP3 inflammasome formation via inhibition of K+ efflux and generation of mtROS.188 β-glucan-induced memory was beneficial for attenuating IL-1β secretion in macrophages from patients with the NLRP3-associated autoinflammatory disease cryopyrin-associated periodic syndrome (CAPS), suggesting that attenuating IL-1β secretion may have therapeutic potential for NLRP3-related diseases.188 It will also be important to determine how innate immune memory affects NLRP3 inflammasome assembly and inhibits activating signals.
Autophagy
Autophagy, an intracellular lysosomal degradation pathway, is classified into canonical and noncanonical autophagy pathways.189,190,191,192 Recent studies have shown that numerous autophagy receptors containing ubiquitin-binding domains and LC3-interacting regions are involved in selective autophagy pathways targeting various types of cargo, including mitochondria, macromolecules such as lipids, aggregated proteins, and intracytoplasmic microbes.193 In addition, LC3-associated phagocytosis (LAP) targets phagocytosed particles, such as dying cells or extracellular pathogens.193,194,195,196 Autophagy acts as the principal inhibitory pathway to limit excessive activation of the NLRP3 inflammasome in the context of various pathological conditions.189,190,191,192 As numerous reviews have summarized the relationship between autophagy and the inflammasome,189,190,191,192,197,198 in this section, we describe recent work regarding the mechanisms by which autophagy pathways, in particular autophagy-related genes (ATGs), regulate NLRP3 inflammasome activation and its physiopathological consequences.
The autophagy protein immunity-related GTPase family M protein (IRGM) functions in the regulation of core autophagy machinery by promoting the formation of autophagy initiation complexes.199 Recent studies have shown that IRGM interacts with NLRP3 and PYCARD/ASC, thus leading to their autophagic degradation via the Sequestosome1 (SQSTM1)/p62-dependent pathway. IRGM impedes inflammasome assembly by blocking the polymerization of NLRP3 and ASC, thus showing protective effects against intestinal inflammation in a murine DSS-induced colitis model.200,201 In addition, aberrant autophagy associated with a truncated UVRAG mutation promotes increased inflammatory responses and colitis-associated tumorigenesis through elevated activation of the NLRP3 inflammasome.202 A recent study showed that microglial Atg5 deletion promoted Parkinson’s disease symptoms in a mouse model through upregulation of NLRP3 inflammasome activation via cAMP signaling.203 Taken together, these findings strongly suggest that defective expression or dysregulation of ATGs is associated with upregulated NLRP3 inflammasome activation, leading to pathological responses in NLRP3-associated diseases. Notably, in bronchial cells of patients with cystic fibrosis, Pseudomonas aeruginosa infection results in impaired autophagy, thereby activating the NLRP3 inflammasome and hyperinflammation in cystic fibrosis pulmonary disease.204 Importantly, defective CFTR channels are associated with decreased capacity for selective autophagic clearance of P. aeruginosa infection in cystic fibrosis bronchial cells.204 The precise mechanism linking CFTR channels to selective autophagy activation remains to be determined. However, it is intriguing to speculate that a persistent mitochondrial unfolded protein response (UPRmt) may be involved in this phenomenon and NLRP3 inflammasome activation bronchial cells in cystic fibrosis.204
Several molecules involved in selective autophagy have been reported to play roles in the regulation of the NLRP3 inflammasome. Autophagic adapter SQSTM1-mediated autophagy leads to degradation of pyruvate kinase muscle (PKM), thereby inhibiting the production of mature IL1B in LPS-ATP-treated macrophages and ameliorating synovial inflammation.205 PTEN-induced kinase 1 (PINK1)/Parkin-mediated mitophagy suppresses mtROS and NLRP3 inflammasome-related renal injury in renal tubular epithelial cells (RTECs) during contrast-induced acute kidney injury.206
Defective mitophagy/autophagy does not always lead to activation of the NLRP3 inflammasome. Deletion of pink1, an essential gene for mitophagy, upregulates NLRP3, brown adipose tissue dysfunction, and acquisition of an obesity-prone phenotype, although the canonical NLRP3 inflammasome is not activated.207 In addition, autophagic flux and inflammasome activation are linked to the promotion of NLRP3 inflammasome-mediated pathological inflammation induced by 1-deoxysphingolipids (deoxySLs), atypical sphingolipids that are elevated in patients with hereditary sensory and autonomic neuropathy (HSAN1) or T2DM.208 Rapidly accumulating evidence regarding the cross talk between autophagy components and the inflammasome that regulates immune responses has provided new insights that are likely to lead to the development of novel therapeutic approaches for treating NLRP3-related diseases.
MicroRNAs (miRNAs)
Several studies have confirmed that miRNAs are among the major regulators of the activation of the NLRP3 inflammasome pathways (Table 2).209 Among the numerous miRNAs that directly target and suppress NLRP3, miR-223-3p is one of the best studied in terms of inflammasome regulation.209,210 Recent preclinical studies support the biological importance of miR-223-3p in the regulation of the NLRP3 inflammasome. For example, the synthetic miR-223 analog miR-223-3p caused remarkable attenuation of inflammation and fibrosis development in mouse models with endotoxin acute hepatitis (EAH) or fibrotic nonalcoholic steatohepatitis (NASH).211 Paeonol, a potential therapeutic agent for atherosclerosis, inhibits inflammatory cytokines (IL-1β and IL-6) and NLRP3 inflammasome activation through the upregulation of miR-223 in rat aortic endothelial cells (RAECs).212
Recent studies have also shown that other miRNAs in addition to miR-223-3p are involved in the negative regulation of the NLRP3 inflammasome. For example, miR-139 targeting c-Jun inhibits nerve injury induced by oxygen-glucose deprivation/reoxygenation (OGD/R) through the inhibition of NLRP3 inflammasome activation and cell pyroptosis.213 In addition, miR-183 targeting TXNIP reduces an inflammatory response triggered by the TXNIP-NLRP3 inflammasome, contributing to neuropathic pain in a rat model of chronic constriction injury and in microglia.214 In contrast, several miRNAs participate in the induction and activation of the NLRP3 inflammasome, although the precise mechanisms remain to be determined. Studies showed that the repair of ischemic injury by human umbilical cord mesenchymal stem cell-derived exosomes is mediated by targeting the miR-421/FOXO3a pathway, thereby inhibiting NLRP3 inflammasome activation and pyroptosis.215 In other words, miR-421 directly targets FOXO3a to upregulate pyroptosis and NLRP3 activation.215 The miRNA miR-21 was reported to be a positive regulator of NLRP3 inflammasome activation in myeloid cells through targeting A20, an inhibitor of the NF-κB signaling pathway.216 Further studies are warranted to understand the functions of individual miRNAs and the mechanisms underlying their regulatory effects on NLRP3 inflammasome activation under homeostatic, immune, and pathological conditions.
Recent studies have also examined the molecular interplay between long noncoding RNAs (lncRNAs) and miRNAs in terms of NLRP3 inflammasome regulation. The lncRNA ADAMTS9-AS2, a tumor suppressor, enhances cisplatin sensitivity in gastric cancer cells by activating NLRP3-mediated pyroptotic cell death by sponging miR-223-3p.217 In addition, knocking down the lncRNA NEAT1 inhibits inflammasome activation through induction of miR-3076-3p targeting NLRP3, thereby expanding the tolerogenic phenotype of dendritic cells.218 One important future direction of study involves investigating the mechanisms of cross talk between miRNAs and lncRNAs in the modulation of NLRP3 inflammasome activation and pyroptotic cell death.
Hormones and nuclear receptors
There are at least 48 members of the nuclear receptor gene superfamily that regulate a variety of pathophysiological functions, including metabolism, inflammation, and circadian rhythm.219,220 A range of nuclear receptors play key roles in the regulation of inflammation and NLRP3 inflammasome activation. A recent study highlighted the circadian oscillation of NLRP3 signaling activation and indicated that the circadian clock is essential for the inhibition of inflammation and optimal activation of the NLRP3 inflammasome.221 Recent data strongly suggest the potential benefit of chronotherapy in the pathology of dysregulated NLRP3 signaling activation.221 In support of this report, the core clock component nuclear receptor subfamily 1 group D member 1 (NR1D1, also called Rev-erbα) was shown to be essential for controlling the activity of the NLRP3 inflammasome pathway. Recent studies in Nr1d1-deficient mice showed that NR1D1 is required for the regulation of NLRP3 expression and activation, thereby inhibiting peritoneal inflammation and fulminant hepatitis.222
Several studies have reported negative regulatory functions of nuclear receptors in terms of the NLRP3 inflammasome pathway impacting, in particular, the priming step of NLRP3 activation. Small heterodimer partner (SHP), an orphan nuclear receptor, physically interacts with NLRP3 and suppresses activation of the NLRP3 inflammasome.223 Nuclear receptor related 1 (Nurr1/NR4A2) ameliorates the activation of Müller cells and the cell death of retinal ganglion cells in a diabetes model through suppression of NF-κB action and inhibition of NLRP3 inflammasome component expression, such as NLRP3 and ASC.224 However, some nuclear receptors may function in the activation of the NLRP3 inflammasome. All-trans-retinoic acid, a derivative of vitamin A, induces the expression of NLRP3 and pro-IL-1β at the priming step and promotes activation of the NLRP3 inflammasome by inducing human macrophages to undergo glycolysis.225 Further investigations of nuclear receptor interactions with NLRP3 inflammasome pathway components are likely to provide an explanation for the molecular mechanisms underlying the priming step being regulated in a gene-specific manner.
A recent study showed that the antifibrotic hormone relaxin attenuates profibrotic TGF-β1/IL-1β signaling through inhibition of TLR4-dependent priming in NLRP3 inflammasome activation.226 Although another study showed that the antifibrotic activity of relaxin is mediated by targeting caspase-1 in human dermal fibroblasts,227 it is unclear whether relaxin directly inhibits caspase-1 activity or whether it attenuates assembly of the NLRP3 inflammasome complex. There is a continuing need to accumulate data to investigate the functions of a variety of hormones and their receptors in the regulatory effects induced upon NLRP3-induced pathologies.
Others: cytokines, adapters, Notch1, cAMP, Foxp1, etc.
A variety of cytokines, signaling molecules, and second messengers are potentially involved in the positive or negative regulation of NLRP3 inflammasome activation. Recent studies have shown that IL-37d, a newly discovered negative immune regulator, inhibits the priming step of NLRP3 expression through suppression of NF-κB signaling activation. IL-37d transgenic mice show increased resistance to DSS-induced acute colitis and inhibition of NLRP3 inflammasome overactivation.228 In addition, peritoneal tissue-resident macrophages lacking the tissue-specific transcription factor GATA6 robustly suppressed IL-1β processing through the action of Gata6-mediated production of prostacyclin and IL-10.229 Further studies will be needed to evaluate the regulatory effect of a wide range of cytokines on NLRP3 inflammasome activation.
Several adapter molecules, the functions of which were identified in immune cell signaling, have been suggested to significantly fine-tune NLRP3 inflammasome activation. For example, B cell adapter for phosphoinositide 3-kinase (PI3K) (BCAP) and its association with interacting proteins, such as the caspase-1 pseudosubstrate inhibitor Flightless-1, delays the recruitment of procaspase-1 within the NLRP3–ASC preinflammasome, thereby inhibiting the activation of the NLRP3 inflammasome in macrophages.230 The Toll-IL-1R protein SARM regulates cell survival and IL-1β release upon inflammasome activation by increasing inflammasome-dependent IL-1β production and reducing pyroptosis when SARM is removed from macrophages.231 Moreover, SARM-mediated mitochondrial depolarization determines whether pyroptosis occurs in cells after NLRP3 inflammasome activation.231
Several established signaling molecules, including Notch1, cAMP, and Foxp1, play crucial negative roles in the regulation of the NLRP3 inflammasome in immune cells. Jagged1 (JAG1)-mediated Notch1 signaling in myeloid cells upregulates heat shock transcription factor 1 (HSF1) expression and Snail activity to control NLRP3/caspase-1 activity.232 As discussed in the section on Ca2+ flux, binding of cAMP to NLRP3 leads to inhibition of inflammasome assembly.105 Activation of the cAMP-PKA signaling pathway is linked to inhibition of NLRP3 inflammasome activity through enhancement of K63-linked ubiquitination of NLRP3.233 Genistein-mediated anti-inflammasome activity is mediated through TGF5-cAMP signaling via increased intracellular cAMP levels234 Foxp1 was reported to have a negative regulatory function on endothelial NLRP3 inflammasome activation, acting as a gatekeeper of vessel inflammation.235 Endothelial Foxp1 is regulated by Krüppel-like factor 2 (Klf2) and further regulates NLRP3 inflammasome activation through direct regulation of endothelial inflammasome components, including NLRP3 and caspase-1.235 Exploring the effects of a variety of signaling molecules and/or second messengers on inflammasome regulation may lead to the discovery of potential therapeutic targets against NLRP3-related pathologic inflammation.
Cellular inhibitor of apoptosis protein (cIAP) 1 and cIAP2, members of the IAP family, act as E3 ligases and modulators of the NLRP3 inflammasome.236,237 Upon overexpression of cIAP1 or cIAP2 in macrophages, the levels of IL-1β and pyroptotic cell death are increased in response to inflammasome activators or bacterial infections. Glomulin (GLMN), originally identified through its association with glomuvenous malformations, acts as an inhibitor of Cullin-truly interesting new gene (RING)-box protein 1 (RBX1) E3 ligases and binds to the RING domains of cIAP1 and cIAP2, thereby inhibiting their functions.238
On the other hand, the human serum factor H-related protein FHR1 binds to necrotic cells via its N-terminus and upregulates NLRP3 inflammasome activation in human monocytes, thereby producing IL-1β, TNFα, IL-18, and IL-6, thus contributing to the pathology of anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV) and atherosclerosis.239 Recent studies have also shown that monoamine oxidase (MAO) catalyzes the oxidative deamination of neurotransmitters and amines, generating mtROS and NLPR3 inflammasome activation through a NF-κB-mediated mechanism.240 A number of mechanisms remain to be addressed before we can gain a full understanding of the multiple molecules/pathways that positively and negatively regulate NLRP3 signaling networks in immune cells.
Small molecules/agents as therapeutics against NLRP3 inflammasome activation
There has been rapid progress in the identification of NLRP3 inflammasome-targeting small molecules/agents for use as therapeutics against NLRP3 inflammasome activation. Accumulating evidence has revealed large numbers of inhibitors of NLRP3 inflammasome activation through various pharmacological approaches used with NLRP3 inflammasome-related disease models.13,241,242 Several extensive reviews summarizing NLRP3 inflammasome activators and inhibitors have suggested that developing new small molecules that directly target NLRP3 seems to be more specific, cost-effective, and safer than an overall cytokine blockade.241,242 Determination of the complexity of the NLRP3 inflammasome structure and interactions among its components holds promise for the development of new molecules targeting specific components or interactions of the NLRP3 inflammasome complex. Here, we briefly describe the most potent and most recently discovered inhibitors according to their known targets (Table 3).
The NACHT domain of NLRP3 is the molecular target of diarylsulfonylurea inhibitors, including MCC950/CRID3,243 which is a potent and selective inhibitor of the NLRP3 inflammasome pathway through its interaction with the Walker B motif within the NACHT domain of NLRP3 by which ATP hydrolysis is blocked.244 The mechanism of action of MCC950 is mediated by changing the active conformation of NLRP3 into an inactive state.245 NLRP3 inhibition with MCC950 was shown to significantly suppress IL-1β production and airway inflammation in the lungs of mice with cystic fibrosis246 and to prevent cognitive deficits in mice with experimental autoimmune encephalomyelitis (EAE).247 However, recent studies have shown that MCC950/CRID3 targets wild-type NLRP3 but not NLRP3 gain-of-function point mutants related to CAPS.248 The CFTR(inh)-172 analog CY-09 also inhibits NLRP3 ATPase activity by directly binding to the ATP-binding motif of the NLRP3 NACHT domain.249 Prominent therapeutic effects were observed in mouse models of CAPS and T2DM and in monocytes of gout patients249 treated with CY-09. A recent study showed that CY-09 treatment is beneficial in ameliorating epileptic progression and neuronal loss through attenuation of NLRP3-dependent IL-1β secretion and astrocyte activation.250 Moreover, a β-sulfonyl nitrile compound, OLT1177, reduces ATPase activity by directly binding to NLRP3, followed by inhibition of ASC speck aggregation,251 and BOT-4-one impairs NLRP3 ATPase activity by alkylating NLRP3, leading to obstruction of NLRP3 inflammasome assembly.252 Tranilast, a tryptophan metabolite used for the treatment of allergies and asthma,253 shows remarkable preventive and therapeutic effects in mouse models of gout, CAPS, and T2DM by hindering NLRP3 oligomerization in an ATPase-independent manner.254
With evidence of the importance of NEK7, there is increasing interest in discovering new drugs targeting NEK7 and its interaction with NLRP3, as most of the small molecules targeting the NLRP3 inflammasome were reported prior to the publication of the cryoelectron microscopy structure of NEK7–NLRP3.10 Oridonin, which is the main ingredient of the traditional Chinese herbal medicine Rabdosia rubescens, blocks the interaction between NLRP3 and NEK7 by forming a covalent bond with cysteine 279 in the NACHT domain.70,255 Oridonin shows both preventive and therapeutic effects in peritonitis, gouty arthritis, and T2DM mice via inhibition of NLRP3 activation.255 Ginsenoside Rg3, a natural product extracted from Panax ginseng, was recently reported to selectively inhibit NLRP3 activation. Rg3 does not regulate the upstream signals of the NLRP3 inflammasome but mechanistically abrogates the NEK7–NLRP3 interaction, thereby subsequently disturbing NLRP3–ASC assembly.256 Hughes et al. reported that deglutathionylation of NEK7 by glutathione transferase omega 1-1 (GSTO1-1), a constitutive deglutathionylating enzyme, is required for activation of the NLRP3 inflammasome.257 They used the GSTO1-1 inhibitor C1-27 to show that inhibition of GSTO1-1 had a protective effect in an ECE mouse model, and a more advanced form of inhibitor, designated 25, was reported in a follow-up study.258 In addition, artemisinin259 targeted NEK7–NLRP3 interactions to suppress inflammasome activity in a T2DM disease model.
Studies of small compounds targeting the NLRP3 inflammasome identified KN3014, which directly targets the PYD and thus inhibits the interaction between NLRP3 and ASC.260 KN3014 was shown to block ASC speck formation effectively and significantly reduced IL-1β secretion from the PBMCs of a patient with Muckle–Wells syndrome (MWS). Another study with β-carotene (provitamin A) demonstrated its direct binding to the PYD of NLRP3,261 inhibiting IL-1β secretion from synovial fluid cells retrieved from patients with gouty arthritis. Recent studies identified several peptides that modulate different stages of NLRP3 inflammasome assembly and inhibit IL-1β release, caspase-1 activation, and ASC oligomerization.262 Among these candidates, a peptide with a sequence corresponding to the H2-H3 segment of ASC PYD showed selective inhibitory activity against NLRP3 but was not absent in melanoma 2 (AIM2) and NLR family CARD domain-containing protein 4 (NLRC4) inflammasomes.262
There is a great deal of research interest in the inflammasome. A wide range of natural and synthetic inhibitors have been reported to have inflammasome-inhibiting activity. However, insufficient understanding of the mechanisms of action and potential off-target effects of these molecules limit their further development for clinical use. With the elucidation of the structure and mechanism of inflammasome formation, the identification and design of new inhibitors targeting specific components of the NLRP3 inflammasome will provide new insights and facilitate the development of therapeutics for various autoinflammatory and autoimmune diseases.
Conclusion
The study of NLRP3 inflammasome activation has many implications for health and disease. Significant progress has been made toward understanding the molecular mechanisms underlying the priming/licensing step of NLRP3 inflammasome activation. However, many questions remain, e.g., how the individual and/or multiple PTM regulation is curated for licensing of the NLRP3 inflammasome, how different NLRP3 stimuli are engaged to converge into the signaling cascades that lead to full activation of the NLRP3 inflammasome, and which signaling pathway(s) play key roles in the ultimate assembly of the inflammasome complex. The spatiotemporal regulation of NLRP3 inflammasome activation has not yet been completely elucidated, but recent studies have highlighted the emerging concept of the involvement of the dispersed Golgi apparatus and MTOC in the formation of the NLRP3 inflammasome complex. Further identification of key players involved in regulating the stepwise engagement of organelles and sequential interorganellar communication is needed to clarify the molecular mechanisms of NLRP3 inflammasome activation. It remains to be determined how multiple positive/negative regulatory mechanisms are involved in NLRP3 inflammasome activation as well as the nature of their cell type-specific and NLRP3 stimulus-specific roles. Moreover, the roles of immunometabolism, miRNAs, and lncRNAs in NLRP3 inflammasome activation have only begun to be elucidated. A wide range of small molecules/reagents are being developed for the regulation of NLRP3 inflammasome activation, although therapeutic outcomes have been limited in terms of clinical trials. Future work may provide a comprehensive interpretation of the highly regulated nature of the formation and activation of the NLRP3 inflammasome complex. These efforts will be critical for the future development of potential therapeutic and preventive agents for NLRP3-related diseases.
References
Tschopp, J. & Schroder, K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215 (2010).
Kanneganti, T. D., Lamkanfi, M. & Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Lamkanfi, M. & Dixit, V. M. Inflammasomes and their roles in health and disease. Annu Rev. Cell Dev. Biol. 28, 137–161 (2012).
Yu, H. B. & Finlay, B. B. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe 4, 198–208 (2008).
Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 287, 11–26 (2020).
Shim, D. W. & Lee, K. H. Posttranslational regulation of the NLR family pyrin domain-containing 3 inflammasome. Front Immunol. 9, 1054 (2018).
Stutz, A. et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 214, 1725–1736 (2017).
Sharif, H. et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343 (2019).
McKee, C. M. & Coll, R. C. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 108, 937–952 (2020).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Caseley, E. A., Poulter, J. A., Rodrigues, F., Immunome Project Consortium for Autoinflammatory, D. & McDermott, M. F. Inflammasome inhibition under physiological and pharmacological conditions. Genes Immun. 21, 211–223 (2020).
Pirzada, R. H., Javaid, N. & Choi, S. The roles of the NLRP3 inflammasome in neurodegenerative and metabolic diseases and in relevant advanced therapeutic interventions. Genes (Basel) 11, 131 (2020).
Krainer, J., Siebenhandl, S. & Weinhausel, A. Systemic autoinflammatory diseases. J. Autoimmun. 109, 102421 (2020).
Moossavi, M., Parsamanesh, N., Bahrami, A., Atkin, S. L. & Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 17, 158 (2018).
Kelley, N., Jeltema, D., Duan, Y. & He, Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20, 3328 (2019).
Sun, X. et al. The NLRP3 inflammasome and its role in T1DM. Front. Immunol. 11, 1595 (2020).
Chauhan, D., Vande Walle, L. & Lamkanfi, M. Therapeutic modulation of inflammasome pathways. Immunol. Rev. 297, 123–138 (2020).
Meyers, A. K. & Zhu, X. The NLRP3 inflammasome: metabolic regulation and contribution to inflammaging. Cells 9, 1808 (2020).
Bezbradica, J. S., Coll, R. C. & Schroder, K. Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell. Mol. Immunol. 14, 118–126 (2017).
Rashidi, M., Wicks, I. P. & Vince, J. E. Inflammasomes and cell death: common pathways in microparticle diseases. Trends Mol. Med. 26, 1003–1020 (2020).
Bai, B. et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 11, 776 (2020).
Zheng, M. & Kanneganti, T. D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 297, 26–38 (2020).
Samir, P., Malireddi, R. K. S. & Kanneganti, T. D. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 10, 238 (2020).
Karmakar, M. et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 11, 2212 (2020).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Speir, M. & Lawlor, K. E. RIP-roaring inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation and autoinflammatory disease. Semin. Cell Dev. Biol. 109, 114–124 (2020).
Zhu, Q. & Kanneganti, T. D. Cutting edge: distinct regulatory mechanisms control proinflammatory cytokines IL-18 and IL-1beta. J. Immunol. 198, 4210–4215 (2017).
Barker, B. R., Taxman, D. J. & Ting, J. P. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr. Opin. Immunol. 23, 591–597 (2011).
Afonina, I. S., Zhong, Z., Karin, M. & Beyaert, R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat. Immunol. 18, 861–869 (2017).
Hou, L. et al. Macrophage sphingosine 1-phosphate receptor 2 blockade attenuates liver inflammation and fibrogenesis triggered by NLRP3 inflammasome. Front. Immunol. 11, 1149 (2020).
Tan, Y. F., Wang, M., Chen, Z. Y., Wang, L. & Liu, X. H. Inhibition of BRD4 prevents proliferation and epithelial-mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. 11, 239 (2020).
Xing, Y. et al. Cutting edge: TRAF6 mediates TLR/IL-1R signaling-induced nontranscriptional priming of the NLRP3 inflammasome. J. Immunol. 199, 1561–1566 (2017).
Fernandes-Alnemri, T. et al. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J. Immunol. 191, 3995–3999 (2013).
Lee, S., Suh, G. Y., Ryter, S. W. & Choi, A. M. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am. J. Respir. Cell. Mol. Biol. 54, 151–160 (2016).
Lin, K. M. et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl Acad. Sci. USA 111, 775–780 (2014).
Gurung, P. et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846 (2014).
Kang, S. et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 6, 7515 (2015).
Zhou, Z. et al. K63 ubiquitin chains target NLRP3 inflammasome for autophagic degradation in ox-LDL-stimulated THP-1 macrophages. Aging 12, 1747–1759 (2020).
Tang, J. et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 217, e20182091 (2020).
Song, H. et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 7, 13727 (2016).
Yan, Y. et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015).
Wan, P. et al. Cullin1 binds and promotes NLRP3 ubiquitination to repress systematic inflammasome activation. FASEB J. 33, 5793–5807 (2019).
Kawashima, A. et al. ARIH2 ubiquitinates NLRP3 and negatively regulates NLRP3 inflammasome activation in macrophages. J. Immunol. 199, 3614–3622 (2017).
Humphries, F. et al. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 9, 1560 (2018).
Juliana, C. et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 287, 36617–36622 (2012).
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H. & Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013).
Ren, G. et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 38, e100376 (2019).
Rao, Z. et al. Vitamin D receptor inhibits NLRP3 activation by impeding Its BRCC3-mediated deubiquitination. Front. Immunol. 10, 2783 (2019).
Palazon-Riquelme, P. et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 19, e44766 (2018).
Wang, W. et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 16, e1008335 (2020).
Li, T. & Chen, Z. J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Chen, Q., Sun, L. & Chen, Z. J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016).
Song, N. et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 68, 185–197 (2017). e186.
Zhang, Z. et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med 214, 2671–2693 (2017).
Mortimer, L., Moreau, F., MacDonald, J. A. & Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 17, 1176–1186 (2016).
Ito, M. et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6, 7360 (2015).
Mao, L. et al. Bruton tyrosine kinase deficiency augments NLRP3 inflammasome activation and causes IL-1beta-mediated colitis. J. Clin. Invest 130, 1793–1807 (2020).
Zhang, A. et al. EphA2 phosphorylates NLRP3 and inhibits inflammasomes in airway epithelial cells. EMBO Rep. 21, e49666 (2020).
Spalinger, M. R. et al. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 13, 1590–1601 (2017).
Spalinger, M. R. et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Invest 126, 1783–1800 (2016).
Huang, Y. et al. Myeloid PTEN promotes chemotherapy-induced NLRP3-inflammasome activation and antitumour immunity. Nat. Cell Biol. 22, 716–727 (2020).
Mao, K. et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 23, 201–212 (2013).
Hernandez-Cuellar, E. et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 189, 5113–5117 (2012).
Mishra, B. B. et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat. Immunol. 14, 52–60 (2013).
Barry, R. et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat. Commun. 9, 3001 (2018).
Shao, L. et al. SUMO1 SUMOylates and SENP3 deSUMOylates NLRP3 to orchestrate the inflammasome activation. FASEB J. 34, 1497–1515 (2020).
He, M. et al. An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab. 31, 580–591 (2020). e585.
Zhao, N., Li, C. C., Di, B. & Xu, L. L. Recent advances in the NEK7-licensed NLRP3 inflammasome activation: Mechanisms, role in diseases and related inhibitors. J. Autoimmun. 113, 102515 (2020).
Schmid-Burgk, J. L. et al. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 291, 103–109 (2016).
He, Y., Zeng, M. Y., Yang, D., Motro, B. & Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016).
Chen, X. et al. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis. 10, 906 (2019).
Hoss, F. et al. Alternative splicing regulates stochastic NLRP3 activity. Nat. Commun. 10, 3238 (2019).
Schmacke, N. A. et al. Priming enables a NEK7-independent route of NLRP3 activation. bioRxiv https://doi.org/10.1101/799320 (2019).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Yoshihara, E. et al. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 4, 514 (2014).
Yoshihara, E. TXNIP/TBP-2: a master regulator for glucose homeostasis. Antioxidants 9, 765 (2020).
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z. & Germain, R. N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013).
Franchi, L. et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 193, 4214–4222 (2014).
Park, S. et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366 (2013).
Ichinohe, T., Yamazaki, T., Koshiba, T. & Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl Acad. Sci. USA 110, 17963–17968 (2013).
Samir, P. et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594 (2019).
Li, X. et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun. 8, 15986 (2017).
Wang, J. et al. High glucose mediates NLRP3 inflammasome activation via upregulation of ELF3 expression. Cell Death Dis. 11, 383 (2020).
Hamilton, C. & Anand, P. K. Right place, right time: localisation and assembly of the NLRP3 inflammasome. F1000Res. 8, 676 (2019).
Misawa, T. et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 (2013).
Magupalli, V. G. et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369, eaas8995 (2020).
Chen, J. & Chen, Z. J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018).
Guo, C. et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity 49, 842–856 (2018). e847.
Franklin, B. S. et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 15, 727–737 (2014).
Franklin, B. S., Latz, E. & Schmidt, F. I. The intra- and extracellular functions of ASC specks. Immunol. Rev. 281, 74–87 (2018).
Perregaux, D. & Gabel, C. A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 269, 15195–15203 (1994).
He, Y., Hara, H. & Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021 (2016).
Munoz-Planillo, R. et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
Chen, K. W., Demarco, B. & Broz, P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur. J. Immunol. 50, 170–177 (2020).
Chen, K. W. et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38, e101638 (2019).
Cao, F. et al. P2X7 receptor: a potential therapeutic target for autoimmune diseases. Autoimmun. Rev. 18, 767–777 (2019).
Di, A. et al. The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49, 56–65 (2018). e54.
Triantafilou, K., Hughes, T. R., Triantafilou, M. & Morgan, B. P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913 (2013).
Laudisi, F. et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1beta release. J. Immunol. 191, 1006–1010 (2013).
Beckwith, K. S. et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat. Commun. 11, 2270 (2020).
Deng, M. et al. Platelet-activating factor (PAF) mediates NLRP3-NEK7 inflammasome induction independently of PAFR. J. Exp. Med 216, 2838–2853 (2019).
Murakami, T. et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl Acad. Sci. USA 109, 11282–11287 (2012).
Lee, G. S. et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 (2012).
Rolfes, V. et al. Platelets fuel the inflammasome activation of innate immune cells. Cell Rep. 31, 107615 (2020).
Yaron, J. R. et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 6, e1954 (2015).
Katsnelson, M. A., Rucker, L. G., Russo, H. M. & Dubyak, G. R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 194, 3937–3952 (2015).
Zhang, Y. et al. A membrane potential- and calpain-dependent reversal of caspase-1 inhibition regulates canonical NLRP3 inflammasome. Cell Rep. 24, 2356–2369 (2018). e2355.
Wang, L., Negro, R. & Wu, H. TRPM2, linking oxidative stress and Ca(2+) permeation to NLRP3 inflammasome activation. Curr. Opin. Immunol. 62, 131–135 (2020).
Zhong, Z. et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 4, 1611 (2013).
Zewinger, S. et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 21, 30–41 (2020).
Zhang, C. et al. ADP/P2Y1 aggravates inflammatory bowel disease through ERK5-mediated NLRP3 inflammasome activation. Mucosal Immunol. 13, 931–945 (2020).
Jager, E. et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 11, 4243 (2020).
Domingo-Fernandez, R., Coll, R. C., Kearney, J., Breit, S. & O’Neill, L. A. J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 292, 12077–12087 (2017).
Tang, T. et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 8, 202 (2017).
Green, J. P. et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl Acad. Sci. USA 115, E9371–E9380 (2018).
Bergeron, C. & Cantin, A. M. Cystic fibrosis: pathophysiology of lung disease. Semin. Respir. Crit. Care Med. 40, 715–726 (2019).
Martin, S. L., Saint-Criq, V., Hwang, T. C. & Csanady, L. Ion channels as targets to treat cystic fibrosis lung disease. J. Cyst. Fibros. 17, S22–S27 (2018).
Scambler, T. et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife 8, e49248 (2019).
Huang, B. et al. Ticagrelor inhibits the NLRP3 inflammasome to protect against inflammatory disease independent of the P2Y12 signaling pathway. Cell. Mol. Immunol. (2020) https://doi.org/10.1038/s41423-020-0444-5
Penna, C. et al. Ticagrelor conditioning effects are not additive to cardioprotection induced by direct NLRP3 inflammasome inhibition: role of RISK, NLRP3, and redox cascades. Oxid. Med Cell Longev. 2020, 9219825 (2020).
Elliott, E. I. et al. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J. Immunol. 200, 3047–3052 (2018).
Liu, Q., Zhang, D., Hu, D., Zhou, X. & Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 103, 115–124 (2018).
Iyer, S. S. et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013).
Holley, C. L. & Schroder, K. The rOX-stars of inflammation: links between the inflammasome and mitochondrial meltdown. Clin. Transl. Immunol. 9, e01109 (2020).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Yu, J. W. & Lee, M. S. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch. Pharm. Res 39, 1503–1518 (2016).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Yabal, M., Calleja, D. J., Simpson, D. S. & Lawlor, K. E. Stressing out the mitochondria: mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 105, 377–399 (2019).
Gross, C. J. et al. K(+) efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 45, 761–773 (2016).
Yeon, S. H., Yang, G., Lee, H. E. & Lee, J. Y. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J. Leukoc. Biol. 101, 205–215 (2017).
Panicker, N. et al. Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med 216, 1411–1430 (2019).
Bedient, L. et al. Lytic cell death mechanisms in human respiratory syncytial virus-infected macrophages: roles of pyroptosis and necroptosis. Viruses 12, 932 (2020).
Alves, J. V. et al. Supraphysiological levels of testosterone induce vascular dysfunction via activation of the NLRP3 inflammasome. Front. Immunol. 11, 1647 (2020).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
Coll, R. C., Holley, C. L. & Schroder, K. Mitochondrial DNA synthesis fuels NLRP3 activation. Cell Res. 28, 1046–1047 (2018).
Li, S. et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep. 30, 4370–4385 (2020). e4377.
Dudek, J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 5, 90 (2017).
Chang, Y. H. et al. The causal role of mitochondrial dynamics in regulating innate immunity in diabetes. Front. Endocrinol. (Lausanne) 11, 445 (2020).
Orlowski, G. M. et al. Multiple cathepsins promote pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J. Immunol. 195, 1685–1697 (2015).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Svadlakova, T. et al. Proinflammatory effect of carbon-based nanomaterials: in vitro study on stimulation of inflammasome NLRP3 via destabilisation of lysosomes. Nanomaterials 10, 418 (2020).
Zhang, Y., Chen, Y., Zhang, Y., Li, P. L. & Li, X. Contribution of cathepsin B-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction. Eur. J. Pharm. 865, 172795 (2019).
Chevriaux, A. et al. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front. Cell Dev. Biol. 8, 167 (2020).
Katsnelson, M. A., Lozada-Soto, K. M., Russo, H. M., Miller, B. A. & Dubyak, G. R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am. J. Physiol. Cell Physiol. 311, C83–C100 (2016).
Correa, R. et al. Lysophosphatidylcholine induces NLRP3 inflammasome-mediated foam cell formation and pyroptosis in human monocytes and endothelial cells. Front. Immunol. 10, 2927 (2019).
Westman, J. et al. Lysosome fusion maintains phagosome integrity during fungal infection. Cell Host Microbe 28, 798–812 (2020).
Liu, L. et al. Endothelial cell-derived interleukin-18 released during ischemia reperfusion injury selectively expands T peripheral helper cells to promote alloantibody production. Circulation 141, 464–478 (2020).
Xie, C. B. et al. Complement membrane attack complexes assemble NLRP3 inflammasomes triggering IL-1 activation of IFN-gamma-primed human endothelium. Circ. Res. 124, 1747–1759 (2019).
Yu, S. et al. The complement receptor C5aR2 promotes protein kinase R expression and contributes to NLRP3 inflammasome activation and HMGB1 release from macrophages. J. Biol. Chem. 294, 8384–8394 (2019).
Zhang, T. et al. The C5a/C5aR2 axis promotes renal inflammation and tissue damage. JCI Insight 5, e134081 (2020).
Gratal, P. et al. Purinergic system signaling in metainflammation-associated osteoarthritis. Front. Med. (Lausanne) 7, 506 (2020).
Li, H. et al. P2Y14 receptor has a critical role in acute gouty arthritis by regulating pyroptosis of macrophages. Cell Death Dis. 11, 394 (2020).
Suzuki, T. et al. Extracellular ADP augments microglial inflammasome and NF-kappaB activation via the P2Y12 receptor. Eur. J. Immunol. 50, 205–219 (2020).
Vince, J. E. & Silke, J. The intersection of cell death and inflammasome activation. Cell. Mol. Life Sci. 73, 2349–2367 (2016).
Conos, S. A. et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl Acad. Sci. USA 114, E961–E969 (2017).
Lawlor, K. E. et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015).
Malireddi, R. K. S. et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 215, 1023–1034 (2018).
Orning, P. et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018).
Malireddi, R. K. S. et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 217 (2020).
Song, S. et al. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 42, 230–241 (2020).
Shi, Y. et al. RIPK3 blockade attenuates kidney fibrosis in a folic acid model of renal injury. FASEB J. 34, 10286–10298 (2020).
Kuriakose, T. et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045 (2016).
Kesavardhana, S. & Kanneganti, T. D. ZBP1: A STARGTE to decode the biology of Z-nucleic acids in disease. J. Exp. Med. 217, e20200885 (2020).
Kuriakose, T. & Kanneganti, T. D. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 39, 123–134 (2018).
Kesavardhana, S. et al. The Zalpha2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 295, 8325–8330 (2020).
Zheng, M., Karki, R., Vogel, P. & Kanneganti, T. D. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell 181, 674–687 (2020). e613.
Kuriakose, T., Zheng, M., Neale, G. & Kanneganti, T. D. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J. Immunol. 200, 1489–1495 (2018).
Russell, D. G., Huang, L. & VanderVen, B. C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 19, 291–304 (2019).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242 (2013).
Lin, H. C. et al. Cbl negatively regulates NLRP3 inflammasome activation through GLUT1-dependent glycolysis inhibition. Int. J. Mol. Sci. 21, 5104 (2020).
Xie, M. et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 7, 13280 (2016).
Vinaik, R., Barayan, D., Auger, C., Abdullahi, A. & Jeschke, M. G. Regulation of glycolysis and the Warburg effect in wound healing. JCI Insight 5, e138949 (2020).
Wolf, A. J. et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166, 624–636 (2016).
Tucey, T. M. et al. Metabolic competition between host and pathogen dictates inflammasome responses to fungal infection. PLoS Pathog. 16, e1008695 (2020).
Anand, P. K. Lipids, inflammasomes, metabolism, and disease. Immunol. Rev. 297, 108–122 (2020).
de la Roche, M. et al. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J. Cell Biol. 217, 3560–3576 (2018).
Qin, W. et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol. 15, 983–991 (2019).
Liao, S. T. et al. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 10, 5091 (2019).
Hooftman, A. et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. 32, 468–478 (2020). e467.
Kim, S. R. et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 11, 2127 (2020).
Youm, Y. H. et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med 21, 263–269 (2015).
Miyauchi, T. et al. Up-regulation of FOXO1 and reduced inflammation by beta-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc. Natl Acad. Sci. USA 116, 13533–13542 (2019).
Chung, I. C. et al. Src-family kinase-Cbl axis negatively regulates NLRP3 inflammasome activation. Cell Death Dis. 9, 1109 (2018).
Camilli, G. et al. beta-Glucan-induced reprogramming of human macrophages inhibits NLRP3 inflammasome activation in cryopyrinopathies. J. Clin. Invest 130, 4561–4573 (2020).
Chung, C., Seo, W., Silwal, P. & Jo, E. K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 13, 100 (2020).
Liu, T. Regulation of inflammasome by autophagy. Adv. Exp. Med. Biol. 1209, 109–123 (2019).
Painter, J. D., Galle-Treger, L. & Akbari, O. Role of autophagy in lung inflammation. Front. Immunol. 11, 1337 (2020).
Takahama, M., Akira, S. & Saitoh, T. Autophagy limits activation of the inflammasomes. Immunol. Rev. 281, 62–73 (2018).
Sil, P., Muse, G. & Martinez, J. A ravenous defense: canonical and non-canonical autophagy in immunity. Curr. Opin. Immunol. 50, 21–31 (2018).
Upadhyay, S. & Philips, J. A. LC3-associated phagocytosis: host defense and microbial response. Curr. Opin. Immunol. 60, 81–90 (2019).
Herb, M., Gluschko, A. & Schramm, M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Semin. Cell Dev. Biol. 101, 68–76 (2020).
Heckmann, B. L. & Green, D. R. LC3-associated phagocytosis at a glance. J. Cell Sci. 132, jcs222984 (2019).
Martins, J. D. et al. Autophagy and inflammasome interplay. DNA Cell Biol. 34, 274–281 (2015).
Saitoh, T. & Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 138, 28–36 (2016).
Chauhan, S., Mandell, M. A. & Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 58, 507–521 (2015).
Mehto, S. et al. The Crohn’s disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol. Cell 73, 429–445 (2019). e427.
Mehto, S. et al. IRGM restrains NLRP3 inflammasome activation by mediating its SQSTM1/p62-dependent selective autophagy. Autophagy 15, 1645–1647 (2019).
Quach, C. et al. A truncating mutation in the autophagy gene UVRAG drives inflammation and tumorigenesis in mice. Nat. Commun. 10, 5681 (2019).
Cheng, J. et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 16, 2193–2205 (2020).
Rimessi, A. et al. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci. Adv. 6, eaax9093 (2020).
Zhang, B. et al. SQSTM1-dependent autophagic degradation of PKM2 inhibits the production of mature IL1B/IL-1beta and contributes to LIPUS-mediated anti-inflammatory effect. Autophagy 16, 1262–1278 (2020).
Lin, Q. et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 26, 101254 (2019).
Ko, M. S. et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy https://doi.org/10.1080/15548627.2020.1753002 (2020).
Lauterbach, M. A. et al. 1-Deoxysphingolipids cause autophagosome and lysosome accumulation and trigger NLRP3 inflammasome activation. Autophagy https://doi.org/10.1080/15548627.2020.1804677 (2020).
Boxberger, N., Hecker, M. & Zettl, U. K. Dysregulation of inflammasome priming and activation by MicroRNAs in human immune-mediated diseases. J. Immunol. 202, 2177–2187 (2019).
Bauernfeind, F. et al. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 189, 4175–4181 (2012).
Jimenez Calvente, C., Del Pilar, H., Tameda, M., Johnson, C. D. & Feldstein, A. E. MicroRNA 223 3p negatively regulates the NLRP3 inflammasome in acute and chronic liver. Inj. Mol. Ther. 28, 653–663 (2020).
Shi, X. et al. Paeonol inhibits NLRP3 mediated inflammation in rat endothelial cells by elevating hyperlipidemic rats plasma exosomal miRNA-223. Eur. J. Pharm. 885, 173473 (2020).
Wang, Q. S. et al. MiR-139 protects against oxygen-glucose deprivation/reoxygenation (OGD/R)-induced nerve injury through targeting c-Jun to inhibit NLRP3 inflammasome activation. J. Stroke Cerebrovasc. Dis. 29, 105037 (2020).
Miao, J., Zhou, X., Ji, T. & Chen, G. NF-kappaB p65-dependent transcriptional regulation of histone deacetylase 2 contributes to the chronic constriction injury-induced neuropathic pain via the microRNA-183/TXNIP/NLRP3 axis. J. Neuroinflammation 17, 225 (2020).
Yan, B. et al. Stem cell-derived exosomes prevent pyroptosis and repair ischemic muscle injury through a novel exosome/circHIPK3/ FOXO3a pathway. Theranostics 10, 6728–6742 (2020).
Xue, Z. et al. miR-21 promotes NLRP3 inflammasome activation to mediate pyroptosis and endotoxic shock. Cell Death Dis. 10, 461 (2019).
Ren, N. et al. LncRNA ADAMTS9-AS2 inhibits gastric cancer (GC) development and sensitizes chemoresistant GC cells to cisplatin by regulating miR-223-3p/NLRP3 axis. Aging 12, 11025–11041 (2020).
Zhang, M. et al. Knockdown of NEAT1 induces tolerogenic phenotype in dendritic cells by inhibiting activation of NLRP3 inflammasome. Theranostics 9, 3425–3442 (2019).
Weikum, E. R., Liu, X. & Ortlund, E. A. The nuclear receptor superfamily: a structural perspective. Protein Sci. 27, 1876–1892 (2018).
Gadaleta, R. M. & Magnani, L. Nuclear receptors and chromatin: an inducible couple. J. Mol. Endocrinol. 52, R137–R149 (2014).
Pourcet, B. & Duez, H. Circadian control of inflammasome pathways: implications for circadian medicine. Front. Immunol. 11, 1630 (2020).
Pourcet, B. et al. Nuclear receptor subfamily 1 group D member 1 regulates circadian activity of NLRP3 inflammasome to reduce the severity of fulminant hepatitis in mice. Gastroenterology 154, 1449–1464 (2018). e1420.
Yang, C. S. et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 6, 6115 (2015).
Li, W. et al. Dysfunctional Nurr1 promotes high glucose-induced Muller cell activation by up-regulating the NF-kappaB/NLRP3 inflammasome axis. Neuropeptides 82, 102057 (2020).
Alatshan, A. et al. All-trans retinoic acid enhances both the signaling for priming and the glycolysis for activation of NLRP3 inflammasome in human macrophage. Cells 9, 1591 (2020).
Caceres, F. T., Gaspari, T. A., Samuel, C. S. & Pinar, A. A. Serelaxin inhibits the profibrotic TGF-beta1/IL-1beta axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. FASEB J. 33, 14717–14733 (2019).
Pinar, A. A., Yuferov, A., Gaspari, T. A. & Samuel, C. S. Relaxin can mediate its anti-fibrotic effects by targeting the myofibroblast NLRP3 inflammasome at the level of caspase-1. Front. Pharm. 11, 1201 (2020).
Li, Y. et al. IL-37d negatively regulates NLRP3 transcription via receptor-mediated pathway and alleviates DSS-induced colitis. Inflamm. Bowel Dis. 27, 84–93 (2020).
Ipseiz, N. et al. Tissue-resident macrophages actively suppress IL-1beta release via a reactive prostanoid/IL-10 pathway. EMBO J. 39, e103454 (2020).
Carpentier, S. J. et al. The signaling adaptor BCAP inhibits NLRP3 and NLRC4 inflammasome activation in macrophages through interactions with Flightless-1. Sci. Signal. 12, eaau0615 (2019).
Carty, M. et al. Cell survival and cytokine release after inflammasome activation is regulated by the toll-IL-1R protein SARM. Immunity 50, 1412–1424 (2019). e1416.
Jin, Y. et al. Jagged1-mediated myeloid Notch1 signaling activates HSF1/Snail and controls NLRP3 inflammasome activation in liver inflammatory injury. Cell. Mol. Immunol. 17, 1245–1256 (2019).
Zheng, S. et al. Posttreatment of Maresin1 inhibits NLRP3 inflammasome activation via promotion of NLRP3 ubiquitination. FASEB J. 34, 11944–11956 (2020).
Chen, Y. et al. Genistein protects against DSS-induced colitis by inhibiting NLRP3 inflammasome via TGR5-cAMP signaling. Int. Immunopharmacol. 71, 144–154 (2019).
Zhuang, T. et al. Endothelial Foxp1 suppresses atherosclerosis via modulation of Nlrp3 inflammasome activation. Circ. Res 125, 590–605 (2019).
Duda, D. M. et al. Structure of a glomulin-RBX1-CUL1 complex: inhibition of a RING E3 ligase through masking of its E2-binding surface. Mol. Cell 47, 371–382 (2012).
Tron, A. E. et al. The glomuvenous malformation protein Glomulin binds Rbx1 and regulates cullin RING ligase-mediated turnover of Fbw7. Mol. Cell 46, 67–78 (2012).
Suzuki, S., Suzuki, T., Mimuro, H., Mizushima, T. & Sasakawa, C. Shigella hijacks the glomulin-cIAPs-inflammasome axis to promote inflammation. EMBO Rep. 19, 89–101 (2018).
Irmscher, S. et al. Serum FHR1 binding to necrotic-type cells activates monocytic inflammasome and marks necrotic sites in vasculopathies. Nat. Commun. 10, 2961 (2019).
Sanchez-Rodriguez, R. et al. Targeting monoamine oxidase to dampen NLRP3 inflammasome activation in inflammation. Cell. Mol. Immunol. https://doi.org/10.1038/s41423-020-0441-8 (2020).
Liu, D. et al. Advances in the molecular mechanisms of NLRP3 inflammasome activators and inactivators. Biochem. Pharm. 175, 113863 (2020).
Liu, G., Chen, X., Wang, Q. & Yuan, L. NEK7: a potential therapy target for NLRP3-related diseases. Biosci. Trends 14, 74–82 (2020).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 (2015).
Coll, R. C. et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 15, 556–559 (2019).
Tapia-Abellan, A. et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 15, 560–564 (2019).
McElvaney, O. J. et al. Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 200, 1381–1391 (2019).
Hou, B. et al. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 11, 377 (2020).
Vande Walle, L. et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 17, e3000354 (2019).
Jiang, H. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214, 3219–3238 (2017).
Shen, K. et al. Molecular mechanism of a Specific NLRP3 inhibitor to alleviate seizure severity induced by pentylenetetrazole. Curr. Mol. Pharmacol. https://doi.org/10.2174/1874467213666200810140749 (2020).
Marchetti, C. et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl Acad. Sci. USA 115, E1530–E1539 (2018).
Shim, D. W. et al. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci. Rep. 7, 15020 (2017).
Darakhshan, S. & Pour, A. B. Tranilast: a review of its therapeutic applications. Pharm. Res. 91, 15–28 (2015).
Huang, Y. et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 10, e8689 (2018).
He, H. et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 9, 2550 (2018).
Shi, Y. et al. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J. 34, 208–221 (2020).
Hughes, M. M. et al. Glutathione transferase omega-1 regulates NLRP3 inflammasome activation through NEK7 deglutathionylation. Cell Rep. 29, 151–161 (2019). e155.
Xie, Y. et al. Development of benzenesulfonamide derivatives as potent glutathione transferase omega-1 inhibitors. J. Med. Chem. 63, 2894–2914 (2020).
Kim, S. K., Choe, J. Y. & Park, K. Y. Anti-inflammatory effect of artemisinin on uric acid-induced NLRP3 inflammasome activation through blocking interaction between NLRP3 and NEK7. Biochem. Biophys. Res. Commun. 517, 338–345 (2019).
Kaneko, N. et al. KN3014, a piperidine-containing small compound, inhibits auto-secretion of IL-1beta from PBMCs in a patient with Muckle-Wells syndrome. Sci. Rep. 10, 13562 (2020).
Yang, G. et al. Direct binding to NLRP3 pyrin domain as a novel strategy to prevent NLRP3-driven inflammation and gouty arthritis. Arthritis Rheumatol. 72, 1192–1202 (2020).
Susjan, P. et al. Selective inhibition of NLRP3 inflammasome by designed peptide originating from ASC. FASEB J. https://doi.org/10.1096/fj.201902938RR (2020).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2017R1A5A2015385) and by the framework of an international cooperation program managed by the National Research Foundation of Korea (2015K2A2A6002008).
Author information
Authors and Affiliations
Contributions
E.-K.J. conceptualized the article. E.-K.J. and S.P. wrote and reviewed the manuscript. S.P., J.K.K. and P.S. constructed the figures and tables. C.S. provided edits and comments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paik, S., Kim, J.K., Silwal, P. et al. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol 18, 1141–1160 (2021). https://doi.org/10.1038/s41423-021-00670-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-021-00670-3
Keywords
This article is cited by
-
cGAS-STING, inflammasomes and pyroptosis: an overview of crosstalk mechanism of activation and regulation
Cell Communication and Signaling (2024)
-
Aiouea padiformis extract exhibits anti-inflammatory effects by inhibiting the ATPase activity of NLRP3
Scientific Reports (2024)
-
METTL14 contributes to acute lung injury by stabilizing NLRP3 expression in an IGF2BP2-dependent manner
Cell Death & Disease (2024)
-
GSNOR negatively regulates the NLRP3 inflammasome via S-nitrosation of MAPK14
Cellular & Molecular Immunology (2024)
-
The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases
Nature Reviews Cardiology (2024)
