
Abstract
It remains largely unclear how thymocytes translate relative differences in T cell receptor (TCR) signal strength into distinct developmental programs that drive the cell fate decisions towards conventional (Tconv) or regulatory T cells (Treg). Following TCR activation, intracellular calcium (Ca2+) is the most important second messenger, for which the potassium channel K2P18.1 is a relevant regulator. Here, we identify K2P18.1 as a central translator of the TCR signal into the thymus-derived Treg (tTreg) selection process. TCR signal was coupled to NF-κB-mediated K2P18.1 upregulation in tTreg progenitors. K2P18.1 provided the driving force for sustained Ca2+ influx that facilitated NF-κB- and NFAT-dependent expression of FoxP3, the master transcription factor for Treg development and function. Loss of K2P18.1 ion-current function induced a mild lymphoproliferative phenotype in mice, with reduced Treg numbers that led to aggravated experimental autoimmune encephalomyelitis, while a gain-of-function mutation in K2P18.1 resulted in increased Treg numbers in mice. Our findings in human thymus, recent thymic emigrants and multiple sclerosis patients with a dominant-negative missense K2P18.1 variant that is associated with poor clinical outcomes indicate that K2P18.1 also plays a role in human Treg development. Pharmacological modulation of K2P18.1 specifically modulated Treg numbers in vitro and in vivo. Finally, we identified nitroxoline as a K2P18.1 activator that led to rapid and reversible Treg increase in patients with urinary tract infections. Conclusively, our findings reveal how K2P18.1 translates TCR signals into thymic T cell fate decisions and Treg development, and provide a basis for the therapeutic utilization of Treg in several human disorders.
Similar content being viewed by others


Introduction
Regulatory T cells (Tregs) are essential mediators of immune tolerance. Alterations in their numbers or function contribute to the pathogenesis of autoimmunity and are central in tumor evasion mechanisms in malignant disorders.1,2,3,4
Thymic Treg (tTreg) and conventional T cells (Tconv) both differentiate from thymocytes in response to T cell receptor (TCR) signaling. tTreg receive a stronger TCR signal than Tconv during thymocyte development,5 and this is essential for the induction of tTreg-specific epigenetic changes and gene expression patterns.6 A sufficiently strong TCR signal results in the expression of proximal IL-2 signaling components, facilitating cytokine-mediated induction of FoxP3, the master transcription factor for Treg development and distinctive feature of Treg.7,8 Data from TCR-transgenic mouse studies suggest that TCR signal strength required for tTreg development lies between the signal strengths driving positive and negative selection.9,10
The induction and stabilization of FoxP3 expression are critical to tTreg fate, and are tightly regulated by a variety of signaling pathways such as NF-κB, PI3K/Akt, STAT5 and NFAT.11 Given that a single thymocyte–antigen presenting cell (APC) encounter is believed to determine tTreg differentiation,11 the regulation of these signaling pathways must be highly efficient and dependent on the integration of various cofactors, although only a few are known so far.12 One well-characterized factor that affects this process is intracellular Ca2+, which is known to regulate NFAT and NF-κB signaling.13 Therefore, an enhanced Ca2+ signal in response to TCR activation might be decisive for tTreg fate, as there is intraclonal competition for a restricted resource (self-antigens).14
Interestingly, previous studies have demonstrated that store-operated Ca2+ entry (SOCE) via Ca2+ release-activated Ca2+ (CRAC) channels is essential for Treg development and function. T cell-specific ablation of stromal interaction molecule 1 and 2 (STIM1 and STIM2), both of which are CRAC channel calcium-sensing proteins, led to a lymphoproliferative phenotype and a selective decrease in Treg numbers in the thymus and lymphoid organs in mice.15 However, pharmacological CRAC channel inhibitors only affected peripherally-induced Treg (pTreg).16 Therefore, CRAC channels are presumably not the major determinant of Ca2+-mediated regulation of tTreg development. Other so far unidentified factors should be involved in the regulation of Ca2+ dynamics related to tTreg development. As potassium (K+) channels are the driving force for sustained Ca2+ influx by mediating a hyperpolarizing outward current, they represent likely candidates that are involved in these processes.17,18 In the family of K+ channels, two-pore domain K+ (K2P) channels are especially suited to serve as such a regulatory factor for tTreg development as they are major determinants of K+ conductance, adjust excitability and counteract membrane potential depolarization. Moreover, K2P channels are regulated by various extra- and intracellular stimuli, thereby integrating diverse signals in multiple circumstances ranging from basic cellular functions to complex pathogenic processes.19 In accordance with their complex and variable functions, we and others have identified an essential role for K2P channels in (auto)immune processes such as T cell activation, modulation of effector functions and regulation of immune cell trafficking across the blood–brain barrier.20,21,22,23
Given that the K2P channel K2P18.1 (also known as TWIK-related spinal cord K+ channel, TRESK; encoded by the Kcnk18 gene) is strongly expressed in the thymus and spleen (as well as the spinal cord and brain),24,25 we speculated that it may play a role in tTreg development and function. Two unique features of K2P18.1 specifically argue for this potential: (1) Ca2+ influx induces calmodulin/calcineurin-mediated dephosphorylation of intracellular K2P18.1 domains, leading to an increased probability of the channel being open,26 and (2) sustained Ca2+ influx also induces calmodulin/calcineurin-mediated activation of the NFAT pathway, which is essential for T cell development.27 Interestingly, the binding affinity of the intracellular domain of K2P18.1 (calcineurin binding motif: PQIVID in mouse and PQIIIS in human K2P18.1) to calcineurin considerably exceeds that of NFAT1 (PxIxIT motif), and is the highest among all natural proteins identified so far.28
Therefore, in this study we sought to determine whether K2P18.1 plays a role in translating TCR signal strength to induce thymocyte differentiation into Treg. We show that TCR signal strength is coupled to K2P18.1 expression in tTreg progenitors. Via modulation of intracellular Ca2+ signals, K2P18.1 facilitates NF-κB- and NFAT-mediated FoxP3 expression, and thereby tTreg development and maturation. Pharmacological modulation of K2P18.1 allows to rapidly and reversibly adjust Treg numbers.
Results
Lymphoproliferative phenotype and reduced Treg numbers in Kcnk18 −/− mice
Ca2+ signaling pathways are essential for T cell development and function and in silico models predicted a critical role for K2P18.1 in these processes.17,29 Therefore, we investigated the immunological consequences of genetic Kcnk18 deletion (Supplementary information, Fig. S1a). Kcnk18−/− mice showed healthy development by histological analyses of central nervous system (CNS) and other organs (data not shown). However, Kcnk18 deletion led to a mild lymphoproliferative phenotype with enlarged spleens and higher absolute cell numbers (Fig. 1a), whereas relative proportions of immune cell subsets were not changed (Fig. 1b). Under TCR stimulation, splenocytes from Kcnk18−/− mice showed augmented proliferation compared to C57BL/6 wild-type (WT) mice (Fig. 1c), potentially related to enhanced immune cell activation and/or insufficient suppression by regulatory immune cells. However, under naïve conditions, CD4+ and CD8+ T cells displayed a similar activation status in WT and Kcnk18−/− mice (Supplementary information, Fig. S2a). A more detailed investigation of T helper (Th) subsets revealed reduced frequencies of CD4+CD25+FoxP3+ Treg in the Kcnk18−/− spleens compared to WT ones, whereas frequencies of Th1, Th2 and Th17 subsets were similar (Fig. 1d, e). In further support of a Treg-specific phenotype, in vitro induction of Th1, Th2 and Th17 subsets was unaltered in Kcnk18−/− mice (Supplementary information, Fig. S2b). Thus, loss of K2P18.1 function is associated with reduced Treg numbers.

a Upper panel: photographs of spleens from WT littermate and Kcnk18−/− mice. Scale bar, 5 mm. Lower panel, absolute numbers of splenocytes in WT and Kcnk18−/− mice (n = 8). b Relative proportions for major immune cell subsets of splenocytes from WT and Kcnk18−/− mice. NK natural killer cells, MM monocytes and macrophages, DC dendritic cells (n = 5 per group). c CFSE (carboxyfluorescein succinimidyl ester) proliferation assay of splenocytes isolated from WT and Kcnk18−/− mice. Splenocytes were stimulated with plate-bound anti-CD3 and soluble anti-CD28 antibodies at the indicated concentrations for 72 h and analyzed by flow cytometry (n = 6). d Frequency of Th1 (CD4+FoxP3–IFNγ+), Th2 (CD4+FoxP3–IL4+), Th17 (CD4+FoxP3–IL17+) and CD4+CD25+FoxP3+ Tregs of splenic CD4+ T cells (n = 6). e Representative flow cytometry plots of Th1, Th2, Th17 and Treg gating. Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Kcnk18 −/− mice show a defect in thymus-derived Treg development
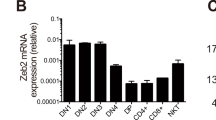
Next, we asked whether the reduction of Treg in Kcnk18−/− mice is a general effect observed in the whole Treg compartment or confined to thymus-derived (tTreg) or peripherally-induced Treg (pTreg). The observation that only tTreg (Helios-positive) but not pTreg (Helios-negative) were reduced (Fig. 2a) points toward a tTreg defect. In support of this hypothesis, (1) we observed significantly reduced Treg numbers in the thymus (tTreg, Fig. 2b, c); (2) in vitro generation of Treg from naïve cells was similar in Kcnk18−/− and WT mice (Fig. 2d); and (3) tTreg reduction was already evident at early developmental stages and was compensated by pTreg during aging (Fig. 2e). K2P18.1 expression was higher on Helios-positive Treg than Helios-negative Treg and positively correlated with Helios expression (Fig. 2f). Moreover, Kcnk18 and K2P18.1 were higher expressed in tTreg compared to other T cell developmental stages in the thymus at mRNA (Fig. 2g) and protein levels, respectively (Fig. 2h; Supplementary information, Fig. S8a). Thus, high K2P18.1 expression is specific for tTreg and functionally relevant to tTreg development. Consistent with this, a CD4+ cell-specific Kcnk18-knockout mouse exhibited a similar phenotype with an age-dependent reduction of tTreg in thymus (Fig. 2i) and spleen (Fig. 2j). Moreover, we observed no differences in major thymocyte or thymic APC subsets (Supplementary information, Fig. S3a, b). Cumulatively, these data show that Treg reduction observed in Kcnk18−/− mice is due to a defect in tTreg development.

a Frequency of Helios-positive and -negative WT and Kcnk18−/− Tregs in CD4+ T cells from spleen evaluated by flow cytometry (n = 8). b Frequency of Treg in thymic single positive (SP) CD4 thymocytes. Left, bar graphs; right, representative dot plots (n = 8). c Immunofluorescence staining for FoxP3 (green) and CD4 (red) in WT and Kcnk18−/− thymus. Scale bars represent 25 µm or 100 µm, respectively. Left panel, representative staining; right panel, quantification of CD4+FoxP3+ cells/mm2 (n = 3). d In vitro induced Treg (iTreg): CD4+CD25− naïve T cells were isolated from spleens by magnetic-activated cell sorting (MACS), stimulated with plate-bound anti-CD3 (1 µg/mL) and soluble anti-CD28 (2 μg/mL) in the presence of TGFβ (5 ng/mL) for 3 days, and then analyzed by flow cytometry. Left, bar graphs; right, representative dot plots (n = 8). e Age-dependent frequency of WT and Kcnk18−/− Tregs in thymus (left) and spleen (right) (n = 8). f Left: K2P18.1 expression in Helios-positive and -negative WT Tregs isolated from spleen; right: correlation of K2P18.1 and Helios expression (n = 8). g qPCR for Kcnk18 expression in double negative (DN), double positive (DP), CD4 SP (Tconv) and Treg isolated from WT thymus. Relative Kcnk18 expression (2–∆∆CT) compared to DN is depicted (n = 8). h Flow cytometry for K2P18.1 expression in Tconv (CD4+CD25−FoxP3−) and Treg (CD4+CD25+FoxP3+) isolated from WT thymus (n = 6). i, j Age-dependent frequency of Treg from WT and Kcnk18fl/fl × CD4cre thymus (i) and spleen (j) evaluated by flow cytometry (n = 8). Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
K2P18.1 deficiency has no impact on Treg phenotype and function
So far, we found reduced tTreg numbers, therefore we next asked whether the observed defect in tTreg development also affects Treg phenotype and function. The phenotypes of Kcnk18−/− and WT Tregs were similar as indicated by the expression levels of various Treg effector molecules (Fig. 3a). Suppression assays of Tconv proliferation by Treg revealed no differences in Treg function (Fig. 3b), which was further substantiated by unchanged Treg-related IL-10 production (Fig. 3c). Thus, loss of K2P18.1 function led to reduced tTreg numbers, whereas the Treg function is not impaired.

a Surface expression of the indicated effector molecules on Tregs isolated from spleens of WT and Kcnk18−/− mice (n = 3–4). b Suppression assay: Tconvs (CD4+CD25– T cells) were isolated from 2D2 (TCRMOG) transgenic mice and Tregs from WT and Kcnk18−/− mice, and then Tconvs were stained with CFSE and co-cultured with Tregs (WT or Kcnk18−/−, 1:1, 2:1, 4:1) and MOG (myelin oligodendrocyte glycoprotein)-loaded APCs for 3 days. Suppression of Tconv proliferation by Treg was calculated as proportion of proliferated Tconv with Treg divided by proportion of proliferated Tconv without Treg (n = 10 per group). c Intracellular cytokine staining for IL-10 (left, bar graphs; right, representative dot plots): MACS-isolated splenic CD4+ T cells were stimulated with plate-bound anti-CD3 (2.0 μg/mL) and soluble anti-CD28 (4.0 μg/mL) for 24 h, then re-stimulated with leukocyte activation cocktail (0.5 µg/mL, containing PMA, ionomycin and Brefeldin A) for 4 h and analyzed by flow cytometry. CD4+ T cells were used for gating on Treg (CD25+FoxP3+); IL-10 expression in Treg was assessed by plotting IL-10 against CD4 (n = 10). Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
tTreg development is regulated by the ion-current function of K2P18.1
To determine whether the impaired development of tTreg observed in response to loss of K2P18.1 function was related to the ion-current function of K2P18.1, we generated a mutant mouse line with a single amino acid change (G339R) eliminating ion-current flow through K2P18.1 (Supplementary information, Fig. S1b).30 We found the same phenotype with reduced tTreg in spleen and thymus (Fig. 4a) and preserved Treg function (Fig. 4b) in Kcnk18G339R mice as observed in Kcnk18−/− mice. Thus, proper tTreg development depends on the ion-current through K2P18.1 and not on interactions with other proteins via its intracellular domain.

a Treg proportions in thymus and spleen from WT, Kcnk18−/− and Kcnk18G339R mice (n = 5 per group). b Suppression assay of proliferation of Tconv from 2D2 mice by Tregs isolated from WT, Kcnk18−/− and Kcnk18G339R mice as described in Fig. 3b (n = 10 per group). Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
K2P18.1 regulates central signaling pathways for tTreg development
Next, we investigated the molecular mechanisms that connect K2P18.1 ion-current and the processes of tTreg development. Consistent with a regular function of tTreg, we found similar demethylation of the FoxP3-TSDR (forkhead box P3 Treg-specific demethylated region) in WT and Kcnk18G339R mice, which corresponds to stability of FoxP3 expression and immunosuppressive function of Treg (Fig. 5a).31 Furthermore, WT and Kcnk18G339R tTregs showed similar expression patterns of FoxP3-regulated tTreg signature genes, indicating that the defect in tTreg development occurs before established FoxP3 expression (Fig. 5b). Unbiased bulk RNA-sequencing revealed that compared to WT tTreg, Kcnk18G339R tTreg were characterized by gene expression patterns for decreased T cell activation and signaling and increased stress responses, cell cycle and apoptosis (Fig. 5c; Supplementary information, Fig. S4). However, cell cycle (Supplementary information, Fig. S5a), proliferation (Supplementary information, Fig. S5b), apoptosis rates (Supplementary information, Fig. S5c) and thymus egress rates (Supplementary information, Fig. S5d) were comparable between WT and Kcnk18G339R tTregs. For T cell signaling, pathways involving PI3K/Akt, NF-κB, NFAT and STAT5 have been implicated as important regulators of FoxP3 expression (Supplementary information, S1d). Therefore, we next investigated whether loss of the K2P18.1 ion-current leads to alterations in these cascades.11 In comparison to WT tTreg, Kcnk18G339R tTreg showed no relevant alterations in PI3K/Akt signaling (Fig. 5d). In contrast, NF-κB translocation to the nucleus was reduced in Kcnk18G339R tTreg, even though external TCR stimulation was able to compensate for those differences (Fig. 5e). Similar to NF-κB, we found decreased NFAT translocation to the nucleus in Kcnk18G339R tTreg (Fig. 5f); however, those alterations were not compensated by external TCR stimulation. K2P18.1 might not only influence the first step of TCR-dependent remodeling of the FoxP3 locus, but also the second cytokine-dependent step inducing FoxP3 expression via IL-2 signaling. However, we detected no differences in phosphorylated STAT5 levels, the major downstream target of IL-2-mediated signaling (Fig. 5g). Thus, loss of K2P18.1 ion-current function is associated with reduced nuclear translocation of NF-κB and NFAT, which are both regulated by distinct intracellular Ca2+ signals.32,33 K2P18.1 might therefore regulate NF-κB and NFAT signaling by fine-tuning intracellular Ca2+ concentration ([Ca2+]i). Consistent with this, live-cell Ca2+ imaging of tTreg revealed reduced Ca2+ influx upon TCR stimulation in cells from Kcnk18G339R mice as compared to WT mice (Fig. 5h). Therefore, K2P18.1 is able to regulate essential signaling pathways upstream of FoxP3 expression via modulation of [Ca2+]i.

a Methylation status of the FoxP3-TSDR in WT-Foxp3RFP and Kcnk18G339R/FoxP3RFP Tregs isolated from thymus or spleen (n = 4 per group). b, c mRNA sequencing of WT-FoxP3RFP and Kcnk18G339R/FoxP3RFP Tregs (CD3+CD4+CD8–CD25+FoxP3+) isolated from thymus: volcano plot of FoxP3-regulated Treg signature genes (b) and heatmap of unbiased analysis for significantly differentially expressed genes (FDR < 0.05), automated gene clustering (hclust) and GO-term annotation (n = 4 per group) (c). d Phospho-Akt (pAkt) expression in WT and Kcnk18G339R tTregs isolated from thymus analyzed by flow cytometry (n = 5). e, f Imaging flow cytometry of NF-κB (e) and NFAT (f) translocation to the nucleus in WT and Kcnk18G339R tTregs isolated from thymus. Cells were either left untreated or stimulated (stim) with plate-bound anti-CD3 (2 μg/mL) and soluble anti-CD28 (4 μg/mL) antibodies for 3 h (n = 4–5). Translocation scores under naïve and stimulated conditions were calculated. For f, right, representative images; BF, bright field; DRAQ5 (nuclear staining, red), NFAT (green), FoxP3 (yellow), merge (DRAQ5, NFAT). g Phospho-STAT5 (pSTAT5) expression analyzed by flow cytometry in naïve and stimulated tTregs from WT and Kcnk18G339R mice (n = 5). h Live-cell Ca2+ imaging of WT-Foxp3RFP and Kcnk18G339R/Foxp3RFP tTregs isolated from thymus. Left, peak maximum of ∆ fura-2 ratio; right, representative fluorescence microscopy images showing RFP expression (n = 4). Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
K2P18.1 expression provides a selection advantage for tTreg and is coupled to TCR signal strength via NF-κB signaling
For thymic selection, tTregs require strong TCR signals and thus also high [Ca2+]i.6,34,35,36 We therefore asked whether K2P18.1 is involved in the TCR-dependent selection process and whether there is a link between TCR signal and K2P18.1 to facilitate the Ca2+ signal in tTreg. Consistent with a specific role of K2P18.1 in the competitive processes of thymic tTreg selection, Kcnk18G339R mice showed a significantly narrowed Treg TCR repertoire in the periphery, whereas the Tconv TCR repertoire was not altered compared to WT mice (Fig. 6a). tTreg that received a strong TCR signal (as indicated by high Nur77 expression level)5 showed high K2P18.1 expression level, whereas this correlation was nearly abrogated in Kcnk18G339R mice (Fig. 6b). This indicates a biological interrelation of TCR signal and K2P18.1 via its ion-current function. In agreement, increasing concentrations of an anti-CD3 antibody led to a dose-dependent enhancement of K2P18.1 expression in tTreg (Fig. 6c), whereas Tconv showed rather decreasing K2P18.1 expression upon stimulation (Fig. 6d). These data indicate a specific link between TCR signal and K2P18.1 expression in tTreg. As the Kcnk18 gene locus shows multiple putative binding sites for the NF-κB-related protein c-Rel and NF-κB1, Kcnk18 expression might be connected to the TCR signal via NF-κB signaling (Fig. 6e; Supplementary information, Table S1). Consistent with this hypothesis, we observed that upregulation of Kcnk18 expression under TCR stimulation was inhibited by pharmacological blockade of NF-κB activation using cardamonin (CDN) or parthenolide (PTN) (Fig. 6f).37,38 These data collectively demonstrate that K2P18.1 expression provides a specific selection advantage for tTreg in the thymus and is coupled to TCR signal strength via NF-κB signaling.

a Clonality of TCR repertoire in WT (WT-Foxp3RFP) and Kcnk18G339R/Foxp3RFP Tconv and Treg in spleen and lymph nodes evaluated by deep sequencing (n = 6). b Correlation between K2P18.1 and Nur77 expression (area) in tTregs isolated from WT and Kcnk18G339R thymus. c, d Flow cytometry analysis for K2P18.1 expression in tTreg (c) and Tconv (d) isolated from thymus and stimulated with the indicated concentrations of anti-CD3 antibody and soluble anti-CD28 (2 µg/mL) (n = 6–8 per group). e Scheme of the region 2520 bp (5′UTR in magenta) upstream of Kcnk18 start codon showing putative transcription factor binding sites including NF-κB1 and p65/c-Rel. Kcnk18 (Entrez gene ID: 332396) was used for transcription factor binding site analysis with ConSite. See Supplementary information, Table S1 for detailed locations of transcription factor binding sites. f qPCR for Kcnk18 expression on WT tTreg isolated from thymus. Naïve or stimulated tTreg (stim, plate-bound anti-CD3 (2 μg/mL) and soluble anti-CD28 (4 μg/mL) antibodies for 3 h) were treated with different inhibitors of NF-κB signaling (20 µM CDN or 5 µM PTN for 3 h) (n = 6). Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
K2P18.1 channel function is important for FoxP3 induction in tTreg progenitors
Recently Owen et al.39 identified two distinct developmental programs for tTreg comprising CD25+ Treg progenitors (CD25+ TregP cells) and Foxp3lo Treg progenitors (Foxp3lo TregP cells). Therefore, we next questioned whether loss of K2P18.1 ion-current affects specific progenitor subsets. We found no differences in the proportions of CD25+ TregP cells between Kcnk18G339R and WT mice and less mature TregP cells in Kcnk18G339R mice, whereas FoxP3lo TregP cells and more mature Treg progenitors already expressing FoxP3 were reduced (Fig. 7a; Supplementary information, Fig. S3c, d), which indicates that K2P18.1 ion-current is especially important at stages of FoxP3 induction in tTreg progenitors. In support of this hypothesis, kinase activity profiles showed substantial alterations in CD25+ TregP cells from Kcnk18G339R mice (compared to WT mice, mainly comprising kinases involved in TCR, Ca2+ and NF-κB signaling), but not in FoxP3lo TregP cells (Fig. 7b). CD25+ tTregP cells showed a stronger correlation between K2P18.1 and Nur77 expression (Fig. 7c) and higher expression levels of K2P18.1 compared to FoxP3lo tTregP and mature tTreg cells (Figs. 7d, 6b). Nur77 expression is highly dependent on the intracellular Ca2+ signal.40 Consistent with an important role of K2P18.1 in Ca2+ signaling in tTreg progenitors, the correlation between Nur77 and K2P18.1 expression was nearly abrogated in Kcnk18G339R mice (Fig. 7c). Moreover, K2P18.1 and Nur77 expression was reduced in Kcnk18G339R mice compared to WT mice (Fig. 7d). The difference in Nur77 expression between Kcnk18G339R and WT mice was pronounced in CD25+ tTregP cells, which supports that K2P18.1 is especially important at stages of FoxP3 induction in tTreg progenitors (Fig. 7d). In accordance, stimulation of TregP with IL-2 and increasing doses of cloxiquine (CXQ) (a K2P18.1 agonist, for further details see below) led to increased mature tTreg only in CD25+ TregP (Fig. 8a) and not in FoxP3lo TregP cells (Fig. 8b). Further, this shows that K2P18.1 ion-current is important for the thymic selection process of tTreg as with increasing CXQ concentrations, tTreg populations with low Nur77 expression were increasingly selected. Thus, K2P18.1 channel function is especially important for tTreg developmental stages, where FoxP3 induction is required to proceed in maturation.

a Frequencies of the indicated developing tTreg cell populations (including FoxP3lo TregP (CD3+CD4+CD8–CD25–FoxP3lo) and CD25+ TregP (CD3+CD4+CD8–CD25+FoxP3−)) in the thymus of WT and Kcnk18G339R/Foxp3RFP mice (n = 5). b Kinase activity profiles of CD25+ TregP and FoxP3lo TregP sorted from Kcnk18G339R/Foxp3RFP thymus compared to the corresponding WT-Foxp3RFP cell subsets and annotation of kinases to the indicated signaling pathways. Color key indicates the mean kinase statistic (meanKS) of the differential kinase activity. A positive meanKS indicates enhanced kinase activity in Kcnk18G339R; negative meanKS shows reduced kinase activity in Kcnk18G339R (n = 6). c Correlation between K2P18.1 and Nur77 expression (area) in ex vivo CD25+ TregP and FoxP3lo TregP isolated from WT (upper panel) and Kcnk18G339R (lower panel) thymus. d K2P18.1 and Nur77 expression (MFI) in these cell types from WT or Kcnk18G339R thymus (n = 6). Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.

a, b CD25+ TregP (a) and FoxP3lo TregP (b) cells were isolated by cell sorting from WT and Kcnk18G339R/Foxp3RFP thymus and stimulated for 72 h with 1 U/mL IL-2 and increasing concentrations of CXQ (n = 8). Left, Frequencies of converted tTreg; middle, representative FACS plots showing the Treg gate of the indicated conditions; right, histograms displaying Nur77 levels amongst converted tTreg cells. Midline represents median of Nur77 expression in WT CD25+ TregP cells. Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Modulation of K2P18.1 function ameliorates autoimmune neuroinflammation in vivo
Reduced numbers and/or impaired function of tTreg are pathophysiologic hallmarks of autoimmune disorders. We thus asked whether activation of K2P18.1 can increase tTreg numbers and whether this is meaningful in the context of autoimmunity in vivo. We generated a mutant mouse line carrying a single amino acid mutation, S276A, which disrupted the phosphorylation of the S276 residue (Supplementary information, Fig. S1c). In contrast to G339R, introducing the S276A point mutation into murine K2P18.1 results in increased (4.2-fold) basal K+ currents.26 Single positive CD4 thymocytes isolated from Kcnk18S276A and Kcnk18G339R mice partly expressed K2P18.1, whereas tTregs were all K2P18.1 positive and showed high K2P18.1 expression levels (Supplementary information, Fig. S6a, b). Kcnk18S276A mice showed increased Treg numbers in thymus and spleen (Fig. 9a) and Treg function was not changed (Fig. 9b). These data further support an ion-current-mediated K2P18.1 effect on tTreg development and open up the possibility of a therapeutic application in the context of autoimmunity. To further investigate this possibility, we performed a detailed electrophysiological characterization of potential K2P18.1 modulators in K2P18.1-transfected HEK293T cells. CXQ and loratadine (Lo) were identified as most suitable activating and inhibiting agents, respectively (Supplementary information, Fig. 7a–k), and showed specific effects on K2P18.1-related potassium outward current in tTreg (Fig. 9c). In agreement with the results obtained in transgenic mice, K2P18.1 activation by CXQ facilitated Treg development, whereas inhibition by Lo reduced Treg numbers in thymic organ cultures (Fig. 9d). In contrast, thymic organ cultures of Kcnk18G339R mice showed no changes of Treg numbers upon drug treatment (Fig. 9d), indicating a specific effect of K2P18.1. Treatment of WT mice with CXQ and Lo for 7 days mimicked the in vitro results and demonstrated that pharmacological modulation of tTreg development is possible under physiological conditions (Fig. 9e). Next, we assessed whether these findings are relevant in the context of autoimmunity in vivo using experimental autoimmune encephalitis (EAE) as a paradigmatic model. In EAE, loss of K2P18.1 ion-current in Kcnk18G339R mice led to an aggravated disease course, whereas the prophylactic treatment with CXQ (starting 7 days before immunization to stimulate tTreg development) ameliorated disease severity (Fig. 9f). CXQ treatment had no impact on EAE disease course in Kcnk18G339R mice, indicating a specific, K2P18.1 ion-current-dependent effect of CXQ (Fig. 9f). Consistent with a Treg-driven and K2P18.1-specific effect, CXQ treatment increased the proportions of Treg in thymus, spleen and CNS only in WT mice, and did not change those in Kcnk18G339R mice (Fig. 9g). Compared to WT mice, Kcnk18G339R mice showed decreased Treg proportions (Fig. 9g) and an increase of pathogenic Th1 and Th17 subsets in spleen and CNS (Fig. 9h). CXQ treatment had no obvious effect on Treg function as indicated by similar IL-10 production in the presence or absence of CXQ (Fig. 9i). Thus, Treg numbers can be dynamically adjusted by pharmacological modulation of K2P18.1 in mice, which can be exploited for the therapy of autoimmunity.

a Treg proportions in thymus and spleen from WT, Kcnk18G339R and Kcnk18S276A mice (n = 8 per group). b Suppression assay of proliferation of Tconv by Treg isolated from WT and Kcnk18S276A mice (n = 6 per group). c Potassium outward currents in tTregs from WT-Foxp3RFP and Kcnk18G339R/Foxp3RFP mice with or without K2P18.1 inhibitor Lo (0.5 µM) or K2P18.1 activator CXQ (3 µM) treatment (n = 3–6). d E18 thymic organ cultures of WT and Kcnk18G339R mice treated with Lo (0.5 µM) or CXQ (3 µM). Flow cytometry of tTreg frequencies in the thymus on day 7 of thymic organ culture (n = 6 per group). e Treg frequencies in the thymus of WT mice on day 7 after daily i.p. injection of Lo (10 mg/kg) or CXQ (25 mg/kg) (n = 5 per group). f Disease course of MOG35–55 EAE in WT and Kcnk18G339R mice prophylactically (starting 7 days before immunization) treated with CXQ or vehicle (PBS) (n = 12 per group, two independent experiments). g Frequency of Treg in thymus, spleen and CNS of EAE mice (n = 8). h Frequency of Th1, Th2 and Th17 in spleen and CNS of EAE mice (n = 8 per group). i Intracellular cytokine staining for IL-10 in Tregs isolated from spleens of EAE mice. Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
K2P18.1 is involved in human tTreg development and autoimmunity
Next, we asked whether the findings in mice can be translated to humans. To test this, we investigated human thymus and blood samples. We found high expression of K2P18.1 and FoxP3 in the medulla of human thymus of healthy donors (HD) (Fig. 10a).41,42 In addition, most FoxP3+ cells in the thymic medulla co-expressed K2P18.1 (Fig. 10b). Moreover, human recent thymic emigrant (RTE) Treg (CD3+CD8−CD4+CD45RA+CD31+CD25−CD127+; Supplementary information, Fig. S8b) collected from peripheral blood mononuclear cells (PBMCs), expressed significantly more KCNK18/K2P18.1 than non-RTE (nRTE) Treg (CD3+CD8−CD4+CD45RA−CD31−CD25hiCD127lo) at both the mRNA and protein levels (Fig. 10c). Next, we investigated whether alterations in tTreg-related K2P18.1 function are relevant in the context of human autoimmunity. To do this, we compared the K2P18.1 protein expression on RTE and nRTE Tregs in relapsing-remitting multiple sclerosis (RRMS) patients versus HD. RRMS patients showed lower expression levels of K2P18.1 on both RTE and nRTE Tregs (Fig. 10d), suggesting that reduced K2P18.1 activity might be related to human autoimmunity. Interestingly, previous studies identified a single nucleotide polymorphism (SNP, rs140325655) coding for a dominant-negative missense K2P18.1 variant (K2P18.1-C110R) in which ion conductance is abolished.43 In a multicentre RRMS cohort, we identified 15 patients carrying the rs140325655 SNP. Cryopreserved PBMCs were available for seven of these patients. In accordance with our mouse data, the rs140325655 RRMS patients had significantly reduced numbers of Treg in the peripheral blood compared to RRMS controls (Fig. 10e). Functional parameters were not different between these two groups (Fig. 10f, g). However, rs140325655 RRMS patients showed more severe disability (EDSS) at baseline and 2-year follow up and significantly higher relapse rates (Fig. 10h). Cohort-specific (Münster versus Mainz) effects were not observed. A pharmacological activation of K2P18.1 might therefore have beneficial effects on human autoimmunity by increasing Treg numbers. For a first feasibility evaluation in humans, we searched for approved drugs with similar molecular structure to CXQ and identified nitroxoline, an antibiotic used to treat urinary tract infections (UTI) (Supplementary information, Fig. S9a).44 Nitroxoline significantly increased the ion-current through human K2P18.1 channels in an oocyte expression system (Fig. 10i). In UTI patients treated with nitroxoline or nitrofurantoin (another antibiotic used to treat UTI) for 7 days, we measured Treg numbers in the peripheral blood before, under and after therapy. Interestingly, only nitroxoline led to a rapid and reversible elevation of Treg numbers of up to 40.2% ± 7.5% compared to baseline (Fig. 10j). In vitro, nitroxoline had no impact on Treg proliferation unless toxic concentrations were reached (Supplementary information, Fig. S9b, c). In addition, Treg phenotype (Supplementary information, Fig. S9d) and function (Supplementary information, Fig. S9e) were similar to untreated conditions. Nitroxoline-treated subjects reported no adverse events except red discoloring of urine.

a Representative immunohistochemical staining of human thymus for K2P18.1 and FoxP3. Scale bars represent 200 µm or 50 µm, respectively (n = 3). b Representative immunofluorescence staining of human thymus for K2P18.1 (green), FoxP3 (red) and DAPI (blue). Inset shows marked area in higher magnification. Scale bars represent 50 µm or 25 µm, respectively (n = 3). c K2P18.1 mRNA and protein expression in sorted RTE Treg (CD4+CD8−CD31+CD25highCD127lo) and nRTE Treg (CD4+CD8−CD31−CD25highCD127lo) (n = 10 + 13). d Expression of K2P18.1 in RTE and nRTE Tregs from RRMS patients (MS, n = 25) and sex- and age-matched healthy controls (HD, n = 31). e Treg proportions in PBMCs from RRMS patients with (MS-SNP, n = 7) or without (MS-ctr, n = 15) the dominant-negative K2P18.1-C110R variant. f IL-10 production in Treg from RRMS patients with (MS-SNP, n = 7) or without (MS-ctr, n = 15) the dominant-negative K2P18.1-C110R variant. Left: bar graphs; right: representative dot plots. CD4+ T cells were used for gating on Treg (CD25+FoxP3+); IL-10 expression in Treg was assessed by plotting IL-10 against CD4. g Effector molecule expression in Treg from RRMS patients with (MS-SNP, n = 7) or without (MS-ctr, n = 15) the dominant-negative K2P18.1-C110R variant. h Clinical parameters of RRMS patients with (n = 15) or without (n = 15) the K2P18.1-C110R variant. EDSS, expanded disability status scale; ARR, annualized relapse rate; BL, baseline. i Normalized K2P18.1 current of oocytes transfected with hK2P2.1 in the presence of 300 µM nitroxoline (NTX, n = 7) or under control conditions (n = 6). Left, normalized hK2P18.1 current over time; Right, quantification of normalized current at the end of NTX application. Currents were normalized to the end of high-potassium buffer application. j Patients with UTI received nitroxoline (n = 4) or nitrofurantoin (n = 8) according to the summary of product characteristics for 7 days. Evaluation of Treg numbers by flow cytometry at the indicated time points. BL, baseline. Data are represented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Overall, these results strongly support a similar role for K2P18.1 in the regulation of tTreg development and autoimmunity in humans, further underlining the clinical relevance of our findings and opening up the possibility to exploit this mechanism in therapeutic applications. Drug repurposing might allow for immediate clinical drug development.
Discussion
tTreg development in thymus is a tightly regulated and highly efficient process. To trigger successful tTreg generation, TCR and consecutive intracellular Ca2+ signal strength need to reach a narrow corridor. However, the tuning mechanisms for those signals are largely unknown.
Here we found that genetic deletion of K2P18.1 induced a mild lymphoproliferative phenotype with reduced numbers of functional tTreg. The reduction of tTreg numbers was related to the loss of K2P18.1 ion-current function, which led to a defect in tTreg generation at developmental stages where FoxP3 expression is induced. K2P18.1 expression provided a specific selection advantage for tTreg progenitors and was coupled to TCR signal strength via NF-κB signaling. This advantage was provided by K2P18.1 via driving sustained Ca2+ influx inducing NF-κB- and NFAT-mediated FoxP3 expression. Pharmacological or genetic amplification of K2P18.1-mediated ion-current increased tTreg numbers and alleviated CNS autoimmunity. We observed a similar role for K2P18.1 in human tTreg development as well as in autoimmunity, and first in-human data showed the therapeutic potential of pharmacological K2P18.1 modulation.
The fact that K2P18.1-knockout mice and mice carrying a loss of ion-current function point mutation (G339R) presented the same phenotype of reduced tTreg, while the S276A point mutation, with augmented ion-current, led to increased tTreg numbers, strongly support our initial hypothesis that K+ conductance through K2P18.1 plays a crucial role in Treg development in the thymus.
Thymic selection of tTregs is limited by intraclonal competition for rare, tissue-specific self-antigens and Treg development seems to require higher TCR signaling strength than Tconv differentiation.5 Therefore, a prolonged Ca2+ signal mediated by K2P18.1 might facilitate TCR signal and Treg development. Similar to members of the tumor-necrosis factor receptor superfamily that couple TCR signal strength and tTreg development,12 Nur77 expression was positively correlated with K2P18.1 expression in tTreg progenitors. Furthermore, higher K2P18.1 expression resulted in a selective advantage to undergo maturation. We identified that the NF-κB pathway directly links TCR signaling and K2P18.1 expression.
Consistent with distinct developmental programs for different tTreg progenitor subsets,39 we found that the correlation between Nur77 and K2P18.1 was much stronger for CD25+ TregP cells than for FoxP3lo TregP cells and loss of K2P18.1 ion-current led to a pronounced reduction of Nur77 expression levels in CD25+ TregP cells. Furthermore, the kinome of CD25+ TregP cells was significantly altered in Kcnk18G339R mice, whereas only slight changes were observed for that of FoxP3lo TregP cells. Pharmacological activation of K2P18.1 increased mature tTreg only in CD25+ TregP cells and not in FoxP3lo TregP cells. Cumulatively, these data indicate a specific selection advantage by high K2P18.1 expression in CD25+ TregP cells. However, as we observed reduced proportions at all tTreg developmental stages that require induction and expression of FoxP3 including FoxP3lo TregP cells, this role of K2P18.1 seems to involve further tTreg progenitors. For FoxP3lo TregP cells, those have not been identified so far.39 Thus, K2P18.1 expression represents an important factor defining the Treg developmental niche decoding TCR signaling strength and facilitating FoxP3 expression.
Previous studies revealed that variations in TCR signaling strength are encoded as specific patterns of [Ca2+]i dynamics and then decoded into differential lymphocyte fates and functions.34,35,45 Ca2+-regulated transcription factors including NF-κB and NFAT decode those signals in lymphocytes and are essential for induction and stabilization of FoxP3 expression in tTreg.11,13,46,47 Loss of K2P18.1 ion-current function reduced nuclear translocation of both NF-κB and NFAT. External TCR stimulation was able to compensate for reduced NF-κB signaling. Thus, K2P18.1 ion-current function is even more important for NFAT-related cellular signals. This might be related to differential tuning of those transcription factors by [Ca2+]i. NF-κB is activated by one or few transient cytoplasmic Ca2+ spikes, whereas NFAT activation requires a sustained increase in [Ca2+]i.32,33 To achieve such high Ca2+ signals for NFAT activation in tTreg, we postulate a positive feedback loop involving TCR, NF-κB and K2P18.1 in which TCR-induced transient Ca2+ spikes activate NF-κB, which in turn facilitates K2P18.1 expression. Upregulation of this K+ conductance hyperpolarizes tTreg and thus enhances the driving force for sustained Ca2+ entry. Following tTreg maturation, loss of K2P18.1 might have no significant impact on Treg function since Treg does not require NFAT to suppress effector immune cells.48,49,50
Although bulk RNA-sequencing of tTreg suggested effects on cell cycle and apoptosis by loss of K2P18.1 ion-current function, subsequent functional experiments did not confirm these findings, which does not exclude discrete alterations that we were not able to detect. In addition, proliferation of tTreg was not obviously affected by loss of K2P18.1 function; however, further experiments might be required to exclude this. Furthermore, despite the observed lymphoproliferative phenotype, markers indicative of T cell activation and proliferation showed only slight alterations in Tconv from Kcnk18−/− mice, while not reaching statistical significance. These data are consistent with the observed lymphoproliferative phenotype, but the used markers seem not to be sensitive enough for the clear detection of effects on immune cell activation.
Treg cell research has impressively progressed over the past two decades. However, most knowledge is based on observations in animal models and studies in humans are sparse.42 To the best of our knowledge, this is the first report of a potential role for K2P18.1 in human tTreg development. We found that FoxP3+ thymocytes co-expressing K2P18.1 were predominantly in the medullary region of the human thymus, which is of particular relevance to Treg development.41,42 Furthermore, we observed higher K2P18.1 expression levels in RTE Treg compared to nRTE Treg. RTEs have been shown to be preferential precursors of Tregs in the periphery.51 In the context of human autoimmunity, such as in MS, K2P18.1 expression was reduced on both Treg subtypes. Whether this is a pathogenic feature and/or the consequence of MS remains elusive, however, these findings further support a central role of ion channels in MS.52 In strong support of a critical role of K2P18.1 in Treg-related immune tolerance and MS, MS patients carrying the missense K2P18.1 variant rs140325655, with abolished ion conductance, presented reduced Treg numbers in peripheral blood and poor clinical outcomes. Although this serves as a proof of principle, it remains elusive whether rs140325655 represents a relevant genetic risk variant in other MS cohorts.
We here showed that pharmacological modulators (activator: CXQ; blocker: Lo) are able to modulate K2P18.1 function, thereby adjusting Treg numbers with clear impact on autoimmunity in vivo. We did not observe alterations of tTreg numbers in Kcnk18G339R mice upon treatment with CXQ and Lo either in vitro or in vivo, arguing for specific effects of K2P18.1. However, as Lo is mainly known as histamine 1 receptor antagonist, further drug development is needed to reduce off-target effects. A pharmacological method capable of dynamically modulating Treg numbers might avoid the shortcomings of cell based-approaches such as high costs, long time required, low cell yield and extensive precautions.53,54,55 Moreover, our results from transgenic mice (e.g., CD4+ cell-specific Kcnk18−/− mice, major immune cell subsets in thymus and spleen, TCR repertoire of Tconv) argue for specific effects of K2P18.1 modulation on the tTreg compartment, which is in contrast to previously used Treg-inducing drugs such as rapamycin that broadly affects the immune system.56
Based on CXQ we searched for approved drugs with similar molecular structures and identified nitroxoline, an antibiotic used to treat UTI. Nitroxoline activated human K2P18.1 channel function in expression models and consistent with to our mouse data, led to a rapid and reversible elevation of Treg numbers with unaltered function in the peripheral blood of patients with UTI. Of note, nitrofurantoin, another antibiotic used to treat UTI, showed no effect on Treg numbers, arguing for a rather specific effect of nitroxoline and no major influence of the infection itself. The therapeutic implications of our findings might be far-reaching since a rapid and dynamic regulation of Treg numbers will be useful in a large variety of human disorders including autoimmunity, malignoma, sepsis, organ transplant or COVID-19 infection characterized by a viral response and a host (hyper)inflammatory disease phase.57 However, the low sample size does not allow for definitive conclusions and we cannot formally rule out off-target effects of nitroxoline on other K2P18.1-expressing cells affecting Treg numbers.
In conclusion, our data support an important role for K2P18.1 in tTreg development by a multistep mechanism: (1) TCR activation leads to upregulation of K2P18.1 expression via NF-κB; (2) high K2P18.1 expression allows for a selection advantage that enables Treg progenitors to undergo maturation; (3) K2P18.1-related intracellular Ca2+ signals facilitate NF-κB and NFAT signaling, forming a positive feedback loop that promotes FoxP3 induction and stabilization (Supplementary information, Fig. S1e). Thus, K2P18.1-related Ca2+ dynamics drive tTreg fate and define the tTreg developmental niche. Pharmacological modulation of K2P18.1-mediated Ca2+ signaling allows for dynamic regulation of tTreg numbers, suggesting that therapeutic administration of K2P18.1-modulating agents could treat a wide range of pathological conditions. First in human experiments with a K2P18.1 activator support further clinical development.
Materials and methods
Software for data acquisition and analysis and a detailed list of antibodies used in this study can be found in Supplementary information, Data S1.
Mice
All animal studies were approved by institutional care committee and state committees for animal welfare (84-02.04.2016_A307, A17.019). Animal experiments were conducted in accordance with the European Union normative for care and use of experimental animals and the German Animal Protection Law. Mice were raised in an in-house animal facility or purchased from Charles River and Jackson Laboratories and kept in individually ventilated cages under specific pathogen-free conditions and fed ad libitum. All mice were on a C57BL/6 background. Transgenic mouse lines were bred to generate homozygous mice. Mice were used at the age of 8–12 weeks for all experiments, if not stated otherwise.
Active MOG-EAE
Induction of EAE was performed in 8–12-week-old female mice as previously described.20,58 Briefly, MOG35–55 peptide was dissolved in phosphate-buffered saline (PBS) (2 mg/mL) and homogenized with complete Freund’s Adjuvant (CFA, 2 mg/mL) at 1:1 ratio and stored for 30 min at 4 °C. 100 µL MOG emulsion was injected into each flank of anesthetized mice (isoflurane). Injection of pertussis toxin (PTX, 1 µg/µL) was performed on day 0 and day 2 after intraperitoneal (i.p.) immunization with MOG (200 µL per dose). Health status (weight, disease score, general appearance and performance) of mice was monitored on a daily basis. Treatment of WT and Kcnk18G339R mice with CXQ was performed by i.p. injection of 25 mg/kg body weight CXQ (in 100 µL PBS, DMSO) or vehicle alone daily, starting 1 week before immunization.
Generation of Kcnk18 −/− mice
CRISPR-Cas9 system was used to generate Kcnk18−/− mice.59 The gRNA target was selected in the protein-coding part of exon 3 of the Kcnk18 gene. This gRNA was cloned into the plasmid gRNA_Cloning Vector (Addgene plasmid# 41824) digested with the AflII restriction endonuclease using Gibson assembly with help of oligonucleotide pairs: KCNK18_Insert_F2/KCNK18_Insert_R2, resulting in plasmid pgRNA_KCNK18cas2. CV19 ES cells (passage 13 (129SV × C57BL/6J)) were expanded and the linearized targeting vector DNA pKCNK18_targ (100 µg) was electroporated together with 70 µg of each plasmid DNA: pgRNA_KCNKcas2 and hCAS9 (Addgene plasmid# 41815) at 25 µF and 400 V in 0.8-mm electroporation cuvettes (Gene Pulser; Bio-Rad). After electroporation, cells were cultivated for 10 min at room temperature (RT) and plated onto ten 100-mm diameter culture dishes containing a γ-irradiated monolayer of mouse primary G418-resistant fibroblast feeder cells. After 32 h, 350 µg/mL G418 and 0.2 µM FIAU (2′-deoxy-2′-fluoro-β-D-arabinofuranosyl-5-iodouracil) were added to the culture medium. The medium was replaced every day, and colonies were picked and analyzed 8 days after plating. Positively-targeted ES cell clones were analyzed using Southern blot. Correctly-targeted ES cells from two independent clones (E1 and D2) were injected into 3.5-day B6D2F1 blastocysts. Routinely, 12–14 ES cells were injected into one blastocoel. After injection, the blastocoel was transferred into the uteri of 2.5-day pseudopregnant CD-1 foster mice. The mice carried pups to term. Positively-targeted F0 and F1 animals were identified by qPCR and sequencing analysis of genomic DNA isolated from tail biopsies. The subjects were male and female Kcnk18-deficient mice (Kcnk18+/−) and the mouse line was established by breeding male with female C57Bl/6J mice to produce heterozygous mice. Subsequently, heterozygous mice were interbred to achieve Kcnk18−/− homozygosity. In experiments WT littermates were used as controls.
Generation of Kcnk18 G339R mice
The G339R mutation, located in the selectivity filter of K2P18.1, demonstrates a loss of ion channel function. The G339R point mutation was obtained by random mutagenesis in C3HeB/FeJ mice as previously described.30 The C3HeB/FeJ-Kcnk18G339R mice were contributed by AG Molecular Electrophysiology (Institute of Physiology Würzburg, Germany), backcrossed to C57Bl/6 for ten generations and kept as homozygous lines. In experiments WT littermates were used as controls.
Generation of Kcnk18 S276A mice
The S276A mutation in murine K2P18.1 leads to highly increased basal K2P18.1 K+ currents (4.2-fold increase) and was proposed to be the main amino acid residue responsible for K2P18.1 Ca2+ sensitivity.26 The Kcnk18S276A mouse line was generated by direct oocyte microinjections using the CRISPR-Cas9 components together with the donor DNA oligo (Kcnk18_mutREV) followed by subsequent chirurgical embryo transfer. For the preparation of CRISPR-Cas9 microinjection solution, commercially synthesized Kcnk18_crRNA4 (CAGATTGCTGAGAATAGAAC), together with the tracrRNA and dCas9 protein were mixed as follows: 200 pmol of crRNA were mixed with 200 pmol of tracrRNA in 10 mM potassium acetate and 3 mM HEPES buffer (pH 7.5) and incubated at 95 °C for 2 min, followed by cooling to RT. The annealed crRNA/tracrRNA complexes were mixed with Cas9 mRNA, Cas9 protein, and Kcnk18_mutREV template DNA oligo. Microinjections were performed in B6D2F1 (hybrid between C57BL/6J and DBA strains) fertilized one-cell oocytes. Oocytes were removed from oviducts of super-ovulated B6D2F1 female mice in M2 media supplemented with hyaluronidase (400 µg/mL). Cytoplasmic microinjections were performed in M2 media using the Transjector 5246 (Eppendorf), and Narishige NT-88NE micromanipulators attached to a Nikon Diaphot 300 inverted microscope. Oocytes that survived microinjections were transferred to oviducts of pseudopregnant CD1 foster mice and carried to term. Positively-targeted F0 and F1 animals were identified by qPCR and sequencing analysis of genomic DNA isolated from tail biopsies. The subjects were males and females of Kcnk18S276A mice. The mouse line was established by breeding male with female C57BL/6J mice to produce heterozygous mice. Subsequently, heterozygous mice were interbred to achieve Kcnk18S276A homozygosity. In experiments WT littermates were used as controls.
Generation of Kcnk18 fl/f mice
To induce a cell type-specific deletion of K2P18.1, we generated a Kcnk18-floxed (Kcnk18fl/fl) mouse line that was then crossbred to a CD4cre mouse line to delete K2P18.1 specifically in CD4+ cells. The Kcnk18 gene targeting construct for insertion of the loxP sites (pKCNK18_targ.) was designed as follows: the 3.5 kb left flanking region containing intron 2 genomic sequences was PCR amplified from mouse genomic DNA using oligonucleotides KCN_FLAdn (GTCTCAAGCGTCTCTTGGAGCGGCCGCAAGGCCTCAAATCCCTGATGTAGT) and KCN_FLArn (GCTCTAGACGTCTCTGAGAGGATCCGACAGGCAGATAAGAAAGAAGAAAGG), and subcloned. The 1.8 kb right flanking region containing non-protein-coding part of exon 3 genomic sequences was PCR amplified using oligonucleotides KCN_FLBdn (GTCTCAAGCGTCTCTTGGACGCGTCTTCCCACACCTTGGTTTTATACAG) and KCN_FLBrn (GCTCTAGACGTCTCTGAGAGTCGACATAGATGATAGATAACCAAGAAAGAAAG) and consequently subcloned. The 1.0 kb protein-coding part of the exon 3 together with intronic sequences was PCR amplified using oligonucleotides KCN_ex3dn (GTCTCAAGCGTCTCTTGGAATTCAGGATGAGTAATTTTTGCTGTGTAA) and KCN_ex3rn (GCTCTAGACGTCTCTGAGACGCGTATAACTTCGTATAATGTATGCTATACGAAGTTATGGTACCTCACATCTGTAGGTCACGGAAA), and subcloned. The protein-coding part of the exon 3 flanking LoxP site together with the KpnI site was introduced by PCR cloning with help of the oligonucleotide KCN_ex3rn. All individual clones were verified by sequencing and assembled into the final targeting construct. The pBluescript-based plasmid backbone together with the negative selection marker (thymidine kinase cassette and diphtheria toxin gene), were added to the left flanking region. The positive selection marker (neomycin cassette flanked by two FRT sites and one LoxP site), was cloned as EcoRI–BamHI DNA fragment between left flanking region and 1.0 kb protein-coding part of exon 3 DNA fragment. The gRNA target was selected from the protein-coding part of exon 3 of the Kcnk18 gene. This gRNA was cloned into plasmid gRNA_Cloning Vector (gift from George Church (Addgene plasmid# 41824)) digested with the AflII restriction endonuclease, using Gibson assembly with help of oligonucleotide pair: KCNK_Insert_F2 (TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGAGATTGCTGAGAATAGAAC)/KCNK_Insert_R2 (GACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACGTTCTATTCTCAGCAATCTC), resulting in plasmid pgRNA_KCNKcas2. CV19 ES cells (passage 13 (129Sv × C57BL/6J)) were expanded in HEPES-buffered Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 15% fetal bovine serum (FBS), non-essential amino acids, L-glutamine, β-mercaptoethanol, 1000 U/mL recombinant leukemia inhibitory factor (MERCK Millipore), and antibiotics (100 U/mL penicillin and 100 U/mL streptomycin). For electroporation, 2 × 107 cells were resuspended in 0.8 mL Capecchi buffer (20 mM HEPES, pH 7.4, 173 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 6 mM dextrose, 0.1 mM β-mercaptoethanol). The NotI-linearized targeting vector DNA pKCNK18_targ. (100 µg) was electroporated together with 70 µg of each plasmid DNA, pgRNA_KCNKcas2 and hCAS9 (cas9 coding plasmid was a gift from George Church (Addgene plasmid# 41815)) at 25 µF and 400 V in 0.8 mm electroporation cuvettes (Gene Pulser; Bio-Rad). After electroporation, cells were cultivated for 10 min at RT and plated onto ten 100-mm diameter culture dishes containing a γ-irradiated monolayer of mouse primary G418-resistant fibroblast feeder cells. Thirty-two hours later, 350 µg/mL G418 (Invitrogen) and 0.2 µM FIAU (Moravek Biochemicals and Radiochemicals, USA) were added to the culture medium. The medium was replaced every day, and colonies were picked and analyzed 8 days after plating. Correctly-targeted ES cells from two independent clones (E1 and D2) were injected into 3.5-day B6D2F1 blastocysts. Routinely, we are injecting 12–14 ES cells into one blastocoele. After injection, blastocysts were kept in KSOM medium and subsequently transferred into the uteri of 2.5-day pseudopregnant CD-1 foster mice. The mice carried pups to term. Chimeras were identified by their agouti coat color contribution. For the germline transmission, high percentage male chimaeras were crossed to the C57BL/6J female mice. Heterozygous agouti offspring (Kcnk18fl/−) were confirmed by Southern blot analysis and further tested by PCR for the presence of the targeted allele. The FLPe-mediated neo cassette excision was performed in vivo by cross-breeding mice with the FLPe transgene, resulting in heterozygous Kcnk18-floxed mice (Kcnk18fl/+). Mice were kept in specific pathogen-free animal facilities. All mouse procedures were performed in compliance with the guidelines for the welfare of experimental animals issued by the Federal Government of Germany. The mouse line was established by breeding male with female C57Bl/6J mice to produce heterozygous mice. Subsequently, heterozygous mice were interbred to achieve Kcnk18fl/fl homozygosity.
Primary cell isolation
Leukocyte isolation
Organs (mouse spleen, thymus and lymph node) were homogenized by a 40 µm cell strainer and washed with 10 mL washing medium (DMEM, 1% FCS, 1% penicillin/streptomycin). Erythrocytes in the splenocyte suspension were lysed with ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3) for 30 s, stopped by addition of washing medium. Single cell suspensions were washed once again and resuspended in the desired buffer for subsequent applications.
Isolation of CD4+ T cells and Treg
To isolate CD4+ T cells and Treg from spleen or lymph node, CD4+ T Cell Isolation Kit (MACS, Miltenyi Biotec) or CD4+CD25+ Regulatory T Cell Isolation Kit (MACS, Miltenyi Biotec) were used according to the manufacturer’s protocol. In brief, single cell suspensions were incubated with the CD4+ T cell biotin–antibody cocktail for 5 min (4 °C), followed by the incubation with anti-biotin MicroBeads for 10 min (4 °C). Labeled cells were separated from the unlabeled CD4+ T cell population by magnetic field separation. For further Treg isolation, cells were labeled by a CD25-PE antibody and incubated with anti-PE MicroBeads (each step 15 min, 4 °C). Alternatively, CD4+ T cells and Treg (CD3+CD4+CD8–CD25+FoxP3+) were isolated by FACS sorting of Foxp3RFP mice, counterstained for CD3, CD4, CD25 and CD8a.
Isolation of thymic APC and mTEC
Thymus of WT and Kcnk18G339R mice was homogenized by enzymatic digestion. Therefore, the thymus was dissected into small pieces by cutting with scissors in 5 mL RPMI containing 2% FCS. 0.5 mg/mL collagenase D and 20 µg/mL DNase I were added to the tissue and incubated for 45 min at 37 °C on an orbital shaker. Digestion was stopped by 5 mL of 10 mM EDTA, followed by incubation for 5 min at RT. Thymus homogenate was then rinsed through a 70 µm cell strainer and washed with 20 mL RPMI + 2% FCS. To isolate CD11c+ cells, CD11c MicroBeads (MACS, Miltenyi Biotec) were used in a MACS separation according to the manufacturer’s protocol. After isolation, cells were stained for flow cytometry with antibodies directed to CD8, CD11b, CD11c, CD45R/B220, CD86, MHC-II, SIRPα. For CD45– mTEC isolation, we used the CD11c– flow-through from CD11c MACS isolation in a negative selection using CD45 MicroBeads (MACS, Miltenyi Biotec) according to the manufacturer’s protocol. After MACS isolation, CD45– mTEC was used for flow cytometry analysis (CD45, CD80, BP-1, EpCAM, MHC-II).
Proliferation assay
Splenocytes were isolated as described before. Then cells were labeled with VybrantTM CFDA SE Cell Tracer (12.5 µM) in 2 mL PBS + 2% FCS for 10 min at 37 °C, followed by addition of 10 mL cold washing buffer and incubation on ice for 10 min. Afterwards splenocytes were washed and seeded into 96-well plates (U-bottom) coated with different concentrations of anti-CD3. Soluble anti-CD28 was added to the splenocyte complete medium (DMEM, 10 mM HEPES, 25 µg/mL gentamycin, 5 µM β-mercaptoethanol, 1% non-essential amino acids, 5% FCS) as indicated in the respective experiments. Cells were cultured for 3 days (37 °C, 5% CO2) prior to FACS analysis.
The proliferation of human CD4+ T cells was assessed by labeling the cells with CFSE as described before.58 Cells were cultured in the presence of plate-bound anti-CD3 (1 µg/mL), soluble anti-CD28 (2 µg/mL) and nitroxoline at different concentrations for up to 7 days (37 °C, 5% CO2) prior to FACS analysis.
Suppression assay
Splenocytes were isolated as described before. CD4+CD25– Tconv and CD4+CD25+ Treg were isolated as described above using the CD4+CD25+ Regulatory T cell Isolation Kit (MACS, Miltenyi Biotec). Tconv were stained with CFSE to track cell proliferation and co-cultured with Treg (4:1 ratio, stimulation with 0.5 µg/mL plate-bound anti-CD3, 1 µg/mL soluble anti-CD28). Cells were cultured for 3 days (37 °C, 5% CO2) prior to FACS analysis. For antigen-specific suppression assays, Treg from the respective mouse line and Tconv from 2D2 mice were isolated and labeled with CFSE as described before. For isolation of APCs from WT mice, the spleen was homogenized by enzymatic digestion. Therefore, 0.1 mg/mL collagenase D was injected into the spleen and incubated for 15 min at 37 °C. Spleen was homogenized by a 70 µm cell strainer. To isolate CD11c+ APCs, CD11c MicroBeads (MACS, Miltenyi Biotec) were used in a MACS separation according to the manufacturer’s protocol. APCs were incubated with 20 µg/mL MOG35–55 for 10 min at 37 °C and washed with PBS before setting up the culture. A co-culture of 5 × 104 APCs and in total 2 × 105 Tconv and Treg (ratios of 1:1, 2:1 and 4:1) was set up for 3 days. Flow cytometry analysis of CFSE-labeled Tconv was used to determine the suppressive capacity of Treg.
In vitro Treg induction
Splenocytes were used for isolation of CD4+CD62L+ naïve T cells by the CD4+CD62L+ T Cell MACS Isolation Kit (MACS, Miltenyi Biotec). Then CD4+CD62L+ T cells were seeded into 96-well plates (U-bottom) in the presence of 2 µg/mL plate-bound anti-CD3 and 1 µg/mL soluble anti-CD28, 10 µg/mL anti-IL-4, 10 µg/mL anti-IFN-γ and 5 ng/mL TGFβ in mouse T cell medium (IMDM, 10% FCS, 1% L-glutamine, 5 µM β-mercaptoethanol) for 3 days (37 °C, 5% CO2) prior to FACS analysis.
In vitro Th1, Th2 and Th17 induction
Splenocytes were used for isolation of CD4+CD25–CD44− naïve T cells using the naïve CD4+ T Cell MACS Isolation Kit (MACS, Miltenyi Biotec). Then CD4+CD25–CD44– T cells were seeded into 96-well plates (U-bottom) in the presence of 2 µg/mL plate-bound anti-CD3 and 4 µg/mL soluble anti-CD28. For Th1 induction, 200 U/mL IL-12, 50 U/mL IL-2 and 10 µg/mL anti-IL-4; for Th2 induction, 200 U/mL IL-4, 50 U/mL IL-2 and 10 µg/mL anti-IFNγ; for Th17 induction, 10,000 U/mL IL-6, 40 U/mL IL-23, 8400 U/mL IL-1b, 10 U/mL human-TGFβ1, 10 µg/mL anti-IL-4, 10 µg/mL anti-IFNγ and 10 µg/mL anti-IL-2 were added to the respective culture in mouse T cell medium (IMDM, 10% FCS, 1% L-glutamine, 5 µM β-mercaptoethanol) for 3 days (37 °C, 5% CO2) prior to FACS analysis.
Thymic Treg induction from progenitor cells
Thymic Treg progenitor cells (CD3+CD4+CD8−CD25+FoxP3− TregP and CD3+CD4+CD8−CD25–FoxP3lo TregP) were isolated by FACS sorting. Cells were then cultured in mouse T cell medium with or without low-dose IL-2 (1 U/mL) in the presence of different concentrations of CXQ (1 µM, 3 µM) for 3 days (37 °C, 5% CO2). Differentiated cells were stained for Treg cell markers (CD3+CD4+CD25+FoxP3+) for quantification of mature tTreg induction and for Nur77 to assess TCR activation by flow cytometry.
Immunophenotyping
Phenotyping of different immune cell subsets was performed using flow cytometry. Therefore, immune cell subsets from spleen and thymus were characterized by staining for CD3, CD4, CD8, CD11b, CD11c, CD19, CD45R/B220 and NK1.1 and CD44, CD62L and CD69 to assess their activation status. For characterization of the T-helper cell subsets Th1, Th2 and Th17, cells were intracellularly stained for their respective signature cytokines IFNγ, IL-4 and IL-17A. Further characterization of Tregs was performed by staining for CD3, CD4, CD8, CD25, FoxP3, Helios, IL-10, CD39, CD73, GITR (CD357), ICOS (CD278), TIGIT, CTLA-4 (CD152) and LAG3 (CD223). Thymic T cell developmental stages were assessed by staining for CD3, CD4, CD5, CD8, CD44, CD45R/B220, CD69 and TCRβ: DN (CD3+CD4−CD8− double-negative), DP (CD3+CD4+CD8+ double-positive), SP4 (CD3+CD4+CD8− single positive) and SP8 (CD3+CD4−CD8+ single positive) cell subsets. Development stages of SP4 cells towards tTreg were evaluated in Foxp3RFP mice by staining for CD3, CD4, CD8, CD25, CD69, CCR7 and CCR9. Thymic APCs were characterized using antibodies directed to CD11b, CD11c, CD45R/B220, CD86, MHC-II and SIRPα; mTEC by CD45, CD80 BP-1, EpCAM and MHC-II.
Cell cycle analysis
Thymic CD4+ T cells were isolated with CD4+ T Cell Isolation Kit (MACS, Miltenyi Biotec) as described above. Cells were analyzed directly after isolation and after 24 h and 48 h of stimulation (plate-bound anti-CD3 (2 µg/mL), anti-CD28 (4 µg/mL) in solution). Then cells were stained for CD4, CD8 and FoxP3 to gate for Treg cells as well as for PCNA and PI to analyze the cell cycle with flow cytometry. Cells in G0-phase were PCNA−/PI−, in G1-phase PCNAlo/PIlo, in S-phase PCNA+/PIint and in G2-phase PCNAlo/−/PI+.
Apoptosis analysis
CD4+ T cells from spleen and thymus were isolated with CD4+ T Cell Isolation Kit (MACS, Miltenyi Biotec). Cells were stained with CD3, CD4, CD8, CD25, FoxP3, fixable viability dye eFluor780 (FVD780) and Fam-FLICA-DEVD (FAM-FLICA Caspase 3 & 7 Assay Kit, LKT Labs) to analyze proportion of apoptotic cells. Caspase3/7+FVD− cells were defined as early apoptotic and Caspase3/7+FVD+ cells as late apoptotic.
Transcription factor staining
CD4+ T cells from thymus were isolated with CD4+ Isolation Kit (MACS, Miltenyi Biotec). Cells were stimulated for 3 h with 2 µg/mL anti-CD3 (plate-bound) and 4 µg/mL soluble anti-CD28 in mouse T cell medium. After harvest, cells were stained for Treg marker and transcription factors (NFAT, NF-κB, pSTAT5, pAkt). Mean fluorescence intensity (MFI), measured by flow cytometry, was used for pSTAT5 and pAkt as a measure for activation of STAT5 and Akt. Translocation of NFAT and NF-κB was assessed by ImageStream (AMNIS INSPIRE acquisition software, Luminex) and translocation score was calculated using nuclear translocation wizard in AMNIS IDEA software (Luminex).
Analysis of K2P18.1 and Nur77 expression
CD4+ T cells were stained for CD3, CD4, CD8, CD25, FoxP3, K2P18.1 (anti-TRESK (E-2) monoclonal antibody (Santa Cruz, sc-514525), followed by secondary antibody staining) and Nur77, and analyzed by flow cytometry. For assessment of K2P18.1 expression upon TCR stimulation, CD4+ T cells were stimulated for 24 h with and without plate-bound anti-CD3 (0, 0.1, 0.5, 1 and 2 µg/mL) in mouse T cell media in the presence of soluble anti-CD28 (2 µg/mL) in all conditions. Thereafter cells were stained for CD3, CD4, CD8, CD25, FoxP3 and K2P18.1 and analyzed by flow cytometry.
Blocking NF-kB signaling
CD4+CD25+ cells were isolated from WT thymus and stimulated with 2 µg/mL plate-bound anti-CD3 and 4 µg/mL soluble anti-CD28 for 3 h at 37 °C, 5% CO2. 20 µM CDN (Sigma-Aldrich), 5 µM PTN (Sigma-Aldrich) or DMSO was added to the cell culture during stimulation and in the unstimulated conditions. Cells were collected after 3 h and prepared for RNA isolation.
Calcium imaging
CD4+ T cells were plated on 18 mm coverslips pre-coated with Poly-D-Lysine (PDL) at a density of 3 × 106 cells per coverslip. Cells were centrifuged at 300 × g for 2 min and subsequently processed for imaging experiments without resting. This allowed attachment of the cells to the coverslip throughout the experiment. No movement of the cells was observed after change of solutions within the recording chamber. T cells were loaded for 30 min with fura-2 AM and 0.005% Pluronic (Sigma-Aldrich) in a HEPES-buffered solution (artificial CSF, containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM glucose, 2 mM MgSO4, 2 mM CaCl2 and 30 mM HEPES, pH 7.35, osmotic concentration of 305 mOsmol/kg). Prior to experiments, PDL was removed and coverslips were washed with the HEPES-buffered solution, which then served as the extracellular solution throughout the experiment. Prior to the Ca2+ imaging experiment, RFP-positive cells were visualized by exciting with a LED lamp at 530 nm. The fluorescence emission was collected at > 560 nm. In addition, a bright field image allowed visualization of all CD4+ T cells and thus calculation of the percentage of RFP-positive cells (FoxP3+ Treg). [Ca2+]i was measured in single lymphocytes using excitation light provided by a LED lamp, with the beam passing through a monochromator at 340 nm and 380 nm with bandwidth of 10 nm (Cairn Research, Kent, UK). Emitted fluorescent light passed through a 515-nm long pass filter to a cooled CCD camera (Retiga; QImaging) and was digitized to 12-bit resolution. Ca2+ imaging data was acquired at a frame interval of 2 s and analyzed using MetaFluor® Fluorescence Ratio Imaging Software (Molecular Devices, LLC). Traces were computed and plotted as fura-2 ratio of excitation acquired at 340 nm and 380 nm, both with emission at > 515 nm. Prior to stimulation of the cells with a mixture of 2 µg/mL biotin anti-CD3, 4 µg/mL biotin anti-CD28 and 40 µg/mL avidin, a 60 s baseline Ca2+ signal was recorded. The CD3/CD28-induced Ca2+ signal was measured in single cells due to the heterogeneous responses obtained. Ionomycin (1 µM) was given at the end of the experiment to assess viability of the cells. Ca2+ responses were visualized using Origin (OriginLab). A positive Ca2+ response was scored and measured if a fluorescence increase of 0.4 fura-ratio within 7.5 min after application of the CD3/CD28 mix was detected. This cutoff was defined to account for spontaneous non-CD3/CD28-driven Ca2+ oscillations which can be seen in lymphocytes. Areas under the curve (AUCs) were computed within a defined region using the AUC analyzer tool in Origin.
Intracellular cytokine staining
For analysis of IL-10 production of Treg with flow cytometry, CD4+ T cells were first stimulated for 2 days with 2 µg/mL plate-bound anti-CD3 and 4 µg/mL soluble anti-CD28 in mouse T cell medium. Then cells were re-stimulated with 0.5 µl/mL leukocyte activation cocktail containing PMA, ionomycin and brefeldin A for 4 h. Cells were stained for CD3, CD4, CD8, CD25, FoxP3 and IL-10 using the FoxP3/Transcription factor staining buffer kit and analyzed by flow cytometry.
Thymic organ cultures
P0 mice were sacrificed by decapitation and thymic lobes were removed, separated and transferred into 24-well Transwell® dishes (pore size, 3 µm). The lower compartment of Transwell® dishes was filled with 600 µL RPMI containing 10% FCS and 1% Pen/Strep. Pharmacological compounds (DMSO, CXQ and Lo) were added into the lower compartment and organ cultures were kept in the incubator (37 °C, 5% CO2) for 4–7 days. Thymic cell analysis was performed by homogenizing thymic lobes and/or collecting emigrated cells from the supernatant and subsequent flow cytometry analysis. T cell egress ratios were calculated as the absolute numbers of T cells that migrated into the lower compartment divided by the numbers of T cells found in the thymic lobes.
TSDR methylation status
FACS-sorted Treg and Tconv from WT-Foxp3RFP and Kcnk18G339R/Foxp3RFP thymus and spleen were used for analysis of DNA methylation at the TSDR. Therefore, genomic DNA was isolated using Quick-DNA Microprep Plus Kit (Zymo Research) following the manufacturer’s instructions. Methylation analysis of the TSDR for low-input samples was performed by using bisulfite sequencing as described recently.60 We used the primers mTSDR-sen-for (5′-AGGTTGTTTTTGGGATATAGAATATG-3′), mTSDR-sen-rev (5′-ACCTATAAAATAAATTATCTACCCCCTTC-3′), mTSDR-sen-seq1 (5′-GTTGTTATAATTTGAATTTGGTTAG-3′) for amplification and pyrosequencing. The TSDR methylation analysis covers CpG motifs on chromosome X position 7450356–7450388 (genome assembly GRCm39).
Transcriptomics
RNA isolation
FACS-sorted T cell subsets from thymus and spleen were used for analyzing the expression of Kcnk18 by qPCR. RNA was isolated with Quick-RNA Microprep Kit (Zymo Research) following the manufacturer’s protocol. Tissue homogenates and cells were lysed in 300 µL RNA Lysis buffer, followed by sample clearing. The supernatant was mixed with 95%–100% ethanol and transferred to the column. In-column DNAse I treatment was performed. After washing and drying the column, RNA was eluted by pre-warmed DNase/RNase-free water (15 µL). RNA quality was assessed with NanoDrop by A260/A280 and A260/A230 ratios.
Real-time qPCR
Reverse transcription was performed with Maxima Reverse Transcriptase (Thermo Fisher Scientific) and random hexamer primers. 50 ng cDNA was used for Real-time qPCR with SYBR Green 2× Master Mix (Thermo Fisher Scientific). Therefore, 1 µM of each primer (mouse samples: mKcnk18fwd_qPCR and mKcnk18rev_qPCR; human samples: Hs_KCNK18_1_SG QuantiTect Primer Assay, Qiagen) or 1 µM housekeeping primer for the respective control (mouse samples: 18s-fwd and 18s-rev; human samples: pbgd-fwd and pbgd-rev), 10 µL SYBR Green Master mix, and 50 ng cDNA were mixed. PCR was performed on a Step-One-Plus Real-Time PCR System (Applied Biosystems) with the following steps: hold 2 min 50 °C, initial denaturation 10 min 95 °C, amplification (50×) 10 s 95 °C—45 s 58 °C—1 min 72 °C. Data were analyzed with StepOne software (Applied Biosystems, v2.1) calculating δCT values and n-fold expression.
RNA sequencing and analysis
FACS-sorted tTreg from WT-Foxp3RFP and Kcnk18G339R/Foxp3RFP thymus were used for bulk RNA sequencing. RNA was isolated with Quick-RNA Microprep Kit (Zymo Research) as described above. Quality and amount of RNA were verified by NanoDrop and Bioanalyzer RNA 6000 Nano Kit (Agilent). Samples with RIN values > 6.5 were used for RNA sequencing. NEBNext® rRNA depletion was performed followed by NEBNext directional Ultra RNA II Library preparation and sequencing on NextSeq500 (Illumina) platform (75 cycles, high output v2 kit). Raw sequencing data were analyzed by Linux bash tools following the analysis pipeline: (1) quality control (fastqc), (2) trimming (Trimmomatic 0.36),61 (3) alignment to mouse genome (Hisat 2.1.0; build: mm10),62 (4) aligned read sorting (Samtools)63 and (5) read counting (HTseq 0.10.0.64) Expression analysis was performed with R/Bioconducter DESeq2.65 Treg signature genes were assigned according to a previous study.66 Significantly regulated genes (FDR < 0.05) were used for further analysis and visualization using pHeatmap (https://cran.r-project.org/package=pheatmap) package in R. GO term gene enrichment analysis was performed using the PANTHER classification system online tool.67,68
TCR sequencing
RNA from FACS-sorted Treg from spleen and lymph node was isolated using Quick-RNA Microprep Kit (Zymo Research) as described above and reverse transcribed into cDNA. The TCRβ chain was then amplified by multiplex PCR using primers specific for all 54 known expressed Vβ and all 13 Jβ regions and then subjected to deep sequencing by ImmunoSEQ™ (Adaptive Biotechnologies Corp., Seattle, WA, USA). The data were analyzed using the ImmunoSEQ™ Analyzer software and measures of diversity were evaluated. Shannon entropy is calculated by summing the frequency of each clone times the log2 of the same frequency over all productive reads in a sample. When this value is normalized based on the total number of productive unique sequences and subtracted from 1, clonality results. Values for clonality range from 0 to 1. Values near 1 represent samples with one or a few predominant clones (monoclonal or oligoclonal samples) dominating the observed repertoire. Clonality values near 0 represent more polyclonal samples.
Analysis of transcription factor binding sites
2 kb upstream sequence including 5′UTR of mouse Kcnk18 (Entrez Gene ID: 332396) was used for analysis of potential transcription factor binding sites. Binding site analysis was performed using ConSite (http://consite.genereg.net/).69 Putative transcription factors were reviewed manually in the context of Treg development. Sequences of binding sites were checked by browsing transcription factors with Jaspar (http://jaspar.genereg.net/) and compared to the predicted site in the 2 kb upstream/5′UTR sequence (Supplementary information, Table S1).
Kinase activity profiliing
For serine-threonine kinase activity profiling, 6 biological replicates were sorted of both FoxP3lo TregP and CD25+ TregP from WT-FoxP3RFP and Kcnk18G339R/FoxP3RFP thymus. Cells were lysed on ice for 15 min by homogenizing with M-PER Mammalian Protein Extraction Reagent supplemented with 1× Halt Protease and 1× Phosphotase Inhibitor Cocktail (Thermo Fisher Scientific). Afterwards, lysates were centrifuged for 15 min at 16,000× g and 4 °C. The supernatants were snap frozen in liquid nitrogen and stored at −80 °C until further processing. On the next day, kinase assays were performed using the serine-threonine kinase (STK) PamChip® microarray system according to the manufacturer’s protocol (PamGene, HH’s-Hertogenbosch, The Netherlands). This multiplex phosphopeptide array-based methodology contains four positive controls and 140 serine/threonine peptides allowing the kinome-wide identification of hyper-activated kinases. Each of these peptides consists of a sequence of 15 amino acids which corresponds to a putative phosphorylation site serving as serine-threonine kinase substrates. As previously described,70,71 visualization of the phosphorylation activity of serine-threonine kinases was performed by a four-step protocol: blocking with 2% BSA (cycles 1–30) is followed by incubation of the protein lysates with a mixture of ATP and primary antibodies (assay mix, cycles 30–90). Next, samples are incubated with a fluorescently (FITC)-labeled secondary antibody (detection mix) and images are recorded (cycles 90–124). As previously stated,72 development of the fluorescence signal was detected by Alexa488 fluorescence. Measurements were performed on a Pamstation12 from PamGene. Unlike the company’s recommendation and due to low cell amounts after sorting, we analyzed only 5000 FoxP3lo TregP and 10,000 CD25+ TregP per array. Kinase activity profiling and data analysis were performed using Evolve software and BioNavigator Analysis tool (PamGene). Signal intensities were analyzed as a function of time. The Upstream Kinase tool was used to identify differentially active kinases between those in Kcnk18G339R and WT cells. The Kinase statistic (mean) score is calculated as the mean “treatment-control” group differences ((LFC)/Standard deviation), for a set of peptides assigned to a given kinase, derived from 6 current databases. The ranking factor is the Final (Median) Kinase Score (meanKS) which is based on a combined specificity score (derived from permutation of a set of peptides for a kinase) and significance score (derived from permutation of samples, in Kcnk18G339R and WT groups).
Immunofluorescence staining
Mouse thymus
Mouse thymus slices (10 µm) were fixed with 4% paraformaldehyde (PFA) for 10 min at 4 °C, washed three times with PBS and permeabilized with Fixation/Permeabilization buffer (eBioscience) for 30 min at RT, followed by blocking with 10% goat serum in PBS for 1 h at RT and a second blocking step using Avidin/Biotin Blocking Kit (15 min Avidin D, 15 min Biotin Solution). Slices were incubated with primary antibody (anti-FoxP3 Biotin, 1:30 in Permeabilization buffer) overnight at 4 °C. After washing, secondary antibody (Streptavidin-AF488, 1:1000 in Permeabilization buffer) was added to the slices and incubated for 3 h at RT. For CD4 staining, slices were washed, incubated first with anti-CD4 antibody (1:100 in PBS + 5% BSA for 2 h at RT) and second with Cy3 anti-rat antibody (1:500 in PBS + 1% BSA), and mounted with ProLong™ gold antifade mountant with DAPI (Invitrogen). Imaging was performed on Zeiss fluorescence microscope; ImageJ was used for cell counting.
Human thymus
Human thymus tissue of immunologically healthy patients was removed during necessary cardiac surgery (partial thymectomy) as a donation from patients for research proposes. The patient gave written informed consent according to national and European law, the declaration of Helsinki and the principles of Good clinical practice (GCP). All were approved by the faculty’s IRB (Ref# 46/00 and 45/00). For immunohistochemistry (IHC), tissue specimens were fixed in 4% PFA and embedded in paraffin. 4-µm-thick sections were used for IHC. IHC was performed using the Dako REALTM Detection System (Dako, #K5001), an automated immunostainer (AutostainerLink 48, Dako), and a biotin-streptavidin technique. Sections were deparaffinized and intrinsic peroxidase activity was blocked by incubation with 5% H2O2 in PBS for 5 min afterwards. We used mouse anti-FOXP3 (Abcam 20034; 1:100) and anti-TRESK (polyclonal, Immunoglobe; 1:100) as primary antibodies, and biotinylated secondary anti-mouse and -rabbit antibodies. For double-IHC, we used the abovementioned primary antibodies (1:50) followed by Cy3- or Alexa488-conjugated antibodies (1:250). Nuclei were stained using DAPI (1:5000, Invitrogen).
Human subjects
All healthy individuals (n = 31, age: 18–52 years, average: 29.4 ± 0.8, 47% female, 53% male) and RRMS patients (n = 55, age: 17–54 years, 30.3 ± 1.7, 52% female, 48% male, diagnosed according to the McDonald criteria 2017)73 were recruited at the Departments of Neurology of the University Hospital Münster and the University Hospital Mainz in Germany. Patients received pre-treatments including β interferons and glatiramer acetate, teriflunomide, fingolimod and natalizumab.
For isolation of human RTE and nRTE populations of CD4+ Tconv and Treg, we used frozen PBMC samples from healthy individuals and patients with RRMS. PBMCs were stained for CD4, CD31, CD45RA, CD25, and CD127 and subsequently sorted by FACS isolating the following subsets: Tconv RTE (CD4+CD25−CD31+CD45RA+), Tconv nRTE (CD4+CD25−CD31−CD45RA–), Treg RTE (CD4+CD25+CD127lo CD31+CD45RA+) and Treg nRTE (CD4+CD25+CD127lo CD31–CD45RA−). For analysis of K2P18.1 expression on RTE and nRTE subsets using flow cytometry, cells were stained for CD3, CD4, CD25, CD31, CD45RA and K2P18.1. In some experiments female patients with uncomplicated UTI received nitroxoline (mean age: 33.5 years, 250 mg 3×/d) or nitrofurantoin (mean age: 31.25 years, 50 mg 4×/d) for 7 days according to the summary of product characteristics. PBMCs were isolated before, under (day 4 and day 6) and after treatment (day 14 and day 21).
For rs140325655 SNP analyses, genomic DNA was isolated from frozen PBMCs using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Genomic DNA was then hybridized to Infinium OmniExpress BeadChip arrays for whole-genome genotyping (Illumina, CA, USA). After processing and quality control of the genotyping data, phasing and imputation were performed using IMPUTE2 (version 2.3.2). Confirmation of SNP status was performed using the TaqMan® Genotyping assay (Assay ID: C_160407463_10; Thermo Fisher, CA, USA). Patients with confirmed major allele were compared to patients with heterozygosity for the minor allele and were matched for sex, age (±3 years) and first immunomodulatory treatment. For evaluation of clinically relevant disease-related outcomes (time from clinical disease onset to definite diagnosis of RRMS, cumulative baseline disability reflected by EDSS at baseline and in year two and annualized relapse rate).
The experiments were performed according to the declaration of Helsinki and approved by the local ethics committee (Ref# 2014-398-f-S). All healthy donors and RRMS patients gave written informed consent.
Molecular biology and oocyte handling
hTRESK wt/pSGEM was a kind gift from Dr. Frank Döring (University of Wuerzburg). cDNA was linearized using NheI and in vitro transcription was performed using mMessage mMaschine T7 Transcription Kit (Invitrogen, Carlsbad, CA, USA). For two-electrode voltage clamp (TEVC) measurements, defolliculated oocytes were purchased from Ecocyte Bioscience (Dortmund, Germany). Oocytes were injected with 1 ng cRNA using Nanoliter Injector 2000 (WPI, Berlin, Germany) and stored at 18 °C in Barth solution for 48 h. Barth solution (containing 88 mmol/L NaCl, 1 mmol/L KCl, 0.4 mmol/L CaCl2, 0.33 mmol/L Ca(NO3)2, 0.6 mmol/L MgSO4, 5 mmol/L Tris-HCl, 2.4 mmol/L NaHCO3) was supplemented with 80 mg/L theophylline, 63 mg/L benzylpenicillin, 40 mg/L streptomycin and 100 mg/L gentamycin.
Electrophysiology
Whole cell recordings from transfected HEK293 cells (48–72 h post transfection) and primary mouse tTreg were performed at RT in a bath solution consisting of 135 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 5 mM HEPES, pH 7.4. Patch pipettes were pulled from borosilicate glass capillaries (Science Products, Hofheim, Gemany) and heat-polished to give input resistances of 3–6 megaohms. The pipette recording solution contained 140 mM KCl, 2 mM MgCl2, 1 mM EGTA, 1 mM Na2ATP, 100 μM cyclic AMP, and 5 mM HEPES, pH 7.3. Currents were recorded with an EPC9 (Heka) patch-clamp amplifier and low-pass filtered at 1–2 kHz. Stimulation and data acquisition were controlled by the Pulse/Pulsefit software package (Heka) on a Macintosh computer, and data analysis was performed with Igor software (WaveMetrics, Lake Oswego, OR).
In further experiments nitroxoline (Sigma-Aldrich) was prepared as 100 mM stock in DMSO and diluted with high-concentrated potassium buffer (containing 17.4 mmol/L NaCl, 80 mmol/L KCl, 1.8 mmol/L CaCl2 and 5 mmol/L HEPES (pH 7.4, adjusted using 1 M NaOH)) to a final concentration of 300 µM resulting in a DMSO concentration of 0.3%. Low-concentrated potassium buffer contained 95.4 mmol/L NaCl, 2 mmol/L KCl, 1.8 mmol/L CaCl2 and 5 mmol/L HEPES (pH 7.4, adjusted by 1 M NaOH). All test solutions were adjusted to a final DMSO concentration of 0.3%. Activity of nitroxoline in hTRESK-expressing Xenopus laevis oocytes was determined by TEVC at RT using previously described pulse protocols and recording pipettes (0.5–1.5 MΩ) backfilled with 3 M KCl.74 In short, oocytes were clamped to a holding potential of 0 mV and the following pulse protocol was applied 75 times (75 sweeps): 0 mV for 2 s, −100 mV for 0.5 s and 0 mV for 2 s. Low-potassium buffer (sweeps 1–15 and 61–75) followed by high-potassium buffer (sweeps 16–30 and 46–60) and 300 µM nitroxoline solution (sweeps 31–45) were sequentially superfused. Currents were normalized to the end of high-potassium buffer application (sweep 30) and effect of nitroxoline was evaluated at the end of compound application (sweep 45).
Statistical analysis
For column-based data, Gaussian distribution was evaluated by D’Agostino-Pearson normality test. Dependent on normality for analysis of two groups, two-tailed t-test (unpaired/paired) or Mann–Whitney rank sum or Wilcoxon paired signed-rank test was used. If more groups were compared, we applied one way ANOVA, complemented by Bonferroni test for multiple comparisons for parametric data, or the Kruskal–Wallis test including Dunn’s post hoc test for non-parametric data. Linear regression analysis was performed using UNIANOVA following graphical inspection of residuals for presence of heteroskedasticity. For datasets with presence of this phenomenon, we used the heteroskedasticity-consistent 3 (HC3)-standard error estimates.75 Comparison of EAE data was performed using two way ANOVA. P values indicated were obtained from column-factor analysis (comparison between groups). Statistical analysis was performed using SPSS 27 (IBM, NY, USA) or GraphPad Prism. P > 0.05 were classified as not significant, P < 0.05 (*) as significant, P < 0.01 (**) and P < 0.001 (***) as highly significant.
References
Najafi, M., Farhood, B. & Mortezaee, K. Contribution of regulatory T cells to cancer: a review. J. Cell. Physiol. 234, 7983–7993 (2019).
Dominguez-Villar, M. & Hafler, D. A. Regulatory T cells in autoimmune disease. Nat. Immunol. 19, 665–673 (2018).
Wing, K. & Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 11, 7–13 (2010).
Sakaguchi, S. et al. Regulatory T cells and human disease. Annu. Rev. Immunol. 38, 541–566 (2020).
Moran, A. E. et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 208, 1279–1289 (2011).
Morikawa, H. & Sakaguchi, S. Genetic and epigenetic basis of Treg cell development and function: From a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol. Rev. 259, 192–205 (2014).
Lio, C. W. J. & Hsieh, C. S. A two-step process for thymic regulatory T cell development. Immunity 28, 100–111 (2008).
Li, M. O. & Rudensky, A. Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 16, 220–233 (2016).
Maloy, K. J. & Powrie, F. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2, 816–822 (2001).
Hogquist, K. A. & Jameson, S. C. The self-obsession of T cells: How TCR signaling thresholds affect fate ‘decisions’ and effector function. Nat. Immunol. 15, 815–823 (2014).
Hsieh, C. S., Lee, H. M. & Lio, C. W. J. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 12, 157–167 (2012).
Mahmud, S. A. et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat. Immunol. 15, 473–481 (2014).
Berry, C. T., May, M. J. & Freedman, B. D. STIM- and Orai-mediated calcium entry controls NF-κB activity and function in lymphocytes. Cell Calcium 74, 131–143 (2018).
Bautista, J. L. et al. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat. Immunol. 10, 610–617 (2009).
Oh-hora, M. et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 9, 432–443 (2008).
Jin, S. et al. Natural regulatory T cells are resistant to calcium release-activated calcium (CRAC/ORAI) channel inhibition. Int. Immunol. 25, 497–506 (2013).
Eichinger, P. et al. Human T cells in silico: Modelling dynamic intracellular calcium and its influence on cellular electrophysiology. J. Immunol. Methods 461, 78–84 (2018).
Feske, S., Wulff, H. & Skolnik, E. Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 33, 291–353 (2015).
Feliciangeli, S., Chatelain, F. C., Bichet, D. & Lesage, F. The family of K2P channels: Salient structural and functional properties. J. Physiol. 593, 2587–2603 (2015).
Bittner, S. et al. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat. Med. 19, 1161–1165 (2013).
Meuth, S. G. et al. Altered neuronal expression of TASK1 and TASK3 potassium channels in rodent and human autoimmune CNS inflammation. Neurosci. Lett. 446, 133–138 (2008).
Bittner, S. et al. Expression of K2P5.1 potassium channels on CD4+T lymphocytes correlates with disease activity in rheumatoid arthritis patients. Arthritis Res. Ther. 13, R21 (2011).
Bittner, S. et al. Upregulation of K2P5.1 potassium channels in multiple sclerosis. Ann. Neurol. 68, 58–69 (2010).
Kang, D., Mariash, E. & Kim, D. Functional expression of TRESK-2, a new member of the tandem-pore K + channel family. J. Biol. Chem. 279, 28063–28070 (2004).
Keshavaprasad, B. et al. Species-spicific differences in response to anesthetics and other modulators by the K2P channel TRESK. Anesth. Analg. 101, 1042–1049 (2005).
Czirják, G., Tóth, Z. E. & Enyedi, P. The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J. Biol. Chem. 279, 18550–18558 (2004).
Macian, F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5, 472–484 (2005).
Czirják, G. & Enyedi, P. Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K+ channel, TRESK. J. Biol. Chem. 281, 14677–14682 (2006).
Lo, W. L., Donermeyer, D. L. & Allen, P. M. A voltage-gated sodium channel is essential for the positive selection of CD4 + T cells. Nat. Immunol. 13, 880–887 (2012).
Dobler, T. et al. TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J. Physiol. 585, 867–879 (2007).
Floess, S. et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5, 0169–0178 (2007).
Dolmetsch, R. E. & Lewis, R. S. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+channels generates [Ca2+]i oscillations in T lyrnphocytes. J. Gen. Physiol. 103, 365–388 (1994).
Dolmetsch, R. E., Lewis, R. S., Goodnow, C. C. & Healy, J. I. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386, 855–858 (1997).
Feske, S., Skolnik, E. Y. & Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 12, 532–547 (2012).
Vig, M. & Kinet, J. P. Calcium signaling in immune cells. Nat. Immunol. 10, 21–27 (2009).
Ruan, Q. et al. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity 31, 932–940 (2009).
Li, Y. Y., Huang, S. S., Lee, M. M., Deng, J. S. & Huang, G. J. Anti-inflammatory activities of cardamonin from Alpinia katsumadai through heme oxygenase-1 induction and inhibition of NF-κB and MAPK signaling pathway in the carrageenan-induced paw edema. Int. Immunopharmacol. 25, 332–339 (2015).
Saadane, A., Masters, S., DiDonato, J., Li, J. & Berger, M. Parthenolide inhibits IκB kinase, NF-κB activation, and inflammatory response in cystic fibrosis cells and mice. Am. J. Respir. Cell Mol. Biol. 36, 728–736 (2007).
Owen, D. L. et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat. Immunol. 20, 195–205 (2019).
Woronicz, J. D. et al. Regulation of the Nur77 orphan steroid receptor in activation-induced apoptosis. Mol. Cell. Biol. 15, 6364–6376 (1995).
Cowan, J. E. et al. The thymic medulla is required for Foxp3+ regulatory but not conventional CD4+ thymocyte development. J. Exp. Med. 210, 675–681 (2013).
Caramalho, Í., Nunes-Cabaço, H., Foxall, R. B. & Sousa, A. E. Regulatory T-cell development in the human thymus. Front. Immunol. 6, 395 (2015).
Andres-Enguix, I. et al. Functional analysis of missense variants in the TRESK (KCNK18) K + channel. Sci. Rep. 2, 237 (2012).
Kranz, J. et al. Clinical practice guideline: uncomplicated bacterial community-acquired urinary tract infection in adults-epidemiology, diagnosis, treatment, and prevention. Dtsch. Arztebl. Int. 114, 866–873 (2017).
Hogan, P. G., Lewis, R. S. & Rao, A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533 (2010).
Bettelli, E., Dastrange, M. & Oukka, M. Foxp3 interacts with nuclear factor of activated T cells and NF-κB to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. USA 102, 5138–5143 (2005).
Kwon, H. K., Chen, H. M., Mathis, D. & Benoist, C. Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat. Immunol. 18, 1238–1248 (2017).
Vaeth, M. et al. Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 109, 16258–16263 (2012).
Bandukwala, H. S. et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 34, 479–491 (2011).
Li, Q. et al. Constitutive nuclear localization of NFAT in Foxp3 + regulatory T cells independent of calcineurin activity. J. Immunol. 188, 4268–4277 (2012).
Paiva, R. S. et al. Recent thymic emigrants are the preferential precursors of regulatory T cells differentiated in the periphery. Proc. Natl. Acad. Sci. USA 110, 6494–6499 (2013).
Bittner, S. & Meuth, S. G. Targeting ion channels for the treatment of autoimmune neuroinflammation. Ther. Adv. Neurol. Disord. 6, 322–336 (2013).
Ferreira, L. M. R., Muller, Y. D., Bluestone, J. A. & Tang, Q. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 18, 749–769 (2019).
Romano, M., Fanelli, G., Albany, C. J., Giganti, G. & Lombardi, G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front. Immunol. 10, 43 (2019).
Elias, S. & Rudensky, A. Y. Therapeutic use of regulatory T cells for graft-versus-host disease. Br. J. Haematol. 187, 25–38 (2019).
Qu, Y. et al. The effect of immunosuppressive drug rapamycin on regulatory CD4+CD25+Foxp3+T cells in mice. Transpl. Immunol. 17, 153–161 (2007).
Pawlitzki, M. et al. Merits and culprits of immunotherapies for neurological diseases in times of COVID-19. EBioMedicine 56, 102822 (2020).
Ruck, T. et al. CD4+NKG2D+ T cells exhibit enhanced migratory and encephalitogenic properties in neuroinflammation. PLoS ONE 8, e81455 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Permanyer, M. et al. Efficient IL-2R signaling differentially affects the stability, function, and composition of the regulatory T-cell pool. Cell. Mol. Immunol. 18, 398–414 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Hill, J. A. et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800 (2007).
Thomas, P. D. et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 (2003).
Mi, H. et al. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204 (2009).
Lenhard, B. et al. Identification of conserved regulatory elements by comparative genome analysis. J. Biol. 2, 13 (2003).
Alack, K. et al. Profiling of human lymphocytes reveals a specific network of protein kinases modulated by endurance training status. Sci. Rep. 10, 888 (2020).
Chirumamilla, C. S. et al. Profiling activity of cellular kinases in migrating T-cells. in Methods in Molecular Biology 1930, 99–113 (Humana Press Inc., 2019).
Schwill, M. et al. Systemic analysis of tyrosine kinase signaling reveals a common adaptive response program in a HER2-positive breast cancer. Sci. Signal. 12, eaau2875 (2019).
Thompson, A. J. et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 17, 162–173 (2018).
Lengyel, M., Dobolyi, A., Czirják, G. & Enyedi, P. Selective and state-dependent activation of TRESK (K2P18.1) background potassium channel by cloxyquin. Br. J. Pharmacol. 174, 2102–2113 (2017).
Hayes, A. F. & Cai, L. Using heteroskedasticity-consistent standard error estimators in OLS regression: An introduction and software implementation. Behav. Res. Methods 39, 709–722 (2007).
Acknowledgements
This work was funded by the Interdisciplinary Center for Clinical Research (IZKF) of the Medical Faculty of Münster (Dan3/012/17 to U.D.), by the Deutsche Forschungsgemeinschaft (SFB BI1822/1–1 and CRC-TR-128 project B12 to S.Bi., project B04 to T.Bo. and project B06 to T.Bu. and S.G.M., and RTG 2515 (Chembion) to B.W., G.S. and T.Bu.), the Else Kröner-Fresenius-Stiftung (2018_A03 to T.R.) and the Hertie Foundation (mylab to S.Bi.). Further, this work was supported by a fellowship of the Graduate School of the Cells-in-Motion Cluster of Excellence (EXC 1003—CiM), University of Münster, Germany to S.Bo. We thank Jeannette Budde for excellent technical assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
T.R., S.Bo., S.Bi. and S.G.M. designed the study and methods; formal analysis was done by N.O., T.H., U.D. and G.S.; T.R., S.Bo., S.P., C.B.S., D.C., P.M., M.Lin., M.Lie., S.K., L.G., L.R., M. P., J.A.S., A.H., T.P., T.K., P.S., F.D., E.W., A.I.C., G.M.z.H., S.F. and J.H. performed the experiments and were responsible for data analysis; resources were provided by F.L., B.W., F.D., E.W. and B.T.; T.R. and S.Bo. wrote the original draft; B.W., L.K., H.W., T.Bu., T.Bo., G.M.z.H., B.T., N.O., T.H., U.D., F.D., E.W., J.R. and T.M. reviewed and edited the manuscript; funding acquisition was done by T. R., S.Bi. and S.G.M.; S.G.M supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruck, T., Bock, S., Pfeuffer, S. et al. K2P18.1 translates T cell receptor signals into thymic regulatory T cell development. Cell Res 32, 72–88 (2022). https://doi.org/10.1038/s41422-021-00580-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-021-00580-z
This article is cited by
-
NF-κB in biology and targeted therapy: new insights and translational implications
Signal Transduction and Targeted Therapy (2024)
-
Cloxyquin activates hTRESK by allosteric modulation of the selectivity filter
Communications Biology (2023)
-
Dietary regulation in health and disease
Signal Transduction and Targeted Therapy (2022)
-
Thinking outside the box: non-canonical targets in multiple sclerosis
Nature Reviews Drug Discovery (2022)
-
The current standing of autologous haematopoietic stem cell transplantation for the treatment of multiple sclerosis
Journal of Neurology (2022)
