
Abstract
Common fragile sites (CFSs) are genomic loci prone to the formation of breaks or gaps on metaphase chromosomes. They are hotspots for chromosome rearrangements and structural variations, which have been extensively implicated in carcinogenesis, aging, and other pathological processes. Although many CFSs were identified decades ago, a consensus is still lacking for why they are particularly unstable and sensitive to replication perturbations. This is in part due to the lack of high-resolution mapping data for the vast majority of the CFSs, which has hindered mechanistic interrogations. Here, we seek to map human CFSs with high resolution on a genome-wide scale by sequencing the sites of mitotic DNA synthesis (MiDASeq) that are specific for CFSs. We generated a nucleotide-resolution atlas of MiDAS sites (MDSs) that covered most of the known CFSs, and comprehensively analyzed their sequence characteristics and genomic features. Our data on MDSs tallied well with long-standing hypotheses to explain CFS fragility while highlighting the contributions of late replication timing and large transcription units. Notably, the MDSs also encompassed most of the recurrent double-strand break clusters previously identified in mouse neural stem/progenitor cells, thus bridging evolutionarily conserved break points across species. Moreover, MiDAseq provides an important resource that can stimulate future research on CFSs to further unravel the mechanisms and biological relevance underlying these labile genomic regions.
Similar content being viewed by others

Introduction
Common fragile sites (CFSs) are chromosome regions prone to forming gaps or breaks (termed CFS “expression”) visible during metaphase, especially following replication stress.1 Since the initial description of the 17 CFSs in human lymphocytes,2 including the widely-studied FRA3B and FRA16D loci, many studies have led to great expansion of this family, which now contains more than 80 members distributed on nearly all chromosomes.3 Interestingly, CFSs are not exclusive to human cells. Instead, they are well documented in a wide range of other species including non-human primates,4 carnivores,5 mice,6,7 and avians,8 suggesting a conserved role for CFSs. CFS expression can be induced by different replication-perturbing agents or conditions, e.g., low-dose aphidicolin (APH) that reversibly inhibits replicative DNA polymerase.9,10 CFSs become even more vulnerable to replication perturbations upon mutation of essential genome caretaker genes, such as the ataxia telangiectasia and Rad3-related (ATR),9,11 ataxia telangiectasia mutated (ATM),12 and Fanconi anemia (FA) proteins,13,14,15 whose deficiency leads to premature aging and cancer predisposition, which also implies a disease relevance of CFSs. Moreover, the FA complementation group D2 (FANCD2) was found to bind to CFSs under replication stress conditions and to persist at these loci until anaphase.13,16,17 Hence, FANCD2 is widely used as a protein marker to define the location of CFSs.
Although breaks and gaps formed at CFSs on metaphase chromosomes have been used to define these genomic loci, the underlying mechanism for their fragility has remained a mystery. In our previous studies, we have revealed that CFS expression under replication stress results from enzymatic cleavage of the under-replicated DNA in order to promote chromatid segregation.18 Moreover, we showed that CFS expression is associated with mitotic DNA synthesis (MiDAS) that occurs via a form of break-induced replication (BIR).19 CFSs are also hotspots of viral gene integration,20,21,22 sister chromatid exchange (SCE),23,24,25 and chromosome rearrangements,26,27 all of which are well implicated in cancer, although the sequence of events is yet to be delineated. Another prevalent characteristic of CFSs is that they are enriched in extremely large genes.8,28 For example, three of the most frequently expressed human CFSs, including FRA3B, FRA16D, and FRA6E, are all located in genes larger than 1 Mb.1 Recently, many of the CFS-associated genes have been shown to harbor copy number variants (CNVs), especially large gene deletions that are recurrent in a diverse range of cancers.1,28,29 In fact, a number of the large genes mapped to CFSs have been implied as tumor suppressors, for example, fragile histidine triad (FHIT) in FRA3B and WW domain-containing oxidoreductase (WWOX) in FRA16D, loss of which was experimentally shown to drive tumorigenesis and has been correlated with poor clinical outcomes.30,31,32,33
In addition to CFSs’ intensively-studied role in cancer, it has also been proposed that they promote neuronal genetic diversity and may impact on neuronal development and functions.34,35 In support of this hypothesis, a number of CFSs were associated with large genes involved in neuronal development and functions, such as activator of transcription and developmental regulator AUTS2, par-3 family cell polarity regulator beta (PARD3B), and limbic system-associated membrane protein (LSAMP).36 Consistent with this, a recent study has shown that 6 out of 27 recurrent double-strand break clusters (RDCs) in mouse neural stem/progenitor cells (NSPCs) can be matched to homologous human CFSs.37 Moreover, considering that CFSs are extremely unstable, but nevertheless conserved across species, arguments have been made that CFSs may play an evolutionary role, perhaps by virtue of the high frequency of chromosome rearrangements that occur at CFSs, which might drive genetic variations.34,38
Given the biological importance of CFSs, numerous efforts have been made to elucidate the mechanisms underlying CFS fragility. Early studies proposed various hypotheses that might explain the hypersensitivity of CFSs to replication stress. For example, abundant AT dinucleotide repeats found within many CFSs, such as FRA16D, have been proposed to be responsible for CFS vulnerability to replication perturbations.39,40,41 This is because such sequences are highly flexible and prone to forming secondary structures that impede DNA replication fork progression.42 Late replication timing has also been repeatedly argued as a determinant of CFS susceptibility to replication perturbations.43,44 In addition, numerous studies have underlined the role of replication origin paucity in CFSs.45,46 For instance, the over 1.5 Mb FRA3B was reported to contain only four putative active replication origins, which were clustered within a 50 kb region.47 The problem might be compounded by the fact that the dormant origins within CFSs seem to be less efficiently activated to compensate for impeded fork progression under replication stress,47 further increasing the chance of replication failures and thus CFS expression. More recently, large transcription units were brought into focus. It is posited that the persistent transcription of large genes throughout the cell cycle can interfere with prereplication complexes (pre-RCs) during the G1 phase,48,49 while creating a high probability of replication-transcription collision during S phase,50 which collectively contribute to the formation of CFSs.
Hitherto, most mechanistic models of CFS fragility have been proposed based on molecular dissection or sequencing of only a very small fraction of the well-characterized CFSs. Therefore, it remains unclear whether these models can reflect a ubiquitous mechanism, if there is one, by which CFS instability arises. It should be noted that data collected from different studies often lead to inconsistent conclusions, suggesting that CFSs may be heterogeneous, and that the labile nature of CFSs is more likely to depend on the interplay between multiple factors whose contributions vary in different genomic contexts. Therefore, to comprehend the mechanisms governing CFS instability, there is a need to perform multi-factor analysis of every individual CFS on a genome-wide scale. However, given the large number of CFSs in the human genome and the low resolution of their locations, it is unrealistic to address this question by conventional molecular analyses. In contrast, by virtue of advances in sequencing technology and accumulating data on the genomic landscape in human cells, computational analysis to identify contributing factors to CFS instability is realistic. Indeed, there have already been some attempts to computationally determine which features of the genomic context are predictive of CFS fragility.51 However, because the current resolution of cytogenetically mapped CFSs is too low to impart the exact sites of breaks or gaps, such computational analysis inevitably encounters considerable noise from “non-fragile” regions flanking the bona fide fragile sites. In this regard, a refined map is necessary in order to zoom in to the key regions of CFSs where replication stress has its impact.
To this end, we have developed a methodology for genome-wide mapping of CFSs by exploiting mitotic DNA synthesis (MiDAS) that predominantly occurs at CFSs.19 By sequencing of the nascent DNA from MiDAS events, in a protocol we term MiDASeq, we obtained a genomic atlas of APH-induced MiDAS sites (MDSs) at base pair-resolution, which covered most of the known CFSs. The accurate coordinates allowed us to zoom into each individual MDS to examine sequence characteristics that may explain the fragility of their associated CFS. Intriguingly, these MDSs overlapped with 21 out of 27 genes whose murine homologs were previously identified as RDCs in mouse NSPCs,37 implying strong conservation of these genomic loci across species. Moreover, through mining available datasets, we were able to simultaneously assess how different genomic features, such as the replication/transcription profile and epigenetic landscape, might contribute to the formation of MDSs, which could help to understand the instability of the associated CFSs. The results of our comprehensive analysis of MDSs tally well with classical theories explaining the cause of CFS fragility, demonstrating that MDSs are representative of CFSs and thus can be used to predict other characteristics of CFSs.
Results
Genome-wide fine mapping of MiDAS sites and CFSs in human cells
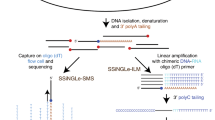
Previous studies have reported that CFSs frequently undergo MiDAS following replication stress.19,52 Therefore, we set out to accurately map CFSs by sequencing nascent DNA generated within the M phase. For this purpose, we established MiDASeq by modifying the 5-ethynyl-2′-deoxyuridine (EdU)-seq protocol53 for use in mitotic cells. For this, we first used EdU to label MiDAS in U2OS cells treated by low dose APH (0.3 µM), and then collected the mitotic cells for the Click-IT reaction that conjugated a biotin molecule to EdU (Fig. 1a). Genomic DNA was isolated and sonicated into 200–400 bp fragments. EdU-containing fragments were then captured by streptavidin beads, followed by end repair, A-tailing, and adapter ligation. A “MiDAS library” was generated off the beads using PCR as previously described,54 and subjected to next generation sequencing (NGS). To assure the validity of this method, we conducted the following controls. First, we demonstrated that streptavidin beads would specifically enrich biotinylated DNA fragments, by showing that only DNA containing dUTP-biotin could be captured and subsequently amplified by PCR (Fig. 1b). Second, we confirmed that the vast majority of the harvested cells were mitotic cells by both flow cytometry analysis and microscopic examination (Supplementary information, Fig. S1a–c). Third, the efficiency of Click-IT reaction within methanol-fixed cells was examined by flow cytometry (Supplementary information, Fig. S1d, e). Fourth, we verified that the sites of EdU incorporation in our experimental setup were representative of CFSs, as revealed by the colocalization of the EdU foci with breaks/gaps on metaphase chromosomes (Fig. 1c), and with FANCD2 in mitotic nuclei (Fig. 1d, e).

a Schematic diagram of MiDASeq. b Validation of the capture & PCR system after incubating streptavidin beads with synthetic DNA containing or not containing dUTP-biotin. c Upper panel, experimental scheme to enrich mitotic cells and to induce MiDAS; lower panel, two representative metaphase spreads showing the EdU signals residing in DAPI-negative gaps or breaks. d Three representative images showing colocalization of EdU (green) with CFS-binding FANCD2 protein (red) in mitotic nuclei after the cells were treated as in c. e Quantification of the percentage of FANCD2 foci that colocalized with EdU. A total of 227 mitotic nuclei were analyzed. Data are means ± SEM, ****P < 0.0001 calculated by paired two-tailed t-test. Scale bars: 20 µm (c), 5 µm (d).
The sequence data obtained were mapped to the human reference genome (version hg38) to identify peaks of MiDASeq. The EdU signal was calculated for each 100 kb genomic window, followed by normalization to the negative control group to acquire a fold change (FC). Only those genomic windows with a signal FC over 8 were output as positive peaks for further analysis. Since EdU peaks clustered in the same CFS gene were found to generally locate within a 100 kb distance to each other, we used 100 kb as the global standard for merging adjacent peaks into one MDS. We conducted three independent experiments to verify the reliability of MiDASeq. In two of the experiments, we labeled cells with EdU for 30 min and then collected them for downstream sequencing, while in the other experiment, we reseeded these collected mitotic cells for another 30 min of EdU labeling before harvesting. In fact, MiDASeq displayed great reproducibility, with an average overlapping rate of 88.1% between datasets (Supplementary information, Fig. S2a). In present study, we analyzed a total of 274 representative MiDASeq peaks from one of the three independent experiments, which were distributed across most human chromosomes (Fig. 2a, b; Supplementary information, Fig. S2b and Table S1). The sizes of these MDSs ranged from 0.1 to 2.3 Mb, with an average size of ~0.5 Mb (Supplementary information, Fig. S2c), which were far smaller than the cytogenetically defined CFSs. 152 of the MDSs were mapped within or adjacent to (< 2 Mb) 87 of the published CFSs, including but not limited to 3p14.2 (FRA3B), 16q23.2 (FRA16D), 7q11 (FRA7J), and 13q32 (FRA13D)36 (Fig. 2c–f; Supplementary information, Fig. S2e), corroborating that MDSs were strongly associated with CFSs in terms of genomic location. Other MDSs resided in the unreported genomic loci (Fig. 2g; Supplementary information, Fig. S2f). Compared to known CFS-associated MDSs, the “anonymous” MDSs were of similar genomic sizes and sequence scores (Supplementary information, Fig. S2c, d), which suggested that, at least in U2OS cells, they could also display as visible breaks or gaps like their counterparts and may represent suspicious CFSs specific for U2OS cells. Interestingly, some cytoband positions for CFSs encompassed multiple MDSs that were clearly separated in our refined map, such as 3p14.2 comprising FHIT- and PTPRG-associated MDSs (Fig. 2d),28 which may represent two neighboring CFSs including FRA3B (FHIT) and an anonymous one. Since FRA3B has been the only nomenclature for 3p14.2, the two neighboring CFSs in that region will lead to the confusion in the boundary of FRA3B when it is characterized in different cell lines that can express 3p14.2 differentially.46,55 Other CFSs associated with two or more MDSs, like 2q22 (FRA2K) and 7q11 (FRA7J) may encounter the same issue.

a Circos plot of the genomic locations of MDSs, with MiDASeq signal, gene density and AT content displayed by circles from outside to inside. Darker blue denotes higher AT content. b Chromosome view of the distribution of MDSs (red bands) mapped by MiDASeq, and RSs (blue bands) defined as the control in this study. c Summary of the number of MDSs identified in different cell lines, their association with previously defined CFSs, and overlap between cell lines. d–f Genomic view of representative MDSs mapped to known CFS locations, including 3p14.2 (FRA3B), 16q23.2 (FRA16D), and 7q11 (FRA7J). The dark blue bar at the top of each view indicates CFS location previously defined at the cytogenetic level. Red, light blue, and green peaks represent MDSs identified in U2OS, HeLa, and HBE cells, respectively. The black line at the bottom of each view represents the CFS finely mapped in that region by FISH previously. g A representative view of MDSs residing in unreported loci on chromosome 12 (q23.1–q23.3). h The size distribution of the genes annotated to MDSs in U2OS cells and HeLa cells.
To investigate whether these MDSs are also DNA synthesis sites during S phase and whether they are only formed in mitosis following replication stress, we profiled the nascent DNA synthesis at these sites both in S phase and M phase by sequencing EdU-incorporated regions. Results showed that DNA synthesis was normally carried out at MDSs without APH treatment, but evidently suppressed in the presence of APH (Supplementary information, Fig. S3a, b). Oppositely, DNA synthesis in mitosis was only seen upon APH treatment (Supplementary information, Fig. S3a, b). These results indicated that MDSs were indeed replication stress-induced DNA synthesis sites specific to M phase, which was further confirmed by quantifying their dependence on MiDAS versus S phase DNA synthesis under replication stress or not (Supplementary information, Fig. S3c).
Using MiDASeq, we also located 85 MDSs in HeLa cells, 48 of which were mapped to 44 known CFSs (Fig. 2c; Supplementary information, Table S2). The majority (75 hits) of the MDSs identified in HeLa cells were present in U2OS cells (Fig. 2c–f; Supplementary information, Fig. S2e, f). Besides cancerous cells with high instability, we also performed MiDASeq on human bronchial epithelial (HBE) cells. Only 27 MDSs were identified, among which 23 overlapped with those in U2OS and 22 mapped to 19 known CFSs (Fig. 2c–e; Supplementary information, Fig. S2e and Table S3). The huge difference in MDS occurrence among different cell lines, especially between normal cells and cancerous cells, might on the one hand inform about the difference in cellular response to replication stress, while on the other hand reflect the cell type specificity of MDSs resembling that of CFSs.
Intriguingly, in many CFSs that overlap ultralong genes, we frequently observed a “twin peak” pattern for the EdU signal (e.g., Fig. 2d). Considering that these loci overlapped with hotspots of CNVs,1,28 there could be two possible interpretations of these results. One is that the region contained in the valley separating the twin peaks of the MiDASeq signal has already been deleted from the genome of the cancer cells used in our study. Indeed, the central location between the MiDAseq signal peaks coincided with the positions of deletions found in various cancers.29,56 The second explanation is that the MiDAS at CFSs is carried out by two converging replication forks initiated at the flanks of CFSs by a process known as BIR, which has been implicated previously in MiDAS.19 This might be the consequence of the so-called “double fork failures” that can occur when convergent replication forks progressing inward origin-poor CFSs are stalled and fail to replicate the DNA in between.1 After entry into mitosis, MiDAS may be activated at both stalling sites, allowing the restart of two new convergent replication forks. To test these possible scenarios, we analyzed the MDS profiles of mitotic cells incubated with EdU for different periods of time. We found that the vast majority of twin peaks tended to merge into one (Supplementary information, Fig. S3d), indicating a “double fork failure” event followed by two converging MiDAS forks to complete the replication of the core region of a CFS. Nevertheless, it should be noted that there also existed very few cases in which the central region between twin peaks was deleted as revealed by whole-genome sequencing (WGS), and thus the two MiDAS forks would never converge there (Supplementary information, Fig. S3e).
To reveal the genetic distribution of MDSs and associated CFSs, we annotated them with the overlapped genes. We found that, in both U2OS and HeLa cells, over half of the MDSs-annotated genes were larger than 100 kb, and that over 35% were larger than 500 kb (Fig. 2h), which is consistent with previous reports that CFSs were enriched in large genes.16,50 Moreover, gene ontology (GO) analysis of the most inclusive set of MDS-annotated genes from U2OS cells revealed that MDSs and associated CFSs were enriched in genes expressed for neuronal development and functions (Supplementary information, Fig. S4), paralleling experimental observations of many frequently expressed CFSs.27,35
Associations of MDSs with other hotspots of genomic instability
The relationship between CFSs and other forms or hotspots of genomic instability such as CNVs,57 RDCs37 and evolutionary breakpoint regions (EBRs)58 has long been intriguing because of their shared association with genomic variation and diversity. RDCs were first brought into our focus because they are hotspots for double-strand breaks (DSBs) forming in mouse NSPCs and mostly harbor large genes carrying neurological functions, which echoes the genetic distribution of MDSs in our study.37 In fact, RDCs, though initially identified in mice, have been linked to human CFSs because of the overlap between the sequences of their associated homolog genes.1,34 They also share some other common features. For example, both RDCs and CFSs have late replication timing and are sensitive to mild replication stress.37 Hence, they are often considered together with regard to genetic variations and disease relevance. However, it remains unclear whether this association reflects a mechanistic link or is merely a coincidence, due largely to the low resolution of previously defined CFSs.
To find out whether MDSs, the surrogate for CFSs, cross with RDCs, we compared the MDSs to RDCs with reference to conserved genes in the human and mouse genomes. Considering the larger sample size of MDSs from U2OS cells could better reveal the nature of these regions and associated CFSs, we hereafter focused on them for comprehensive analysis. As shown in Fig. 3a, we first assigned MDSs to their associated genes, which were frequently involved in neurological processes. Using these genes as coordinates, we were able to match RDCs to CFSs. Strikingly, 21 out of 27 RDCs reliably overlapped with MDSs, and 2 RDCs were in close proximity to MDSs (< 1 Mb) (Fig. 3b; Supplementary information, Table S4). This strong correlation was further validated by visual inspection of several genes in the human genome that corresponded to mouse RDCs, including PARD3B, protein kinase CGMP-dependent 1 (PRKG1), adhesion G protein-coupled receptor B3 (ADGRB3, BAI3 in mice) and catenin delta 2 (CTNND2) (Fig. 3c–f). These data provide strong evidence that RDCs are a subgroup of neuronal-specific MDSs/CFSs, which would probably break at a high frequency under replication stress in rapidly proliferating neuronal cells during neuronal development.34 This, in turn, would likely contribute to the great genetic diversity in brain cells. The strong conservation between MDSs and RDCs also suggested a mechanistic role for MiDAS in RDCs formation, perhaps in a manner similar to how it contributes to CFS expression.

a Cnetplot depicting the eight most enriched gene ontology terms annotated to MDSs. Specific genes are also displayed. b Venn diagram showing the overlap between human MDSs and RDCs identified in mouse NSPCs. c–f Genomic view of the representative genes conserved in human MDSs and mouse RDCs. g Aggregation plots showing the distribution of CNVs in a 400 kb window centered around the middle of MDSs and RSs. h Circos plot displaying distributions of MDSs and CNVs on chromosome 2, chromosome 7, chromosome 9, and chromosome 13. i Violin plot showing the frequency of SNVs in MDSs and RSs. P value was determined using the Wilcoxon rank-sum test. The solid purple line means the median value.
Next, we aligned MDSs with a human EBR map acquired from a recent study.59 For comparison, we defined a control group comprising random genomic sites (RSs) that were matched in size to the test group of MDSs after exclusion of Y chromosome (Fig. 2b). Although both CFSs and EBRs have been reported to be hotspots of chromosome rearrangements, we did not observe any significantly better association between MDSs and human EBRs than between RSs and EBRs (Supplementary information, Fig. S5a, b), probably due to the random distribution of the vast number of EBRs covering nearly 40% of the human genome. Additionally, a comparison of the MiDASeq signal intensity between MDSs overlapped or not overlapped with EBRs also did not show any evident differences (Supplementary information, Fig. S5c). Furthermore, we examined whether the locations of MDSs correlated with the sites of CNVs. We derived CNV maps from WGS data of U2OS cells treated with APH, and profiled the distribution of CNVs including amplifications and deletions across all MDSs and RSs. We observed an apparent peak of CNVs near the center of MDSs, but not in RSs (Fig. 3g). Such peaks were evident only in a small subgroup of MDSs (Fig. 3h). These results agree with some previous findings that most CNVs are distributed randomly across the genome,28,57 but some clusters of CNVs, especially large gene deletions, are enriched in CFSs.1,28 Considering that MDSs are generally replicated in late S phase to M phase, we questioned whether such late replication timing might give rise to an altered probability of nucleotide variations in the human genome, as suggested in a previous study.60 Therefore, we compared the frequency of single nucleotide variants (SNVs) in MDSs and RSs, but found no significant difference (Fig. 3i).
The counterintuitive fact that CFSs are hotspots for deletions, and yet retained in evolution, leads to the question of whether CFSs are an unavoidable cost of some other biological events that are essential for our genome organization and integrity. One such event that we consider is higher-order chromosome regulation, e.g., loop extrusion61 and topologically associating domain (TAD) organization,62 which create structurally vulnerable points such as loop anchorage points and TAD boundaries. In our work, we aligned MDSs with TAD boundaries generated from U2OS nucleolar Hi-C data (courtesy of Cheng Li’s lab), but found no substantial overlap (Supplementary information, Fig. S5d). In consistence, the MiDAS signal was not enriched in the adjacent regions of TAD boundaries (Supplementary information, Fig. S5e).
Sequence characteristics and epigenomic features of MDSs
Because the MDSs were mapped at nucleotide-resolution, we were able to analyze their sequence characteristics. We first assessed the distribution of MDSs across different gene regions, which showed over 70% of the hotspots mapped to introns (Fig. 4a). It has long been proposed that CFSs were AT-rich sequences that are inclined to form secondary structure.39,41 We therefore examined whether this is a general property of all MDSs. As expected, the overall AT content of MDSs was significantly higher than that of RSs (Fig. 4b). We then searched for AT-dinucleotide minisatellites, which have been proposed to be particularly problematic for CFS replication.41,42 Results showed that AT-dinucleotide minisatellites were significantly enriched in MDSs, as compared to their abundance in RSs (Fig. 4c). However, it should be noted that MDSs displayed strong heterogeneity regarding the abundance of these sequences, and therefore the contribution of AT content to the difficulty in replication of some MDSs can not be generalized. We also assessed other repeat sequences, including Alu elements, long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs), which had been associated with replication impediments or early replicating fragile sites.51,63,64 However, we found no evident enrichment of any of them (Supplementary information, Fig. S6a–c). Next, we inspected MDSs for conserved DNA sequences that may contribute to their hypersensitivity to replication stress. Although no consensus sequence present in all MDSs, some significantly conserved DNA motifs were identified (Fig. 4d).

a Distribution of MDSs across the genetic elements. b Overall AT content in MDSs and RSs. Data are shown as means ± SEM, ****P < 0.0001 calculated by Student’s t-test. c The number of AT-dinucleotide minisatellites in MDSs and RSs. Data are shown as means ± SEM, **P < 0.01 calculated by Student’s t-test. d Five most significant sequence motifs identified in MDSs. Numbers at the right are P values indicating statistical significance. e–g Violin plots comparing the levels of H3K4me3, H3K36me3, and H3K27me3, respectively. P value was determined using the Wilcoxon rank-sum test. The solid purple line indicates the median value.
In addition to the nucleotide sequence, the epigenomic landscape represented by histone modifications also plays a pivotal role in determining the texture of a genomic region, for example, by shaping chromatin accessibilities and structural organizations.65,66,67 Indeed, diverse histone modifications can regulate various DNA metabolic processes including DNA replication, transcription, and DNA repair,65 and the collective epigenomic features shape unique chromatin domains or compartments such as heterochromatin.68 To investigate whether MDSs might reflect specific chromatin domains with a common epigenomic profile, we analyzed histone modifications at RSs and MDSs. By mining publicly deposited data for histone modifications reported in U2OS cells, including H3K4me3,69 H3K27me3,69 H3K9me2,70 H3K27ac,70 H3K36me3 and H3K9me3 (source data generated by ENCODE project71), we revealed that the levels of H3K4me3 and H3K36me3 at MDSs were much lower than those at RSs (Fig. 4e, f), indicating poor chromatin accessibility for transcription factor binding and transcription elongation.72 Consistent with this, H3K27me3, a negative regulator of transcription,73 was significantly enriched at MDSs (Fig. 4g). There was no significant change in the level of other types of histone modifications and epigenomic features analyzed (Supplementary information, Fig. S6d–g). Collectively, these data suggested that MDSs not only are difficult to replicate, but also may be difficult to transcribe, making it even more challenging to coordinate the replication and transcription of the generally large genes associated with MDSs.
Replication profiles of MDSs
Numerous studies have suggested that CFSs are late replicating regions with a low density of replication origins,43,46,74,75,76 but this hypothesis is largely based on imprecise CFS coordinates and low sample numbers. Given the strong colocalization of CFSs and MDSs, we were able to indirectly test those longstanding arguments on a genome-wide scale. For this, we retrieved genomic datasets from previous studies on DNA replication timing77 and replication origin usage78 in U2OS cells, and examined their correlations with the MiDASeq signal. This analysis revealed that the vast majority of MDSs were indeed replicated in the late S phase and showed a paucity of active replication origins (Fig. 5a–c). This global trend was confirmed by zooming into several genomic regions, including FRA3B, FRA16D, and two MDSs unrelated to known CFSs on chromosome 3 and chromosome 14, respectively (Fig. 5d, e).

a Volcano plot displaying the characteristics of each RS (left) or MDS (right). The replication origin density and the size of each site are indicated by dot color and size, respectively. b, c Quantitative comparison of the replication timing and replication origin density between MDSs and RSs. P values were determined by Wilcoxon rank-sum test. The solid purple line indicates the median value. d Genomic view of the MiDASeq, replication timing and replication origin density at representative known CFSs including FRA3B and FRA16D. Yellow and green peaks in the track of replication timing indicate early-replicating and late-replicating regions. e Genomic view of the same parameters as in d at two MDSs residing in unreported loci.
Contribution of large gene transcription to MDS formation
As discussed above, multiple studies have suggested that the transcriptional status of the large gene encoded within that region will determine whether a CFS will form.16,28,50 To investigate whether this model applies to MDSs in this study, we first examined whether these regions were actively transcribed in the U2OS cell line using RNA sequencing (RNA-seq). RNA transcription was consistently seen in MDSs (Fig. 6a, b; Supplementary information, Fig. S7a), though the overall RNA level was much lower than that in RSs (Supplementary information, Fig. S7b). However, when we investigated MDSs, we did not find any clear correlation between overall RNA transcription level and MiDASeq signal intensity indicating MDS expression frequency (Fig. 6c). This was also the case for the subgroup of MDSs that spanned over 500 kb (Supplementary information, Fig. S7c). These results differed from our prediction that more active transcription would create a higher probability of replication-transcription collisions subsequently leading to more frequent MDSs expression, and implied that transcription activity, in terms of creating R-loops,50 might not be critical for promoting CFS expression. These data were consistent with a recent study showing that inhibition of transcription in the S phase could not prevent CFS instability.76 To better elucidate the role of transcription, especially large gene transcription, in promoting genomic fragility, we analyzed the transcription profile of large genes (using > 200 kb or > 500 kb as a cutoff) and divided these genes into those overlapped with MDSs (MDS-OV) and those did not (MDS-NO). This analysis revealed that the MiDASeq signal was enriched only in the gene bodies of MDS-OV genes (Fig. 6d; Supplementary information, Fig. S7d). Next, we focused only on genes larger than 500 kb, and compared the transcription level between MDS-OV and MDS-NO genes. We expected that MDS-NO genes would be more likely to be transcriptionally silent than their actively transcribed counterparts, but the results showed no significant difference in overall RNA level between these two subgroups (Supplementary information, Fig. S7e). Nevertheless, a caution to consider is that most large CFS genes have more than one transcription isoforms and primarily express their short isoforms, which would be the overwhelming species in RNA-seq data, while their long isoforms that have been reported to directly affect CFS fragility are not represented in RNA-seq data.1,28 Therefore, we employed deposited nascent RNA transcription data for U2OS cells.79 Genomic view of nascent RNA at MDSs again showed that MDS-OV large genes were actively transcribed (Fig. 6e), though the overall nascent RNA level was no different from that of RSs (Supplementary information, Fig. S7f). We then derived transcription units (TUs) on a global scale by a method as previously described,28,80 which can represent active transcription of long isoforms of large genes (illustrated in Fig. 6f). We compared MDSs to RSs for their overlap with large TUs, and found that MDSs displayed a much higher average fraction in large TUs (> 500 kb or > 200 kb) than RSs did (Fig. 6g; Supplementary information, Fig. S7g). It was the same case for MDS-OV genes versus MDS-NO genes (Fig. 6h; Supplementary information, Fig. S7h), suggesting that MDS-OV genes were more likely to be transcribing their long isoforms. The seemingly contradicting conclusions from transcription level and TUs implied that transcription can impinge on replication in other ways rather than conflicting with replication forks. In fact, it has been hypothesized that when large genes transcribe their long isoforms, they may erase regional origin licenses and further delay replication timing.1,28 In consistence, we observed that MDS-OV genes harbored fewer replication origins and experienced much later replication timing (Fig. 6i, j). To investigate how exactly replication and transcription might interplay in promoting MiDAS occurrence, we conducted multiple regression analysis on the correlation of MiDASeq signal with major replication and transcription parameters for MDSs. Results revealed that only large TUs and late replication timing were significantly affecting the sequencing score (Fig. 6k), suggesting that late replication timing and large genes transcribing long isoforms were the key determinants of whether the locus will undergo MiDAS.

a, b Aggregation plots of the distribution of the MiDASeq signal (red) and RNA-seq signal (dark) over RSs (a) or MDSs (b) in a 2 Mb window centered around the midst of RSs or MDSs. c Scatter plot displaying the MiDASeq signal and RNA-seq signal of every MDS. d Aggregation plots of the MiDASeq signal distribution across the gene bodies of MDS-OV or MDS-NO genes > 500 kb. e Representative view of nascent RNA signal at three MDSs mapped to known CFSs and one MDS residing in unreported genomic loci. f Schematic diagram of transcription of short and long isoforms of large genes, and definition of TUs. g Fraction of RSs and MDSs in large TUs > 500 kb. Data are shown as means ± SEM, ****P < 0.0001 calculated using a Wilcoxon rank-sum test. h Fraction of MDS-NO and MDS-OV genes > 500 kb in large TUs > 500 kb. Data are shown as means ± SEM, ****P < 0.0001 calculated using a Wilcoxon rank-sum test. i Quantitative comparison of origin density between MDS-OV and MDS-NO genes > 500 kb. Data are shown as means ± SEM, ****P < 0.0001 calculated using a Wilcoxon rank-sum test. j Quantitative comparison of replication timing between MDS-OV and MDS-NO genes > 500 kb. Data are shown as means ± SEM, ***P < 0.001 calculated using the Wilcoxon rank-sum test. k Multiple regression analysis of MiDASeq signal and other characteristics including the RNA-Seq signal, nascent RNA level, origin density, replication timing, and fraction in large TUs > 500 kb. Regression coefficient and significance are indicated. Error bars indicate the confidence interval (95%, α = 0.05) of the regression coefficient.
Discussion
In this work, we utilized the sites of APH-induced MiDAS as a surrogate marker for CFSs, and sequenced the nascent DNA from MiDAS events to seek a genome-wide map of CFSs at a base pair-resolution. This methodology captured most of the known human CFSs, providing a much more accurate map of the chromosome locations of each CFS in comparison with previous usage of cytogenetic assays. As expected, MDSs displayed typical features reported for many CFSs including abundant AT content, late replication timing, origin density scarcity, colocalization with large genes, etc. The geographical connection and genetic similarities strongly suggest that MDSs can be representative of CFSs and may be used to investigate and predict other properties of CFSs. It should be noted, however, MDSs identified by MiDASeq are in essence the sites of mitotic DNA synthesis and do not address whether the locus is structurally intact or not. In addition, not all MDSs can be matched to known CFSs, necessitating direct experimental evidence from, e.g., molecular probing. Last but not least, the definition and locations of CFSs are still evolving and under debate, therefore, the relationship between MDSs and CFSs deserves further validation and updating. Taken together, MDSs cannot be equated to CFSs at current stage. Future analysis, combing MiDASseq with cytogenetic assays and other DNA breakage sequencing techniques like End-seq81 and BLESS,82 will be needed to confirm whether MDSs will be present as breaks and gaps in the cell line where they are identified, such that we can ensure that the conclusions drawn from MDSs are truly revealing the nature of CFSs, and meanwhile gain a deeper insight into CFS processing in the M phase.
The number of MDSs we identified varies among different cell lines, though there is a strong inclusive relationship between datasets. This cell type dependence of MDS formation emphasizes the epigenetic layer of MDSs as seen for CFSs.28 Amongst the three cell lines we used, U2OS cells exhibited the most MDSs, comprising the majority of those identified in HeLa and HBE cells, which also provided larger sample size. Therefore, we mainly focused on U2OS cells in this study. However, U2OS cells are notorious for their high instability, which on one hand may play a role in permitting the large number of unreplicated regions to pass through cell cycle checkpoint to form MDSs, while on the other hand brings a limitation to the accuracy and generalization of the data acquired.
Because of the strong association between MDSs and CFSs, we revisited many of the hypotheses for CFS instability by analyzing MDSs, including sequence characteristics, late replication timing, poor replication origin usage, transcription, etc. Our comparative analysis reemphasized the importance of AT-rich sequences, late replication timing, and replication origin paucity. While CFSs are defined as replication stress-sensitive regions, their mechanistic connection to transcription has recently attracted more research interest. Cell-type specificity of CFSs and experimental observations that some CFSs are expressed only in cells transcribing a long isoform of the associated large gene suggest that transcription activity may be a prerequisite for CFS formation.28,45,83 Our data also demonstrated that MDS-associated large genes are actively transcribed with their long isoforms in the cell line used, but the transcription level did not contribute greatly to the frequency of MDS formation, which was instead strongly affected by the replication timing of MDS-associated genes.28 Notably, although replication origin paucity was prevalent in MDSs, this feature was not contributing to the MiDAS signal in a multivariate correlation analysis. Instead, overlapping with large transcription units and replication timing were the significant contributing factors. It raised the possibility that poor origin density could be a consequence of other parameters, e.g., the transcription activity prevents late origin firing at that region as previously proposed by Wilson et al.,28 but not an independent factor contributing to MDS formation.
Some epigenomic features also showed a global trend toward transcription suppression at MDSs, indicating a difficult-to-transcribe context at MDSs. However, it should be noted that the epigenomic landscape is under constant regulation throughout the cell cycle,84 and therefore their relevance to MDS instability is likely to be complex. Considering that the epigenomic data we employed in this study were acquired from asynchronous cells, there may be some underestimation of those factors that act only in a certain phase. To address this problem, cell cycle-specific analysis of the histone modifications at MDSs may be required.
It is clear that the evolutionary relevance of CFSs deserves to be investigated in greater detail. It has been proposed that the chromosome rearrangements at CFSs can provide a genetic driver of evolution. In support of this hypothesis, we found that human MDSs as the surrogate for human CFSs reused most of the RDCs in mouse NSPCs that were hotspots for chromosome translocations and rearrangements.37 Though actively debated, the evolutionary role of CFSs has never been experimentally tested. One conceivable challenge is the difficulty involved in manipulating the genome in the scale of cytogenetically defined CFSs. However, the much more accurate sequence information of CFSs potentially informed by MDSs and the recent breakthroughs in the use of synthetic yeast chromosomes85,86 prompted us to reconsider the possibility of examining the function of CFS in evolution. It will be interesting to investigate how the incorporation of a whole CFS or a part of it will impact the stability of the yeast genome. Moreover, the antibody-free MiDASeq can easily be extended to other species, which provides an approach to characterizing their evolutionary role by looking at their sequences across species.
In conclusion, we pursued a refined map of CFSs in human cells by sequencing CFS-specific mitotic DNA synthesis. The nucleotide-resolution of MDSs informed the accurate coordinates of most of the known CFSs, and allowed us to characterize their genomic context in detail, and to gain a deeper insight into their biological relevance and mechanistic underpinnings. Future studies can further utilize this map for functional studies at specific CFSs or associated genes to validate their pathophysiological implications, which may also unravel therapeutic opportunities for CFS-associated diseases.
The accompanying article by Macheret et al. reports similar findings to ours.87
Materials and methods
Cell culture and harvest of mitotic cells
U2OS and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (Gibco). HBE cells were grown in RPMI Medium Modified (HyClone) with 10% fetal bovine serum (Gibco). All media were supplemented with 1% penicillin-streptomycin (Invitrogen). Cells were grown at 37 °C under 5% CO2. Cells were grown to 20%–30% confluence before being treated with 2 mM thymidine (Sigma, T9250). Following APH (Sigma, A0781, 0.3 µM) and RO3306 (Selleck, S7747, 7 µM) treatments, cells were washed three times with PBS (pre-warmed to 37 °C) within 5 min, and then released into warm media with or without (as the negative control group) 20 µM EdU for incubation for another 30 min. Afterwards, mitotic cells were then manually collected by mitotic shake-off. For longer EdU incubation time, the collected mitotic cells were reseeded into a new plate with the original medium containing 20 μM EdU, and incubated for another 30 min before harvesting for MiDASeq.
Immunofluorescence
Cells were fixed with 4% formaldehyde at room temperature for 15 min, and then permeabilized with 0.5% Triton X-100 in PBS for 20 min. After being blocked with 3% BSA in PBS for a minimum of 1 h at room temperature, cells were incubated with the primary antibody in 3% BSA in PBS at 4 °C overnight. Primary antibody was then removed, cells were washed three times with PBS and then incubated with secondary antibody in 3% BSA in PBS for 1.5 h at room temperature, protected from light. EdU staining was performed with Click-iT™ Imaging Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Nuclear DNA was stained with DAPI. A Nikon Eclipse Ni microscope with Nikon Elements software (Nikon Instruments) was used for imaging. Antibodies used in this study were as follows: rabbit anti-FANCD2 primary antibody (Abcam, ab108928, 1:1000), rabbit anti-histone H3 (phosphor-S10) primary antibody (Abcam, ab47297), Alexa Fluro 555 goat anti-rabbit secondary antibody (Thermo Fisher Scientific, A-21429, 1:1000).
Metaphase spreads
Collected mitotic cells were centrifuged at 400× g at 4 °C for 5 min and washed with PBS once and then swollen in 75 mM KCl in ddH2O (pre-warmed to 37 °C) for 15 min. Cells were centrifuged again and then fixed by the dropwise addition of 5 mL of the methanol: glacial acetic acid (3:1) fixative solution during constant vortexing. Fixed cells were dropped onto slides and dried to make metaphase spreads for EdU staining with the Click-iT™ Imaging Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Metaphase spreads were further stained with DAPI.
Validation of the capture & PCR system
DNA fragments with or without biotin were generated by PCR of a plasmid DNA containing a 300 bp random sequence in the presence or absence of dUTP-biotin. Different amounts of random DNA with or without biotin were added (10 ng, 100 ng, 1000 ng) to the same volume of Dynabeads MyOne Streptavidin C1 Beads (Thermo Fisher Scientific, 65001) and incubated for 15 min while rotating. The beads were washed three times with the TE buffer (10 mM Tris-HCl, pH 8, and 1 mM EDTA) to remove nonspecifically bound DNA, followed by PCR to generate the library off the beads. Gel analysis of the DNA product was used to evaluate the specific binding efficacy between biotin and streptavidin. The random DNA sequence was as follows:
5′GCCGGTGCGTTGCAAGCTTCGGTATGGAAGGACCCTCCGACCGCGAATTGCGATACTTCAGGATTCCCGGAATAGTGCGCACAAGAAGGGTACCCCAGGTGTCCTGTTAACGGGTCGAATATCAAAATCGCGAAAAGCAACTGTGGCAGACGTTGTTCGTATCGTTTGAAGACATGCGCTTCGGACAGCATCCGAGAGTAACGGTACCCCATGATGGCACAGAAGCCTGCATCCAATGAGACCCAGTTTGCATTCGCAAGCCATTGGCGAGAACCTGCAATCCAAGCGACCCGACATCGA3′.
MiDASeq
Fixation and permeabilization
Mitotic cells were fixed in 90% ice-cold methanol on ice for 20 min and permeabilized with 0.5% Triton X-100 in PBS for 20 min.
Click reaction
The cocktail was prepared according to the following formulation: 87 µL PBS, 1 µL biotin-azide (Thermo Fisher, B10184, 10 mM), 10 µL sodium ascorbate (Sigma, A4034, 100 mM), and 2 µL CuSO4 (Sigma, 100 mM), to a 100 µL volume. The cells were resuspended in a proper amount of cocktail (1 mL/2 × 107 cells), followed by Click-IT reaction in a 37 °C water bath for 30 min.
DNA extraction and sonication
Cells were lysed in the Lysis buffer (50 mM Tris-HCl, pH 8.0, 1% SDS, 20 mg/mL RNase A, 20 mg/mL protease K) in a 37 °C water bath for over 6 h. The DNA was extracted using the phenol/chloroform method and sonicated to an average size of 300–400 bp using a Bioruptor (Diagenode).
Biotin-DNA enrichment
An appropriate volume of the Dynabeads MyOne Streptavidin C1 Beads (Thermo Fisher Scientific, 65001, 10 ug/µL) was transferred to a tube and washed with 1× BW buffer (5 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 1 M NaCl) three times, then re-suspended in 2× BW buffer to a final concentration of 5 µg/µL. An equal volume of DNA was added and the mixture was incubated for 15 min at room temperature, with gentle rotation. The beads were washed with 1× BW buffer three times and then re-suspended in EB buffer (10 mM Tris-HCl, pH 8.5) to a final volume of 50 µL.
Library construction
DNA library was constructed with KAPA Hyper Prep Kit (KK8502). Briefly, 3 µL of Enzyme Mix and 7 µL of corresponding buffer were added to beads adsorbed with biotin-DNA. End repair and an A-tailing reaction were performed in a thermocycler according to the manufacturer’s protocol. Adapter stock was diluted to 300 nM and adapter ligation was accomplished at 20 °C for 0.5 h. The beads were washed with a double volume of the TE buffer (10 mM Tris-HCl, pH 8, and 1 mM EDTA) at least three times to remove unligated adapter and adapter-dimer molecules. Each library amplification reaction contained KAPA HiFi HotStart ReadyMix and the Primer Mix (10×) and was carried out using amplification for 15 cycles. Reactions were cleaned up using AMPure XP (Beckman Coulter, A63880).
Sequencing
The DNA library was sequenced at Gene Denovo Biotechnology Co., Ltd (Guangzhou, China) and AnnoroadGene Technology Co., Ltd (Beijing, China) using the HiSeq Xten (Illumina platform).
Flow cytometry
To confirm that vast majority of the harvested cells were mitotic cells, cells were stained with phosphor-H3 antibody (Alexa Fluor® 647 Rat anti-Histone H3 (pS28) (BD Biosciences, 558217, 1:200)), followed by EdU staining and DAPI staining. At least 15,000 cells were analyzed for each sample on a Cytoflex (Beckman Coulter). To validate click reaction, cells treated with 10 µM EdU for 15 h were fixed in 90% ice-cold methanol and subjected to the Click-IT reaction (DMSO or biotin-azide) as described above. Cells were incubated with PE Streptavidin (BD Biosciences, 554061, 1:500) at 4 °C for 0.5 h. Analysis was carried out on a Cytoflex (Beckman Coulter). For each sample, at least 20,000 cells were recorded.
DNA synthesis analysis for MDSs in mitosis and S phase
For S phase DNA synthesis analysis, asynchronous U2OS cells were treated with or without 0.3 µM APH for 18 h and then pulse labeled with EdU for 30 min. Then the cells were collected for fixation with 90% ice-cold methanol on ice for 20 min. To proceed, cells were stained with DAPI, followed by cell sorting based on DNA content (Beckman moflo Astrios EQ) S phase cells were collected and subjected to the same downstream processing of MiDASeq. M phase DNA synthesis analysis was performed following the same procedure of MiDASeq of mitotic cells treated with or without APH in S phase.
Bioinformatics analysis
Detailed information about the source of the deposited data, software and algorithms used in this study can be found in Supplementary information, Table S5.
Identification of MDSs
Raw reads of MiDASeq were trimmed using Trim Galore with default settings and mapped to the human genomes (hg38) by Bowtie2 both for the experimental group and the negative control group. We used SAMtools to remove PCR duplicates and transformed clean reads to BigWig format using deepTools2. Through visualization with IGV, we manually picked out potential MiDAS regions based on the height of the peaks. These regions were then divided into 100 kb-width bins. The read coverage was quantified by using reads per kilobase (RPK) for each bin, which was calculated using bedtools and R. To eliminate the background noise, the read coverage of an experimental group was divided by that of the negative control group to get the MiDASeq signal FC for each bin. The threshold for defining positive genomic bins was first restrained in an approximate range by referring to the FC at most frequently expressed landmark CFSs such as FRA3B, FRA16D and FRA7J, and then determined manually by visual inspection across the whole genome using IGV. Finally, the adjacent positive bins (within 100 kb) were merged using bedtools and defined as MDSs.
Definition of RSs, MDS-OV genes, and MDS-NO genes
RSs are random sites across the genome, with Y chromosome sequences excluded. The size and number of RSs were paired to MDSs. To define MDS-OV and MDS-NO, we first attained the list of all human genes including their Entrez ID and location information from UCSC.hg38.knownGene (version 3.10.0). We calculated the overlapped length of each gene adjacent to each MDS. Genes were defined as MDS-OV genes if they meet any of the following criteria: 1) overlapped length was more than 50% of the width of MDS or the width of the gene itself; 2) the overlapped length was more than 250 kb; 3) the gene contains at least two MDSs. The remaining genes were then defined as MDS-NO genes.
Calculation of overlap between MDSs identified in experimental repeats and in different cell lines
The MDSs in one experimental repeat were divided into four groups according to whether they overlapped with MDSs in other two experimental repeats. For each experimental repeat, the percentage of each group of sites in total MDSs was calculated and visualized with a pie chart. The number of CFSs hit by MDSs in each cell line, the number of MDSs in each cell line hitting CFSs, and the number of MDSs in HeLa and HBE hitting MDSs in U2OS were counted, respectively.
Genomic annotations and gene ontology analysis
MDSs were annotated to the closest genes using R package ChIPseeker. The reference genome for annotation was from UCSC.hg38.knownGene (version 3.10.0). Gene ontology analysis was applied to the annotated genes using R package clusterProfiler, and the reference database was from R package org.Hs.eg.db (version 3.10.0). To access the distribution of MDSs in genetic elements, all the MDSs were cut into 10-bp bins and annotated to genetic elements using R package ChIPseeker. The percentage of bins annotated to each type of genetic elements was calculated and visualized with pie chart. To access the length distribution, annotated genes were divided into four groups: < 100 kb, 100–200 kb, 200–500 kb and > 500 kb. The percentage of each group was calculated and visualized with pie charts.
Calculation of nascent DNA reads density for profiling DNA synthesis at MDSs
Raw reads from MiDASeq for M phase cells and EdU-seq for S phase cells treated with or without APH were trimmed using Trim Galore, and mapped to the human genomes (hg38) by bowtie2 after deduplication, generating bam files containing uniquely mapped reads. The nascent DNA reads density was calculated as the number of mapped reads per 50 kb genomic bin after subtraction of the background reads in negative control using Bamcoverage tool integrated in Galaxy. The reads density was further normalized to the sum of raw reads in mega, after which the resultant BigWig files were used for visualization. To calculate the nascent DNA reads density at each MDS (or RS), the number of reads mapped to that region was counted by bedtools and converted to number of reads per 50 kb, followed by normalization as described above. The dependence on M phase DNA synthesis versus S phase DNA synthesis in the presence or absence of APH was defined for each MDS as log2(M phase reads density in M phase/S phase reads density).
Processing of deposited data
The datasets of Repli-seq, replication origins, DNase-seq and histone modification ChIP-seq were obtained from GEO. For each raw data, we performed adapter trimming, genome mapping and PCR duplicate removal using Trim Galore, Bowtie2 and SAMtools, respectively. Then the bam files were transformed into BigWig format by deepTools2. As for Repli-seq datasets, genome-wide replication-timing profiles were constructed, scaled, and pooled for analysis as described previously.88 Briefly, log2 ratios of the late vs early read counts were calculated for each nonoverlapping 5-kb window for IGV visualization. For each MDS, RS, MDS-OV and MDS-NO, replication timing was calculated as follows: first we summed the late and early read counts of each region, and the log2 ratios of late vs early read counts were calculated as the replication timing value.
Calculation of MiDASeq signal, histone modification signal, origin density, and DNase I hypersensitivity
For all the signal calculations of MDSs, RSs, MDS-OV and MDS-NO, we first obtained the read coverage by calculating the RPK. The RPK values or its logarithmic form (log2RPK) were used to represent origin density and DNase I hypersensitivity derived from nascent strand sequencing and DNase-seq, respectively. As for MiDASeq and histone modification ChIP-seq, the read coverage of the experimental group was divided by that of the negative control group to get signal FC. The signal FC or its logarithmic form (log2FC) was used to represent the signal intensity of MiDAS or histone modifications.
Analysis of the correlation between MDSs and EBRs
The human genomic coordinates for EBRs were downloaded from https://github.com/bioinfoUQAM/RECOMB-CG-2019_supp.59 The NCBI Genome Remapping Service was used to remap EBRs from hg19 to hg38. For each MDS, the length overlapped with EBRs were calculated. The fraction in EBRs were defined as the overlapped length divided by the length of MDS itself. If the fraction in EBRs of an MDS was zero, we defined the MDSs as EBRs non-overlapped (EBRs-NO). Otherwise, we defined the MDSs as EBRs overlapped (EBRs-OV).
RNA-seq data analysis
RNA from asynchronously growing U2OS cells being treated with 0.3 µM APH for 18 h was sequenced commercially by BGI Genomics (BGI-Shenzhen). Reads were trimmed using Trim Galore with default settings and mapped to the human genome (hg38) by HISAT2. We counted reads of each BAM file using FeatureCounts for whole exons according to the latest version of human gene annotation (Release 32, GRCh38.p13) by the GENCODE database. For MDSs and RSs, the reads were counted by bedtools with default settings. The reads per kilobase per mega reads (RPKM) were calculated to represent the transcription signal for MDSs, RSs, MDS-OV and MDS-NO.
Nascent RNA data processing
Nascent RNA raw data were downloaded from GEO.79 We performed adapter trimming by Trim Galore with default parameters. Alignment to the human genome (hg38) was performed by HISAT2 with default settings. For MDS-NO and MDS-OV, we counted reads of each BAM file by featureCounts for full length of genes according to the latest version of human gene annotation (Release 32, GRCh38.p13) by GENCODE database, with “-p -t gene -g gene_id -s 1” parameters. For MDSs and RSs, we counted reads of each BAM file by bedtools for each site with default settings. The RPKM were calculated to represent the nascent RNA transcription signal for MDSs, RSs, MDS-OV and MDS-NO.
Definition of TU
The TUs were defined as previously described80 with some modifications. The reads mapped to each 1 kb bins of genome were counted using bedtools with default settings. Then RPKM were calculated. The index for each 1 kb bin were calculated with RPKM 0.05 and 5000 as the minimum and maximum thresholds, respectively. The index for each gene were calculated with RPKM 0.2 and 100 as the minimum and maximum thresholds, respectively. The emission possibility matrix for the hidden Markov model was calculated manually with R. Genome segmentation was run by segment.pl with “-z 0.6 -p 0.995” parameters. Adjacent bins with the same state were fused into genome segments of sustained contiguous expression. We filtered the fused segments, remaining the segments whose indices were between 4 and 14, which were defined as transcriptional units.
Analysis of the correlation between MDSs and transcription units
For MDSs, the length overlapped with TUs was calculated. The fraction in TUs was defined as the overlapped length divided by the width of the MDS. The same analysis was performed as for RSs, MDS-OV and MDS-NO.
WGS analysis
Whole genomes from asynchronously growing U2OS cells being treated with 0.3 µM APH for 18 h were sequenced commercially by AnnoroadGene Technology Co., Ltd, Beijing, China. Reads were trimmed using Trim Galore with default settings and mapped to the human genomes (hg38) by BWA. Estimated copy numbers were called by Control-FREEC with the default configuration file. CNVs were filtered by the criteria that the estimated copy numbers were not in the range of 1 to 3. SNVs were called and filtered by GATK4 with default settings. The number of SNVs per megabase was calculated to represent SNV frequency for each MDS (or RS).
Analysis of the correlation between MDSs and TAD
The TAD of U2OS nucleolar Hi-C data were the courtesy from Cheng Li’s Lab. The average length of TADs was 560 kb. The length of TAD boundary (TADb) was 140 kb. For each MDS (or RS), the length overlapped with the TAD boundaries was calculated. The fraction in TAD boundaries for each MDS (or RS) was defined as the overlapped length divided by the width of the MDS (or RS).
Calculation of AT content and searching for repeat sequences
As for AT content, we used bedtools to extract sequences and calculate the percentage of overall A and T contents for each RS and MDS. AT or TA dinucleotide minisatellites (> 10 repeats) in each RS and MDS were found using SPADE89 with default settings. The coordinates of Alu elements, LINEs and SINEs were obtained from the UCSC genome database (http://genome.ucsc.edu/), their abundance in each MDS or RS was then calculated using R. Enriched motifs were found using findMotifsGenome.pl of homer.
Data visualization
All the representative genomic profiles were drawn using Gviz. The Circos plots were made using Circos to present the whole genome, and the aggregation plots were drawn using deepTools2. The scatter diagrams showing the length, replication timing and origin density were created using ggplot2. The dotplot of gene ontology analysis was visualized using DOSE.
Statistical analysis
Statistical analysis for violin plot was performed using a Wilcoxon rank-sum test using ggpubr. Regression analysis was performed with Stats using default settings. The forest plot was drawn according to the estimate and the confidence interval (95%, α = 0.05) of the regression coefficient. Student’s t-test was implemented using the software Prism 8 software.
Data availability
The data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE149376.
References
Glover, T. W., Wilson, T. E. & Arlt, M. F. Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501 (2017).
Glover, T. W., Berger, C., Coyle, J. & Echo, B. DNA polymerase? inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 67, 136–142 (1984).
Glover, T. W., Arlt, M. F., Casper, A. M. & Durkin, S. G. Mechanisms of common fragile site instability. Hum. Mol. Genet. 14 Spec No. 2, R197–R205 (2005).
Ruiz-Herrera, A. et al. Conservation of aphidicolin-induced fragile sites in Papionini (Primates) species and humans. Chromosome Res. 12, 683–690 (2004).
Stone, D. M., Jacky, P. B., Hancock, D. D. & Prieur, D. J. Animal model: chromosomal fragile site expression in dogs: I. Breed specific differences. Am. J. Med. Genet. 40, 214–222 (1991).
Matsuyama, A. et al. Fragile site orthologs FHIT/FRA3B and Fhit/Fra14A2: evolutionarily conserved but highly recombinogenic. Proc. Natl. Acad. Sci. USA 100, 14988–14993 (2003).
Helmrich, A., Stout-Weider, K., Hermann, K., Schrock, E. & Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 16, 1222–1230 (2006).
Pentzold, C. et al. FANCD2 binding identifies conserved fragile sites at large transcribed genes in avian cells. Nucleic Acids Res. 46, 1280–1294 (2018).
Casper, A. M., Nghiem, P., Arlt, M. F. & Glover, T. W. ATR regulates fragile site stability. Cell 111, 779–789 (2002).
Durkin, S. G. & Glover, T. W. Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192 (2007).
Durkin, S. G., Arlt, M. F., Howlett, N. G. & Glover, T. W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 25, 4381–4388 (2006).
Ozeri-Galai, E., Schwartz, M., Rahat, A. & Kerem, B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 27, 2109–2117 (2008).
Madireddy, A. et al. FANCD2 facilitates replication through common fragile sites. Mol. Cell 64, 388–404 (2016).
Wang, H. et al. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 9, 2791 (2018).
Özer, Ö. & Hickson, I. D. Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol. 8, 180018 (2018).
Okamoto, Y. et al. Replication stress induces accumulation of FANCD2 at central region of large fragile genes. Nucleic Acids Res. 46, 2932–2944 (2018).
Chan, K. L., Palmai-Pallag, T., Ying, S. & Hickson, I. D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760 (2009).
Ying, S. et al. MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007 (2013).
Minocherhomji, S. et al. Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290 (2015).
Gao, G. et al. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer 56, 59–74 (2016).
Walline, H. M. et al. Integration of high-risk human papillomavirus into cellular cancer-related genes in head and neck cancer cell lines. Head Neck 39, 840–852 (2017).
Wentzensen, N., Vinokurova, S. & Doeberitz, Mv. K. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 64, 3878–3884 (2004).
Feichtinger, W. & Schmid, M. Increased frequencies of sister chromatid exchanges at common fragile sites (1)(q42) and (19)(q13). Hum. Genet. 83, 145–147 (1989).
Glover, T. W. & Stein, C. K. Induction of sister chromatid exchanges at common fragile sites. Am. J. Hum. Genet. 41, 882–890 (1987).
Waisertreiger, I., Popovich, K., Block, M., Anderson, K. R. & Barlow, J. H. Visualizing locus-specific sister chromatid exchange reveals differential patterns of replication stress-induced fragile site breakage. Oncogene 39, 1260–1272 (2020).
Wang, H. et al. BLM prevents instability of structure-forming DNA sequences at common fragile sites. PLoS Genet. 14, e1007816 (2018).
Zheglo, D. et al. The FRA14B common fragile site maps to a region prone to somatic and germline rearrangements within the large GPHN gene. Genes Chromosomes Cancer 58, 284–294 (2018).
Wilson, T. E. et al. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 25, 189–200 (2015).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Chang, R. et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat. Commun. 9, 3486 (2018).
Mare, S. D. et al. WWOX and p53 dysregulation synergize to drive the development of osteosarcoma. Cancer Res. 76, 6107–6117 (2016).
Wu, D. W. et al. FHIT loss confers cisplatin resistance in lung cancer via the AKT/NF-κB/Slug-mediated PUMA reduction. Oncogene 34, 2505–2515 (2014).
Lee, J. I. et al. Loss of Fhit expression is a predictor of poor outcome in tongue cancer. Cancer Res. 61, 837–841 (2001).
Voutsinos, V., Munk, S. H. N. & Oestergaard, V. H. Common chromosomal fragile sites—conserved failure stories. Genes 9, 580 (2018).
Smith, D. I., Zhu, Y., McAvoy, S. & Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 232, 48–57 (2006).
Kumar, R. et al. HumCFS: a database of fragile sites in human chromosomes. BMC Genom. 19, 985 (2019).
Wei, P.-C. et al. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 164, 644–655 (2016).
Wilhelm, K. et al. Fragile sites as drivers of gene and genome. Evol. Curr. Genet. Med. Rep. 6, 136–143 (2018).
Zhang, H. & Freudenreich, C. H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 27, 367–379 (2007).
Zlotorynski, E. Poly(dA:dT) make it and break it. Nat. Rev. Mol. Cell Biol. 19, 619–619 (2018).
Zlotorynski, E. et al. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 23, 7143–7151 (2003).
Irony-Tur Sinai, M. et al. AT-dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res. 47, 9685–9695 (2019).
Palumbo, E., Matricardi, L., Tosoni, E., Bensimon, A. & Russo, A. Replication dynamics at common fragile site FRA6E. Chromosoma 119, 575–587 (2010).
Hellman, A. et al. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol. 20, 4420–4427 (2000).
Tallec, B. L. et al. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 18, 1421–1423 (2011).
Letessier, A. et al. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123 (2011).
Palakodeti, A. et al. Impaired replication dynamics at the FRA3B common fragile site. Hum. Mol. Genet. 19, 99–110 (2010).
Kelly, T. & Callegari, A. J. Dynamics of DNA replication in a eukaryotic cell. Proc. Natl. Acad. Sci. USA 116, 4973–4982 (2019).
Lõoke, M. et al. Relicensing of transcriptionally inactivated replication origins in budding yeast. J. Biol. Chem. 285, 40004–40011 (2010).
Helmrich, A., Ballarino, M. & Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977 (2011).
Fungtammasan, A., Walsh, E., Chiaromonte, F., Eckert, K. A. & Makova, K. D. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 22, 993–1005 (2012).
Bhowmick, R., Minocherhomji, S. & Hickson, I. D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 64, 1117–1126 (2016).
Tubbs, A. et al. Dual roles of Poly(dA:dT) tracts in replication initiation and Fork Collapse. Cell 174, 1127–1142 (2018).
Palozola, K. C. et al. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122 (2017).
Becker, N. A., Thorland, E. C., Denison, S. R., Phillips, L. A. & Smith, D. I. Evidence that instability within the FRA3B region extends four megabases. Oncogene 21, 8713–8722 (2002).
Veerappa, A. M. et al. Global patterns of large copy number variations in the human genome reveal complexity in chromosome organization. Genet. Res. 97, e18 (2015).
Redon, R. et al. Global variation in copy number in the human genome. Nature 444, 444–454 (2006).
Larkin, D. M. et al. Breakpoint regions and homologous synteny blocks in chromosomes have different evolutionary histories. Genome Res. 19, 770–777 (2009).
Vitae, G., Remita, A. M. & Diallo, A. B. Revisiting the landscape of evolutionary breakpoints across human genome using multi-way comparison. bioRxiv https://doi.org/10.1101/696245 (2019).
Woo, Y. H. & Li, W. H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun. 3, 1004 (2012).
Geoffrey, F. et al. Formation of chromosomal domains by loop extrusion. Cell Rep. 15, 2038–2049 (2016).
Suhas, S. P. R. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Jacqueline, H. B. et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632 (2013).
Mirkin, E. V. & Mirkin, S. M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 71, 13–35 (2007).
Zentner, G. E. & Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266 (2013).
Venkatesh, S. & Workman, J. L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 16, 178–189 (2015).
Bell, O., Tiwari, V. K., Thomä, N. H. & Schübeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 12, 554–564 (2011).
Richards, E. J. & Elgin, S. C. R. Epigenetic codes for heterochromatin formation and silencing: Rounding up the usual suspects. Cell 108, 489–500 (2002).
Easwaran, H. et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 22, 837–849 (2012).
Ibarra, A., Benner, C., Tyagi, S., Cool, J. & Hetzer, M. W. Nucleoporin-mediated regulation of cell identity genes. Genes Dev. 30, 2253–2258 (2016).
Farnham P, S. M., Cayting P. Histone modifications by ChIP-seq from ENCODE/Stanford/Yale/USC/Harvard https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=wgEncodeSydhHistone (2011).
Matthew, G. G., Stuart, S. L., Laurie, A. B., Rudolf, J. & Richard, A. Y. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88 (2007).
Tianyi, Z., Sarah, C. & Neil, B. The interplay of histone modifications—writers that read. EMBO Rep. 16, 1467–1481 (2015).
Pelliccia, F., Bosco, N., Curatolo, A. & Rocchi, A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res. 121, 196–200 (2008).
Sarni, D., et al. Replication timing and transcription identifies a novel fragility signature under replication stress. bioRxiv https://doi.org/10.1101/716951 (2019).
Brison, O. et al. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun. 10, 5693 (2019).
Moiseeva, T. N. et al. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl. Acad. Sci. USA 116, 13374–13383 (2019).
Wang, Y. et al. Temporal association of ORCA/LRWD1 to late-firing origins during G1 dictates heterochromatin replication and organization. Nucleic Acids Res. 45, 2490–2502 (2017).
Xiong, W., Li, J., Zhang, E. & Huang, H. BMAL1 regulates transcription initiation and activates circadian clock gene expression in mammals. Biochem. Biophys. Res. Commun. 473, 1019–1025 (2016).
Paulsen, M. T. et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 67, 45–54 (2014).
Canela, A. et al. DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell 63, 898–911 (2016).
Crosetto, N. et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 10, 361–365 (2013).
Hosseini, S. A. et al. Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes Chromosomes Cancer 52, 1017–1029 (2013).
Probst, A. V., Dunleavy, E. & Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 10, 192–206 (2009).
Sarah, M. R. et al. Design of a synthetic yeast genome. Science 355, 1040–1044 (2017).
A fully designed yeast genome. Nat. Methods 14, 469–469, https://doi.org/10.1038/nmeth.4272 (2017).
Macheret, M. et al. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. https://doi.org/10.1038/s41422-020-0358-x (2020).
Marchal, C. et al. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 13, 819–839 (2018).
Mori, H., Evans-Yamamoto, D., Ishiguro, S., Tomita, M. & Yachie, N. Fast and global detection of periodic sequence repeats in large genomic resources. Nucleic Acids Res. 47, e8 (2019).
Acknowledgements
This work was supported by grants to Songmin Ying from the Ministry of Science and Technology of China (2016YFA0100301), the National Natural Science Foundation of China (81870007, 81920108001), the Zhejiang Provincial Natural Science Foundation (LD19H160001), and the Zhejiang Provincial Program for the Cultivation of High‐Level Innovative Health Talents (2016‐63). We thank Prof. Ian Hickson (University of Copenhagen, Denmark) for advice on the analysis of MiDAS, and Dr. Jianming Zeng (University of Macau) for valuable suggestions on bioinformatics. We deeply thank Prof. Cheng Li (Peking University) for the courtesy of data on TAD. Thanks for the technical support by the Core Facilities, Zhejiang University School of Medicine.
Author information
Authors and Affiliations
Contributions
F. Ji and H.L. designed the experiments. F. Ji, H.L., F.Z., X.G., S.L., M.Z.S. and H.C. conducted the experiments. S.P., L.O., F. Jia and Z.F. performed bioinformatics analysis. H.L. and F. Ji wrote the manuscript. X.W. and T.L. generated and analyzed the Hi-C data. W.L. and Z.C. analyzed, interpreted the data. S.Y. and H.S. initiated the study, instructed the project and interpreted the data. All authors edited the manuscript and proofread it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Ji, F., Liao, H., Pan, S. et al. Genome-wide high-resolution mapping of mitotic DNA synthesis sites and common fragile sites by direct sequencing. Cell Res 30, 1009–1023 (2020). https://doi.org/10.1038/s41422-020-0357-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-020-0357-y
This article is cited by
-
RECQL4 is not critical for firing of human DNA replication origins
Scientific Reports (2024)
-
Mitotic DNA synthesis in response to replication stress requires the sequential action of DNA polymerases zeta and delta in human cells
Nature Communications (2023)
-
Replication stress causes delayed mitotic entry and chromosome 12 fragility at the ANKS1B large neuronal gene in human induced pluripotent stem cells
Chromosome Research (2023)
-
DEtail-seq is an ultra-efficient and convenient method for meiotic DNA break profiling in multiple organisms
Science China Life Sciences (2023)
-
Replication stress generates distinctive landscapes of DNA copy number alterations and chromosome scale losses
Genome Biology (2022)
