
Abstract
Adaptive immunity is essential for pathogen and tumor eradication, but may also trigger uncontrolled or pathological inflammation. T cell receptor, co-stimulatory and cytokine signals coordinately dictate specific signaling networks that trigger the activation and functional programming of T cells. In addition, cellular metabolism promotes T cell responses and is dynamically regulated through the interplay of serine/threonine kinases, immunological cues and nutrient signaling networks. In this review, we summarize the upstream regulators and signaling effectors of key serine/threonine kinase-mediated signaling networks, including PI3K–AGC kinases, mTOR and LKB1–AMPK pathways that regulate metabolism, especially in T cells. We also provide our perspectives about the pending questions and clinical applicability of immunometabolic signaling. Understanding the regulators and effectors of immunometabolic signaling networks may uncover therapeutic targets to modulate metabolic programming and T cell responses in human disease.
Similar content being viewed by others
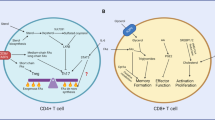
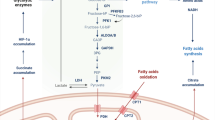
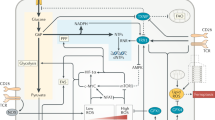
Introduction
The adaptive immune system is comprised of T and B cells and is critical for host responses to invading pathogens and tumors. Upon T cell receptor (TCR) recognition of the cognate antigen in the presence of co-stimulatory and cytokine signals, signaling networks are activated in naïve T cells to facilitate clonal expansion, effector cell differentiation and immune function. Following antigen eradication, most effector cells die via programmed cell death, but some cells persist long-term as memory cells and are primed for rapid recall response. Activation states and subpopulations of T cells account for marked cellular heterogeneity. In addition, T cell functional states have shared and distinct transcriptional programs, along with differences in protein expression, activity and interactions that are only beginning to be understood.1
In recent years, the importance of cellular metabolism in dictating T cell development and function has become increasingly realized. Quiescent cells, such as naïve or memory T cells, favor mitochondria-driven catabolic metabolism, including oxidative phosphorylation (OXPHOS).2 Upon activation, T cells undergo quiescence exit driven in part by the catalysis of nutrients, such as glucose and glutamine, which serves to generate sufficient macromolecular supply for cellular growth and support the increased energy demands associated with such growth. This metabolic reprogramming not only includes elevated aerobic glycolysis and glutaminolysis, but also a marked upregulation of mitochondrial biogenesis and function.2 Furthermore, these changes in metabolic programs must be properly regulated to maintain immune homeostasis and functional specificity of T cell subsets.1,2 How metabolic programs are modulated by upstream signaling networks and the mechanistic role of metabolic rewiring in T cells therefore remain critical to address.
In this review, we discuss cellular signaling pathways involved in immunometabolic regulation, including relevant molecules, upstream and downstream targets and their cell type-specific effects. Specifically, we focus our discussion on phosphoinositide 3 kinase (PI3K) — protein kinase A, G and C (AGC) kinases, mechanistic target of rapamycin (mTOR) and liver kinase B1–5′ AMP-activated protein kinase (LKB1–AMPK) signaling. We also comment on emerging research interests within these pathways and the potential areas for future investigation. Thus, this review provides necessary information on molecular signaling, contextualized for the immunometabolism field, especially in T cells, and highlights how this field may drive advancements in therapies for human disease.
PI3K–AGC signaling
Phospholipids are key second messengers that influence downstream immunometabolic pathways and are thus highly regulated with respect to turnover. One central mediator of phospholipid turnover is PI3K, which converts phosphatidylinositol-(4,5)-bisphosphate (PIP2) into phosphatidylinositol-(3,4,5)-trisphosphate (PIP3). Generation of PIP3 allows for plasma membrane recruitment and functional modulation of proteins containing pleckstrin homology (PH) domains. Thus, PI3K activity can generate a subcellular hub of signaling by recruiting numerous PH domain-containing effector proteins into close proximity. PI3K activity induces diverse signaling pathways involved in regulating cellular function, including Akt (also called protein kinase B), phosphoinositide-dependent protein kinase 1 (PDK1), mTOR complex 1 (mTORC1) and mTORC2 (mTORC1 and mTORC2 discussed in a separate section below). In this section, we will describe the PI3K-dependent signaling networks, especially those related to activation of the AGC kinases, in T cells and how they influence cellular metabolism (Fig. 1).

Activation of TCR, CD28 and IL-2R induces phosphorylation and activation of PI3K and also inactivation of PI3K-suppressing molecules, such as PTEN and PIK3IP1. PIP2 is converted to PIP3 via the activity of PI3K, and PIP3 facilitates plasma membrane recruitment and activation of downstream signaling molecules including PDK1 and Akt. mTORC2 further activates Akt and promotes increased metabolism and T cell effector function.
PI3K
Class I PI3Ks are heterodimeric proteins consisting of a catalytic p110 subunit (p110α, p110β, p110δ, or p110γ) and one of five regulatory subunit isoforms (p50α, p55α, p55γ, p85α, or p85β). In the absence of stimulation, the kinase domain of the catalytic subunit is suppressed by the regulatory subunit.3 The concentrations of PI3K substrates are closely linked to cellular activation state, which, in T cells, is dictated by TCR engagement, CD28 family-mediated co-stimulation and cytokine signaling, such as downstream of IL-2 receptor (IL-2R).2 Following upstream activation, the Src-homology-2 (SH2) domain of PI3K can bind specific phosphorylated YXXM motifs on upstream receptors or adapter proteins, causing the release of inhibitory connections of the regulatory subunit and the translocation of the catalytic subunit to the substrate-rich plasma membrane.4,5 T cell activation is associated with a shift in signaling and transcriptional programs, which enhances anabolic metabolism, fueled by catabolic processes (e.g., glycolysis and glutaminolysis), to meet the increased demand for cellular nutrients and energy. This exit from quiescence is crucial for the development of effector T cell populations.1 Indeed, PI3K activation is a seminal event in this process, as its downstream signaling cascades result in elevated aerobic glycolysis and mitochondrial biogenesis,2,6 as discussed more below.
PI3K signaling is negatively regulated by phosphatase activity. Specifically, PIP3 is converted to PIP2 or PI-(3,4)-P2 by phosphatase and tensin homolog (PTEN) and SH2 domain-containing inositol 5′-phosphatase (SHIP), respectively. SHIP plays an essential role in promoting Th1 cell responses,7 whereas PTEN deficiency leads to hyperactivation of T cells, especially under suboptimal stimulation.8 PTEN is repressed upon strong TCR stimulation or CD28 co-stimulation, in part, due to the Tec family kinase IL-2-inducible T cell kinase (Itk),8,9,10,11 and is also negatively regulated by acylglycerol kinase (AGK)-mediated phosphorylation during CD8+ T cell activation.12 Mice with T cell-specific PTEN deficiency develop spontaneous autoimmunity associated with hyperactivation of T cells as noted above, as well as lymphoma.13 Regulatory T (Treg) cell-specific PTEN deficiency also results in autoimmunity due to destabilized Foxp3 expression (also called instability) and diminished suppressive function of Treg cells.14,15 PTEN deficiency in Treg cells is associated with altered glycolytic and mitochondrial metabolism, as well as changes in DNA methylation at conserved noncoding sequences that are essential for Treg cell stability.14,15 T cells lacking Itk, and thus the ability to suppress PTEN function upon T cell activation, display increased Treg cell differentiation in vitro and in vivo.9 Thus, negative regulation of PI3K signaling is associated with proper metabolic regulation for control of immune homeostasis and suppression of tumorigenesis.
The p110 catalytic subunit of PI3K can be directly inhibited by the transmembrane protein PI3K-interacting protein 1 (PIK3IP1). T cells downregulate PIK3IP1 expression following activation, suggesting its importance in restraining PI3K signaling until proper activation signals are received. Indeed, T cells lacking PIK3IP1 display increased activation and IL-2-driven Th1 cell proliferation.16,17 PI3K signaling is also regulated at the mRNA level by Roquin proteins, which limit translation of target mRNAs encoding co-stimulatory receptors, such as ICOS.18 Mice with Roquin deficiency develop lupus-like autoimmunity with uncontrolled T follicular helper (Tfh) cell responses.18 Interestingly, Treg cell-specific loss of Roquin causes increased T follicular regulatory cells, which are capable of limiting germinal center (GC) responses but not colitis.19 Thus, PIK3IP1 and Roquin restrain T cell responses under selective contexts, unlike the more generic suppression mediated by PTEN.
The role of the class III PI3K (catalytic subunit vacuolar protein sorting 34 (Vps34)) in immunometabolic signaling is less understood, although it is important for autophagy (discussed more below) and vesicle trafficking in certain cell types.3 Interestingly, T cell development does not require Vps34, but survival of naïve T cells is severely impaired in its absence, likely due to improper recycling of the pro-survival cytokine receptor for IL-7 (IL-7R).20,21 Also, Vps34-deficient T cells display enhanced mitochondrial accumulation due to reduced mitochondrial clearance,22 which may also contribute to reduced survival. Together, these results demonstrate that PI3K signaling supports metabolic programming to orchestrate the homeostasis and function of T cells.
AGC kinases
Among the most notable PH domain-containing proteins are members of the AGC kinases, which include PDK1, Akt, ribosomal S6 kinase (RSK, also called p90) and serum/glucocorticoid-regulated kinase 1 (SGK1).3 PDK1 is a serine/threonine kinase with important roles in activating other AGC kinases, including Akt and SGK1 (discussed below). In the absence of PDK1, T cell activation, but not survival, is reduced, in part due to defective NF-κB activation.23 TCR and CD28 activation promotes PDK1 recruitment to the plasma membrane and induces its phosphorylation, which is antagonized by PIP2 or inhibition of PI3K activity. Protein kinase C-θ (PKC-θ)-dependent phosphorylation of PDK1 at Thr513, rather than the auto-phosphorylation of the kinase domain residue (Ser241), is essential for driving T cell activation.23 In activated CD8+ T cells, PDK1 is involved in metabolic reprogramming that sustains glucose uptake and glycolysis downstream of IL-2 stimulation; this process requires the mTOR–HIF-1α (hypoxia-inducible factor-1α) axis, but not PI3K or Akt activity,24,25 although Akt is essential for programming glucose uptake in other T cell subsets as discussed below. In addition, Treg cells deficient in the autophagy protein Atg7 have increased PI3K–PDK1 signaling, which is associated with heightened mTORC1 activity, glycolysis and the development of an autoimmune disorder.26 On the other hand, deficiency for PDK1 in T cells leads to chronic intestinal inflammation, due to an imbalance of CD8+ γδ T cell accumulation; colitis in these mice is attributed to reduced Treg cell function in the absence of PDK1.27 Thus, the proper regulation of PDK1 activity is essential for enforcing appropriate T cell activation, as well as Treg cell function for the control of inflammation.
Akt is the best-studied AGC kinase in immune cells, whose activation is induced upon its recruitment to the plasma membrane. Maximal Akt activation is achieved following phosphorylation by mTORC2 at Ser473, which allows Akt binding to the substrate dock, termed the ‘PIF pocket’, of PDK1, which promotes phosphorylation of Akt at Thr308.28 mTORC2–Akt signaling coordinates T cell activation, differentiation and trafficking.24,29,30,31 In addition, Akt activates processes associated with cellular metabolism, such as by increasing glycolysis during the later stages of TCR and co-stimulatory receptor activation.32,33 This function may be mediated by increased phosphorylation of glucose transporter 1 (GLUT1) by Akt, which promotes its transport to the plasma membrane.34,35 One notable observation is that Akt is essential for programming initial activation of glycolysis in memory, but not naïve, T cells,32,36 but how Akt signaling differentially regulates the timing of glucose metabolism among T cell subsets or during different states of activation is unknown.
One important role for Akt in T cells is to regulate the activity of forkhead box O (FoxO)37 transcription factors by phosphorylation, which leads to their exclusion from the nucleus and termination of target gene transcription. FoxO proteins are more transcriptionally active in quiescent cell populations such as naïve T cells, where they promote expression of the homeostatic cytokine receptor IL-7R and also cell trafficking molecules (e.g., CD62L, CCR7 and S1PR1) via KLF2.37,38,39 Mice with T cell-specific FoxO1 deficiency develop mild autoimmunity, in contrast to the severe, lethal autoimmunity in mice with Treg cell-specific FoxO1 deficiency.40 Interestingly, more recent evidence suggests that proper downregulation of FoxO1 activity is crucial for T cell homeostasis, as mice harboring a constitutively active FoxO1 mutant in T cells also develop severe autoimmunity.41 Mechanistically, FoxO1 downregulation in activated T cells is important for the coordination of cellular growth and proliferation by allowing sustainable mTORC1 signaling and anabolic metabolism.41 In Treg cells, ‘graded’ increases in FoxO1 activity has context-specific functions; constitutive FoxO1 activity impairs Treg cell-dependent suppression of lymphoproliferative disease driven by CD8+ T cells, whereas partial enhancement of FoxO1 activity reduces Treg cell accumulation in non-lymphoid tissues, including tumors, leading to increased anti-tumor T cell activity.42 Thus, the Akt–FoxO1 axis dynamically regulates T cell responses.
Another serine/threonine kinase involved in Akt signaling is glycogen synthase kinase 3 (GSK-3), which is constitutively active in resting cells, but is inactivated via phosphorylation by Akt.43 Active GSK-3 plays an important functional role in T and B cells to promote their survival and to restrict activation, by limiting nuclear factor of activated T cells (NFAT) activity.44 However, GSK-3 function is not limited to naïve or quiescent cells, as it is also a scaling factor for anabolic metabolism in cells with ongoing or elevated metabolic profiles, such as GC B cells.45 The context-specific roles of GSK-3 will therefore be interesting to investigate.
The precise roles of AGC kinases such as RSK, as well as their substrates, in T cell metabolic programming is of continued interest. The role of SGK1 is discussed in a later section.
Emerging perspectives
PI3K–AGC signaling remains an important area of immunology research, as we continue to illuminate its potent influences on immune cell differentiation and magnitude of effector/memory T cell responses. This information has been a central focus of translational immunology research. Specifically, how is T cell-intrinsic PI3K signaling affected within the tumor microenvironment, and can modulation of certain PI3K factors improve the efficacy of treatments, such as adoptive cell therapy?
The transcription factor TCF-1 (encoded by Tcf7) has garnered considerable attention recently due to its role in preserving a stem cell-like state (termed ‘stemness’) in T cell populations during chronic inflammation or in the tumor microenvironment.46,47,48,49,50 In fact, asymmetric PI3K distribution during cell division can impart memory- or effector-like functional identity in daughter cells through differential regulation of TCF-1.51 This PI3K–TCF-1 axis plays a major role in tumor-infiltrating lymphocyte function; elevated extracellular potassium levels within the tumor microenvironment can stifle PI3K signaling, resulting in reduced T cell effector function needed for tumor clearance.46,47 Interestingly, this effect is also associated with elevated TCF-1 expression and stem cell-like phenotypes, including self-renewal and persistence.46
Chimeric antigen receptor T (CAR-T) cells are a promising anti-cancer therapeutic strategy, yet issues with relapse and non-responders to CAR-T cell therapy highlight that additional work is needed to optimize this treatment. Inhibition of PI3K–Akt signaling during CAR-T cell generation ex vivo results in a more central memory phenotype, and these types of CAR-T cells display superior anti-tumor efficacy.52,53 Furthermore, targeting PI3K for anti-tumor therapy is not reserved for CD8+ T cells, as specific inhibition of the PI3K isoform p110δ affects Treg cells more than conventional CD4+ and CD8+ T cells.54 Finally, myeloid cell-specific PI3K modulation may also become an important strategy in combination with checkpoint therapy, as targeting p110γ limits the suppressive activity of tumor infiltrating myeloid cells,55,56 which may indirectly impact T cell function in the tumor microenvironment.
The downstream target repertoire of PI3K-dependent kinases is a topic of renewed interest, especially for cell type or context specificity. For example, PDK1 and Akt dictate differences in transcriptional programs in activated CD8+ T cells, with PDK1 being essential for IL-2-induced glucose metabolism and proliferation and Akt being critical for cellular trafficking and IFN-γ production.24 Also, TCR signal strength differentially regulates Akt activity in CD4+ T cells, and, more importantly, the induction of Akt-dependent targets, which is associated with their differentiation into pro-inflammatory effector cells or Treg cells in vitro.57 In GC B cells, Akt has altered activity compared to non-GC B cells due to phosphorylation site dominance and a preferential induction of negative regulators of B cell receptor (BCR) signaling58; this study helps explain how GC B cells have repressed BCR signaling, despite utilizing the same signaling molecules as non-GC B cells. The concept of “same molecules, new pathways” is an exciting idea for the field of immunometabolic signaling and may help shed light on existing conflicts in the literature. Moreover, it may help to define how similar receptor-mediated signaling pathways can impart different functional programs in distinct immune cell subsets, which may also be mediated by alterations in metabolite composition driven by these pathways.
In addition to PI3K, other phospholipid-modifying enzymes modulate T cell function. For example, phospholipase C-γ1 (PLC-γ1), which is activated upon TCR engagement, enzymatically cleaves PIP2 into diacylglycerol (DAG) and inositol-(1,4,5)-trisphosphate (IP3). IP3 binds to receptors on the endoplasmic reticulum (ER), causing Ca2+ efflux from the ER and subsequent opening of Ca2+ influx channels on the plasma membrane. Elevated cytosolic Ca2+ concentrations directly promote activation of the phosphatase calcineurin, which dephosphorylates NFAT, allowing for its nuclear translocation. This signaling cascade is an important regulator of T cell proliferation by promoting glycolysis and OXPHOS.59 In turn, the induction of glycolysis can feedforward enhance Ca2+–NFAT signaling to further support T cell function.60 The turnover of DAG is also an important regulator of T cell responses and is mediated by the DAG kinases (DGK), which converts DAG to phosphatidic acid; deficiency for DGK is associated with activation of mTOR activity in T cells.61,62 Thus, how other lipid-related signaling networks control metabolic reprogramming will be an important area to address in future studies.
mTOR signaling
mTOR is a serine/threonine protein kinase present in two signaling complexes, mTORC1 and mTORC2, which are composed of distinct protein binding partners.63 mTORC1 is defined by the mTOR kinase, the adapter protein regulatory-associated protein of mTORC1 (Raptor) and mammalian lethal with SEC13 protein 8 (MLST8; also called GβL), as well as the inhibitory subunits proline-rich Akt substrate-1 (PRAS1) and its target DEP domain-interacting protein (DEPTOR). In contrast, mTORC2 is composed of mTOR, rapamycin-insensitive companion of mTOR (Rictor) and DEPTOR, as well as the regulatory proteins, mammalian stress-activated protein kinase-interacting protein 1 (mSIN1) and protein observed with Rictor-1 or -2 (Protor1/2).63 Due to its ability to bind to the FKBP12–rapamycin complex, mTORC1 activity is more sensitive to inhibition by rapamycin than mTORC2, although both complexes are inhibited upon chronic or high-dose rapamycin treatment.64
mTORC1 signaling is essential for T cell development in the thymus, homeostasis in the periphery and differentiation into effector CD4+ Th1, Th2 and Th17 cells, as well as cytotoxic CD8+ T cells.2,29,30,31,65,66,67,68,69 By contrast, mTORC2 activity is more selectively required for Th1 and Th2 cell differentiation,29,30 but also regulates migration of Tfh and Treg cells.70,71 mTOR signaling plays an additional role in the antagonism of conventional T cell responses, by regulating the activation, lineage stability and suppressive function of Treg cells in vivo.14,15,26,72,73,74,79,75 Finally, mTOR signaling antagonizes the differentiation of long-lived memory CD8+ T cells in lymphoid tissues but promotes their development in non-lymphoid tissues.76,77,78,79,80 Thus, mTOR signaling is a central regulator of T cell responses.
mTOR signaling is involved in many cellular processes, with a notable role in promoting protein synthesis through its phosphorylation of ribosomal protein S6 kinase (S6K; upstream kinase for ribosomal protein S6 (S6)) and eukaryotic translation initiation factor 4E (eIF4E) binding proteins (4E-BPs). On the other hand, mTORC2 induces Akt Ser473 phosphorylation, as well as the phosphorylation of PKC and other AGC kinases. Through these actions, mTORC2 promotes cell proliferation and survival63 and also has reported roles in cellular migration.63,70,71 Among its functions, the role of mTOR as a central regulator of cellular metabolism is of high interest in T cells. In this section, we will highlight the mechanisms and functions of mTOR signaling in reprogramming cellular metabolism in T cells (Fig. 2).

The discrete mTOR complexes, mTORC1 (consists of mTOR, Raptor, PRAS1, DEPTOR and MLST8) and mTORC2 (consists of mTOR, Rictor, Protor1/2, mSIN1 and DEPTOR), are activated by immunological receptors (TCR, CD28 and IL-2R) and growth factors. mTORC1 activation is also sensitive to nutrients such as amino acids. Amino acids promote mTORC1 activation through the Rag complex, which also plays a permissive ‘licensing’ role to allow for TCR and CD28 co-stimulatory signals to induce mTORC1 activation. The Tsc complex, whose activity is suppressed by immune and growth factor signals, inhibits the activation of the small G protein Rheb, which promotes mTORC1 activation. mTORC1 induces cell growth and protein translation through S6K and eIF4E, as well as lipid synthesis through PPARγ and SREBP1. mTORC1 inhibits autophagy through ULK1 under nutrient-replete conditions. In contrast, mTORC2 is activated by growth factors and it mainly regulates survival and actin reorganization via Akt, SGK1 and PKC. mTORC2 also plays critical roles in context- or subset-specific metabolic reprogramming for cell growth, proliferation and survival.
Metabolic and immunological inputs for mTORC1
Lysosomes are a key signaling hub for driving mTORC1 activity. Several protein activators of mTORC1 are localized to the lysosome, including the small G proteins Ras homolog enriched in brain (Rheb) and Rag (obligate heterodimers comprised of RagA or RagB paired with RagC or RagD), which associate with the lysosomal membrane via the LAMTOR complex.81,82,83 Growth factors or receptor tyrosine kinases induce mTORC1 activity by inhibiting the GTPase activating protein (GAP) function of the tuberous sclerosis complex (Tsc; comprised of Tsc1, Tsc2 and Tbc1d7); the inhibitory phosphorylation of Tsc2 is usually mediated by Akt, but may be driven by RSK or extracellular signal-related kinase-1 or -2 (Erk1/2).84,85,86,87 Consequently, Rheb retains its GTP-bound state, which, when transiently recruited to the lysosomal membrane, allows Rheb to promote mTORC1 activation.88,89 TCR and CD28 co-stimulatory signals act as a major network that allows for inhibitory phosphorylation of Tsc2, whose deletion leads to spontaneous T cell activation and altered T cell homeostasis.68 However, other pathways, including insulin or Wnt signaling, may also promote mTORC1 signaling for T cell activation, differentiation or function.63,90,91 The precise mechanisms that dictate mTORC2 activity are unclear, although its activation is mediated by PI3K-induced PIP3 binding to mSIN1,92,93 thus allowing for increased mTORC2 activation under physiological conditions where AGC kinase function is likely to be essential.
Amino acids are a major nutrient signal that induces mTORC1 activation. Selective amino acids are stored in lysosomes, where under nutrient-sufficient conditions, they efflux into the cytoplasm via lysosomal v-ATPase, whose activity is regulated by SLC38A9 when SLC38A9 binds to the amino acid arginine.94,95 Upon amino acid stimulation, the Rag proteins convert to their active state (RagA- or RagB-GTP with RagC- or RagD-GDP), thus allowing binding to Raptor and recruitment of mTORC1 to the lysosome where it is engaged by Rheb.82,96 Amino acids are ‘sensed’ by upstream complexes that activate the Rag complex, including sestrin (binds leucine) and CASTOR1 (binds arginine), which trigger inactivation of the GATOR2 complex that suppresses Rag function.97,98,99,100,101,102 Leucyl-tRNA synthetase (LRS) and folliculin (Flcn), which binds folliculin-interacting protein (Fnip; isoforms Fnip1 and Fnip2), sensing systems have also been reported to promote Rag complex activation, by serving as GAPs for RagC or RagD103,104; notably, LRS can also activate Vps34 to stimulate the phospholipase D-dependent activation of mTORC1105 and may also promote the GTP binding of RagA or RagB via its GTPase GATOR1, although this effect has thus far only been identified in yeast.106 Glutamine can also indirectly induce Rag complex activity upon its conversion into α-ketoglutarate (α-KG) during glutaminolysis, which may be driven by leucine-dependent activation of glutaminase (GLS).107 Other nutrients, such as glucose and cholesterol, can prime mTORC1 signaling in a Rag complex-dependent manner, in part, through communication with SLC38A9 or the Neiman-Pick C1 complex,96,108 which requires further investigation in T cells.
Amino acids are also reported to regulate mTORC1 activation independently of the active Rag complex. For example, glutamine activates mTORC1 via ADP-ribosylation factor 1.109 Acetyl co-enzyme A (acetyl-CoA) derived from leucine can also modify Raptor via protein acetylation, such that its expression is stabilized to allow for mTORC1 activation.110 The ability of glutamine to promote mTORC1 activation upon TCR stimulation requires the CBM (CARMA1–Bcl10–MALT) complex, although the mechanisms are unclear as this is independent of canonical NF-κB signaling.111 In addition, the Tsc complex is displaced from the lysosome in the presence of amino acids, especially arginine, in activated Treg cells (and possibly conventional T cells), thus allowing for activation of mTORC1 signaling.112 Therefore, there appears to be coordinated communication between amino acid sensing and regulation of the GAPs that inhibit mTORC1 activation.
Recent work has established baseline differences in the expression of nutrient transporters between naïve CD4+ and CD8+ T cells.113 Upon stimulation by immunological signals, expression of nutrient transporter proteins increases to facilitate nutrient influx (with the degree of upregulation displaying differences in CD4+ and CD8+ T cell subsets113), including upregulation of ASCT2 (transporter for glutamine) and the LAT1–CD98 complex (transporter for leucine or other neutral amino acids).111,114 The increased expression of these transporters is essential for not only driving metabolic reprogramming as discussed below, but also inducing or maintaining mTORC1 signaling. Indeed, the induction of leucine- and glutamine-driven mTORC1 activation is impaired in LAT1- and ASCT2-deficient T cells, respectively,111,114 and mTORC1 activation is also reduced in CD98-deficient Treg cells, probably due to reduced isoleucine influx.115 Intriguingly, amino acids are also critical for promoting TCR- and CD28-dependent mTORC1 signaling in Treg cells,112,116 suggesting that amino acids and, possibly other nutrients, not only promote mTORC1 activation, but are also a prerequisite for immunological receptors to induce (a process we have termed ‘licensing’112) and sustain activity of mTORC1. Whether other nutrients also prime immune receptor signaling pathways at the level of mTORC1 or other signaling pathways remains unexplored.
Glycolysis and mitochondria-associated metabolism
Cellular metabolism is a critical mediator of T cell responses.2 mTORC1 signaling promotes metabolic reprogramming toward increased aerobic glycolysis, glutaminolysis and remodeling of mitochondrial metabolism.2 Upon T cell activation, mTORC1 upregulates expression of the transcription factor c-Myc, whose activity is essential for promoting expression of genes involved in glucose and glutamine metabolism, including hexokinase 2 (rate-limiting enzyme for glycolysis) and GLS; the induction of these c-Myc-dependent programs is crucial for effector T cell proliferation and differentiation.17,31,117 In addition, mTORC1 sustains glycolysis that supports Th17 cell (but antagonizes Treg cell) differentiation by upregulating HIF-1α expression,118,119 which requires upstream PDK1 but not PI3K signaling.25
By regulating the activation of AGC kinases, mTORC2 activity is also linked to metabolic reprogramming, although its function seems to be less important for early metabolic programming (i.e., that occurs during initial quiescence exit) that promotes T cell activation.31 As such, mTORC2 can be considered as an amplifier of mTORC1-related metabolic reprogramming. As an example, mTORC2 activity supports the optimal induction of glycolysis, by inducing Akt activity that promotes surface expression of GLUT1, which regulates Th1 and Tfh cell differentiation.29,34,35,70 Interestingly, mTORC2 activity also promotes Th2 cell differentiation, which also requires glycolysis,31 via the PKC-dependent induction of GATA3 expression.29 mTORC2 signaling can also regulate the Akt-dependent inactivation of GSK-3 at ER–mitochondria contacts to enable pyruvate oxidation in mitochondria.120 The metabolic programs shaped by extracellular minerals, such as sodium, are also likely to be regulated by mTORC2. For example, SGK1 is activated by mTORC2, and SGK1 expression is induced under conditions of elevated salinity and promotes the generation of pathogenic Th17 cells by maintaining expression of IL-23 receptor.121,122 SGK1 also positively regulates Th2 cell differentiation while suppressing Th1 cell development.123 The molecular mechanisms for selectivity of mTORC2 target activation in T cells await discovery.
mTOR signaling also plays pleiotropic roles in regulating mitochondrial metabolism. Mitochondrial metabolism in naïve T cells is catabolic, which supports cellular homeostasis.2 Under these conditions, mTOR signaling is actively retained at lower levels, which establishes the quiescence of these cells.68 Naïve T cells also have low mitochondrial content that increases as the cells differentiate into effector T cells.66,124 mTORC1 activation promotes mitochondrial biogenesis via the induction of several mitochondrial proteins, including mitochondrial ribosomal subunits.66 These effects may also be mediated by the mTORC1–4E-BP1-dependent upregulation of Tfam (transcription factor A, mitochondrial), which promotes expression of mitochondrial DNA-derived genes (e.g., selective components of the electron transport chain).125 Mitochondrial function is further increased as cells transition from effector to memory T cells; this enhanced mitochondrial function is mediated, in part, by mitochondrial fusion driven by CD28 co-stimulation.126,127,128 This event is also likely associated with decreased mTOR signaling,76,77,78,79 but the precise mechanisms that govern downregulation of mTOR signaling for the effector-to-memory T cell transition remain unknown.
The mTORC1-dependent upregulation of mitochondrial metabolism promotes efficient OXPHOS, as well as the generation of different epigenetic-regulating metabolites (e.g., α-KG, 2-hydroxyglutarate and acetyl-CoA) to regulate T cell functional programming.17,66,129,130,131,132,133,134,135 Another consequence of increased mitochondrial biogenesis is augmented serine- and folate-dependent one-carbon metabolism, which regulates the generation of the methyl-donor S-adenosylmethionine (SAM), as well as glutathione and nucleotides important for T cell activation.124,136,137,138,139,140 In T cells, the upregulation of one-carbon metabolism-associated enzymes requires mTORC1 activation,66 which may be mediated by the mTORC1-dependent activation of ATF4.141 In summary, mTOR activity coordinates transcriptional and post-transcriptional programming to mediate anabolic metabolism for control of T cell responses.
Lipid and cholesterol synthesis
Lipids and cholesterol are integral components of cellular membranes and are important regulators of T cell responses. De novo lipid and cholesterol synthesis promotes activation-induced proliferation and differentiation of T cells. This metabolic shift is partially controlled by mTORC1 downstream of TCR activation, as Raptor-deficient naïve CD4+ T cells have lower expression of lipid synthesis-related genes (e.g., Hmgcr (encodes HMG-CoA reductase, HMGCR; rate-limiting enzyme for mevalonate synthesis), Fasn and Scd), associated with reduced proliferation and cell growth.31 These effects also extend to Raptor-deficient Treg cells.72 Unlike mTORC1, the role of mTORC2 in lipid metabolism is largely unknown in immune cells, but some studies suggest a role in the de novo synthesis of sphingolipids by regulating the activity of ceramide synthase.142 Interestingly, ceramide synthesis is involved in promoting efficient TCR signaling, activation and effector responses in human T cells,143,144,145 which appears to be linked with mitochondrial respiration through unknown mechanisms.135
Fatty acid synthesis is initiated by the cytosolic enzyme acetyl-CoA carboxylase-1 (ACC1), which promotes the conversion of mitochondria-derived acetyl-CoA into malonyl-CoA, the precursor for fatty acid synthesis. ACC1 deficiency in T cells inhibits Th17 and induces Treg cell differentiation in vitro, which is recapitulated using the ACC1 inhibitor soraphen A.146 Notably, both pharmacologic or genetic inhibition of ACC1 not only blocks fatty acid synthesis, but also impedes metabolic flux through glycolysis and the tricarboxylic acid cycle,146 possibly due to accumulation of metabolic intermediates (e.g., citrate or others) that enforce feedback inhibition of these pathways. Deficiency for ACC1 also impairs Th1 and Th2 cell development.146 On the other hand, ACC1 deletion in T cells does not affect CD8+ T cell function, even though it enhances cell death and reduces effector T cell expansion in response to bacterial infection.147 These findings suggest that fatty acid synthesis is important for CD8+ T cell persistence. The mitochondrial isoform ACC2 is involved in balancing lipid metabolism in the mitochondria through inhibition of carnitine palmitoyl-transferase a (CPT1a)-dependent fatty acid β-oxidation (FAO),148 but this effect is not essential for CD8+ T cell function.149
In T cells, mTORC1 induces de novo lipid and cholesterol synthesis by increasing the respective expression of sterol regulatory element-binding protein-1 (SREBP1; isoforms SREBP1-a and SREBP1-c) and SREBP2, which may be connected to the cholesterol-dependent activation of mTORC1.31,108 mTORC1 also phosphorylates lipin-1, which promotes the nuclear localization of lipin-1 and allows for the induction of SREBP expression and function.150 The activity of SREBPs is also directly controlled by the cholesterol-sensing protein SREBP cleavage-activating protein (SCAP). Specifically, when cholesterol concentrations are high, SCAP sequesters SREBP in the ER. Upon cholesterol depletion, the SCAP–SREBP complex moves to the trans-Golgi, where SREBPs are cleaved by the proteases S1P and S2P to allow for SREBP translocation from the Golgi complex to the nucleus, where they regulate the expression of lipogenesis-inducing genes and transporters.151,152 Studies have shown important roles for SREBP function in T cells. Deletion of SCAP in T cells does not affect homeostatic proliferation, but impairs activation-induced cell growth, proliferation and differentiation, which promotes clearance of viral infections in vivo.153 The reduction of cell growth and proliferation is restored upon addition of exogenous cholesterol, which supports membrane biogenesis.153 Through c-Myc, HIF-1α and AMPK-independent mechanisms, SCAP deficiency also inhibits metabolic reprogramming towards glycolysis, glutaminolysis and mitochondrial function,153 which may further impede lipid synthesis by limiting the concentrations of upstream, pro-lipogenic metabolites (e.g., acetyl-CoA). Thus, SREBPs are multifactorial regulators of metabolic programming in activated T cells.
How do activated T cells maintain SREBP function when the accumulation of intracellular cholesterol may impede SREBP activity? First, the expression of SULT2B1, which is an oxysterol-metabolizing enzyme, is upregulated upon T cell activation.154 Consequently, oxysterols that may be metabolized from cholesterol are unable to activate the liver X receptor (LXR) transcription factors, which promote cholesterol efflux by increasing expression of the ABCG1 transporter.154,155 LXRβ-deficient T cells have increased cellular proliferation, whereas deletion of ABCG1, but not the related protein ABCA1, reduces T cell proliferation to the same extent as LXR activation,154 demonstrating functional antagonism of SREBP and LXR in T cell proliferation. These effects are probably linked to ABCG1-dependent suppression of mTOR activity.156 Second, cholesterol derivatives can modulate the strength or duration of TCR signaling, with cholesterol esters and cholesterol sulfate decreasing TCR signal strength.157,158 That reducing cholesterol esters does not alter glycolysis or mitochondrial oxygen consumption, despite the increase of TCR signaling,158 may suggest inhibitory roles for cholesterol derivatives on mTOR activation, which remains to be addressed.
Fatty acids also play signaling roles to sustain pro-lipogenic programming apart from SREBP. For instance, fatty acids activate peroxisome proliferator-activated receptors (PPARs), which are nuclear hormone receptors that are important for lipid metabolism, as well as glucose homeostasis in cells. CD28 co-stimulation augments mTORC1 activation, as well as fatty acid uptake, to increase PPARγ expression and activity, respectively, in T cells.159 Further, PPARγ-deficient CD4+ T cells have reduced proliferation, survival and Th17 cell differentiation, which is associated with attenuated autoimmune and graft-vs.-host disease responses in mice.159,160 These features are also associated with reduced metabolic reprogramming upon TCR stimulation, and the defects in activation are restored by exogenous fatty acids.159
Fatty acids differ by length, with those of total carbon atom numbers from 1 to 6 usually categorized as short-chain fatty acids (SCFAs), whereas those of 7–12 carbon atoms are defined as medium-chain fatty acids; long-chain fatty acids (LCFAs) have more than 12 carbons.161 The microbiota-derived SCFA butyrate promotes cellular metabolism, by enhancing the memory differentiation potential of activated CD8+ T cells; butyrate also promotes optimal recall responses upon antigen re-stimulation,162 an effect that has also been observed with the glycolysis-inducing SFCA acetate.163 Intestine-derived SCFAs also support Treg cell generation in non-lymphoid tissues.164,165,166 Dietary LCFAs alter the composition of the gut microbiome.167 Further, LCFAs enhance Th1 and Th17 cell differentiation whereas SCFAs promote Treg cell formation,168 demonstrating unique phenotypes driven by different fatty acids.
Recent work has indicated that de novo-synthesized lipids can also drive catabolic functions of certain T cell subsets. For example, memory CD8+ T cells display less LCFA uptake than effector CD8+ T cells.169 Instead, upon IL-7 stimulation, memory CD8+ T cells can increase expression of aquaporin 9 for glycerol import to promote synthesis of triacylglycerides (TAGs) and sustain ATP production via FAO.170 Thus, TAG synthesis is an important process for modulation of memory T cell survival and self-renewal, which could possibly be exploited for therapeutic manipulation of these processes.
Autophagy
Autophagy is an evolutionarily-conserved pathway that promotes degradation of cytoplasmic components. This process is initiated through the formation of a double-membraned, intracellular organelle called an autophagosome, which, through a series of biochemical reactions, fuses with the lysosome for degradation and recycling of internal proteins and organelles.63 When nutrient concentrations are abundant, mTORC1 impairs autophagic flux171 by directly inhibiting the activity of the ULK complex (ULK1, Atg13 and FIP200), which is required for autophagy.172,173 mTORC1 can also indirectly affect autophagy at the level of lysosome biogenesis. Specifically, mTORC1 suppresses lysosome biogenesis via inhibition of transcription factor EB (TFEB) family transcription factors, which induce expression of numerous lysosome- and autophagy-specific genes.174 On the other hand, under nutrient restriction, mTORC1 activity is suppressed, leading to the activation of the ULK complex and TFEB transcription factors, thus permitting the induction of autophagy.
Autophagy is essential for T cell homeostasis, function and differentiation. For example, deletion of Atg7 in mature T cells reduces cell survival. In addition, compared to naïve T cells, autophagic flux is higher in antigen-specific CD8+ T cells.175 During acute infection of LCMV, autophagy is downregulated in CD8+ T cells undergoing proliferative expansion, whereas autophagy is upregulated before the contraction phase of the response. Deletion of the autophagy proteins Atg5 or Atg7 in effector CD8+ T cells is associated with reduced cell survival and memory CD8+ T cell generation.175,176 Autophagy also supports T cell function in anti-tumor immunity.177 In addition, autophagy is upregulated in Treg cells compared to naïve CD4+ T cells, and it promotes Treg cell responses for suppression of autoimmunity, anti-tumor immunity and other inflammatory disorders.26,178 These effects are attributed, in part, to reduced Treg cell survival in the absence of autophagy. Of note, we have recently found that suppression of autophagy in Treg cells is associated with an increase of mTORC1 activity and glycolysis, partially due to defects in downregulating expression of PI3K-related signaling components.26 Heightened glycolysis has also been observed in autophagy-deficient CD8+ T cells.177 Thus, autophagy supports many aspects of T cell fitness and function.
Emerging perspectives
Several proteins, such as the Tsc complex, Rheb, Rag, Raptor and Rictor, are conserved signaling nodes that influence mTOR activation in T cells.29,30,31,68,76,112,116 What remains to be determined is whether unique protein sensors of TCR-independent signaling networks (e.g., ‘signal 3′ cytokines) or other nutrients aside from amino acids exist, which may directly promote mTORC1 activation in T cells. In addition, besides the Tsc complex,68,76 the T cell-specific proteins that directly antagonize mTOR activity in response to immunological signals and nutrients are largely unknown. These may also include sensors of metabolites, such as those recently identified for SAM,179 which may suppress (or activate) mTORC1 to impart specificity of T cell responses. Another aspect that has remained unclear is how mTORC1-dependent signals are transmitted to functional T cell responses. For example, how can complete abrogation of mTORC1 signaling via Raptor deletion inhibit mitochondrial biogenesis, while incomplete suppression of mTORC1 signaling via Rheb deletion increase mitochondrial synthesis?66,76 One possible explanation for how ‘graded’ reductions in mTORC1 signaling alter metabolic programs, and ultimately cellular fate decisions, may lie in the discrete regulation of downstream targets. Indeed, we have shown that the phosphorylation of 4E-BP1 is more resistant to Rheb deletion than S6K, which is associated with milder defects in T cell activation than Raptor deficiency;31 these observations are consistent with those showing that the 4E-BP1 axis more selectively promotes the growth and proliferation of lymphocytes than the S6K pathway.180 Similarly, how mTORC2 targets affect T cell function in different physiological contexts will be interesting to define, as studies have implicated unique roles of Akt, PKC and SGK1 in T cell responses.29,121,122,123 Finally, the identification of novel mTOR targets in T cells may be achievable using mass spectrometry-based approaches.66,181,182 Such discoveries may uncover new therapeutic targets wherein selective aspects of mTOR signaling may be inhibited without detriment to others, such as dictating suppression of pro-inflammatory T cell responses without suppressing Treg cell function that is important for establishing immunological tolerance.29,30,31,72,73,76,112,116
LKB1–AMPK signaling
The LKB1–AMPK signaling pathway plays a central role in regulating cellular metabolism, proliferation and survival in response to altered nutrient and energy demands. LKB1–AMPK signaling promotes catabolic pathways that produce ATP, and enables metabolic plasticity in T cells in response to energy stress. By regulating metabolic reprogramming, LKB1 and AMPK contribute to T cell differentiation and function. In this section, we describe how LKB1 and AMPK activities are regulated, their effects on metabolism and roles in T cell-mediated immunity (Fig. 3).

The energy stress pathway kinases LKB1 and AMPK are activated by TCR and CD28 co-stimulatory signals, with AMPK activity being mediated, in part, by the Ca2+–CAMMK2 pathway. Energy stress, such as deprivation of glucose or glutamine or an imbalance of AMP/ADP-to-ATP ratio, can also promote LKB1–AMPK signaling. Upstream nutrient-sensing proteins, such as the Fnip–Flcn complex and Roquin, can restrain AMPK function, although the contribution of Fnip and Flcn to AMPK signaling in T cells is still unknown. The activation of LKB1 is associated with changes in mitochondrial metabolism and fitness, as well as increased mevalonate metabolism under certain contexts. By regulating the activity of several downstream targets, AMPK signaling can impede metabolic programming toward glycolysis, glutaminolysis and fatty acid synthesis, while promoting catabolic processes, such as mitophagy and autophagy. AMPK also supports mitochondrial fitness by driving mitochondrial biogenesis and mitochondrial dynamics by promoting mitochondrial fission.
Regulation of LKB1 and AMPK activities
LKB1 is a serine/threonine kinase that functions as a tumor suppressor and is involved in the regulation of cellular metabolism and proliferation. LKB1 has many downstream targets, including the most well-defined target AMPK and AMPK-related kinases, such as BRSK, NUAK and MARK.183,184,185 The upstream regulation of LKB1 is mediated by cellular localization and post-translational modifications. LKB1 forms a complex with STE20-related adapter (STRAD) and MO25, which promotes the cytoplasmic localization and kinase activity of LKB1, respectively.185,186 Further, Akt can inhibit LKB1 by promoting LKB1 nuclear retention.187 LKB1 undergoes many post-translational modifications, including phosphorylation, mevalonate pathway-related protein farnesylation, ubiquitination, SUMOylation and acetylation; however, how many of these modifications regulate LKB1 activity and the modifying enzymes required are unknown.185 Further, the contribution of many upstream regulators to LKB1 signaling in T cells remains underexplored.
AMPK is a conserved serine/threonine kinase that is comprised of α-, β- and γ-subunits. AMPK signaling is regulated by intracellular energy levels by sensing of the AMP/ADP:ATP ratio.188 AMP competes with ATP for binding to the cystathionine β-synthase 1 (CBS1) repeat in the γ-subunit of AMPK, which allosterically activates AMPK. Moreover, AMP and ADP binding to the CBS3 repeat limits access of phosphatases to Thr172 on the AMPK catalytic α-subunit, the critical phosphorylation site that promotes AMPK kinase activity.188,189 Altered nutrient availability can also regulate AMPK. Glucose- and glutamine-deficient conditions induce acute AMPK activation in T cells without altering intracellular ATP levels.190 Glucose starvation also promotes AMPK activation independently of AMP/ADP by reducing intracellular concentrations of fructose-1,6-bisphosphate (FBP).191 Low FBP levels allow the FBP enzyme aldolase to promote association of LKB1–Axin with the lysosomal/endosomal SLC38A9-v-ATPase-Ragulator network for the subsequent activation of AMPK;95,191 whether FBP deprivation leads to AMPK activation in T cells is unknown. Ultimately, the intricate regulation by metabolites allows for a ‘graded’ response of AMPK activity under varying energy stress conditions.
At least three upstream kinases mediate AMPK Thr172 phosphorylation: LKB1, calcium calmodulin kinase kinase 2 (CaMKK2) and TGF-β-activating kinase 1 (TAK1), with roles for LKB1 and CaMKK2 being most understood. As previously mentioned, LKB1 serves as the major kinase acting on Thr172.192,193,194 Further, through regulation by CaMKK2,195,196,197 AMPK may play a role in Ca2+-mediated signaling. In T cells, both TCR and Ca2+ signaling, along with CD28 co-stimulation, coordinate to rapidly and transiently activate AMPK through CaMKK2.198,199,200 AMPK activity is also regulated through protein-protein interactions. Fnip1 and Fnip2 form a complex with AMPK, and a loss-of-function mutation in Fnip1 elevates AMPK activity in B cells, establishing Fnip1 as a negative regulator of AMPK.201 The consequence of impaired Fnip1 function is a profound loss in B cells due to dysregulated AMPK and mTOR signaling.201,202 Additionally, Fnip1 promotes invariant natural killer T cell development in the thymus, potentially through maintaining intracellular ATP levels.203 The Fnip-binding protein Flcn also interacts with AMPK and may serve as a negative regulator of AMPK signaling, and there is some evidence of reciprocal regulation of Fnip1 and Flcn by AMPK.204,205 Beyond the Fnip–Flcn complex, Roquin directly binds the AMPKα1 subunit and limits AMPK signaling, likely through the sequestration of AMPK to stress granules,206 and Roquin-mediated regulation of AMPK is required for Tfh cell responses.206 The contribution of the Flcn–Fnip1–AMPK signaling axis in T cell biology remains poorly defined.
Catabolic programming mediated by LKB1–AMPK
Consistent with its induction by low nutrient abundance, AMPK activity is often associated with the programming of catabolic metabolism to generate ATP. AMPK can directly phosphorylate Raptor at two serine sites (Ser722 and Ser792 of human Raptor), which promotes 14-3-3 binding to Raptor and disrupts mTORC1 signaling.207 Further, the LKB1–AMPK kinase cascade phosphorylates Tsc2 at a site that promotes its GAP activity and thus inhibits mTORC1 signaling.208 In contrast to the inhibitory role of mTORC1 described earlier, AMPK activates autophagy via the phosphorylation of ULK1.209 ULK1 can contribute to autophagy-mediated mitochondrial homeostasis to promote T cell survival.210 Further, independent of mTOR regulation, AMPK also mediates cell cycle arrest through phosphorylation of p53 at Ser15 when glucose is limiting.211 Thus, LKB1–AMPK signaling promotes cell survival by limiting anabolic metabolism and cellular growth during energy stress.
AMPK can also inhibit the synthesis of fatty acids and cholesterol by regulating the enzymes ACC1, ACC2 and HMGCR.212 AMPK phosphorylation of ACC1 Ser79 and ACC2 Ser212 inhibits the conversion of acetyl-CoA to malonyl-CoA, the precursor for fatty acid synthesis.212,213 Mice with mutated Ser79 and Ser212 sites on ACC1 and ACC2, respectively, have elevated lipogenesis and reduced FAO,213 demonstrating that AMPK-dependent inhibition of ACC1 and ACC2 promotes FAO. Further, disruption of the LKB1–AMPK pathway by the metabolite ribulose-5-phosphate, generated through the pentose phosphate pathway, promotes lipogenesis.214 However, LKB1 can also promote mevalonate metabolism to support Treg cell homeostasis in an AMPK-independent manner.215 Thus, LKB1–AMPK signaling primarily acts to suppress lipid synthesis pathways, but AMPK-independent LKB1 signaling may promote de novo lipogenesis in selective contexts.
As noted above, catabolic programs driven by mitochondria support quiescent T cell homeostasis, which can be fueled, in part, by FAO.2 Some subsets of memory CD8+ T cells express high levels of CPT1a, which is a mitochondrial transporter of LCFAs.127 AMPK induces FAO in memory CD8+ T cells, and deficiency for tumor necrosis factor (TNF)-associated factor-6 (TRAF6; signals downstream of TNF receptor superfamily and other immunological receptors) severely impairs AMPK signaling and FAO in CD8+ T cells after IL-2 withdrawal, which induces unchecked metabolic stress and corresponds to impaired memory CD8+ T cell development.216 Importantly, in vivo treatment of TRAF6-deficient mice with the AMPK agonist metformin partially restores memory CD8+ T cell generation.216 Further, AMPKα1 is essential for memory CD8+ T cell recall responses.217 Thus, AMPK signaling promotes lipid metabolism for the generation of a functional memory CD8+ T cell population.
More recent studies using genetic mouse models have questioned the role of FAO in T cell subsets. In contrast to shRNA knockdown or pharmacological inhibition of CPT1a by etomoxir, mice with T cell-specific deletion of Cpt1a do not exhibit defective generation of memory CD8+ T cells or Treg cells.218 Further, mice with Treg cell-specific CPT1a deficiency show normal immune homeostasis, suggesting that CPT1a-dependent FAO is dispensable for Treg cell function for establishment of immune tolerance in vivo.218,219 These discrepancies may be, in part, attributable to off-target effects of high-dose etomoxir, such as depletion of coenzyme A levels that are essential for driving induction of fatty acid synthesis among other functions.220
Memory T cell responses are also important for anti-tumor immunity. The induction of HIF-1α activity via deletion of the von Hippel-Lindau protein promotes glycolysis, which induces effector memory T cell generation and function.221 Studies in tumor cells have demonstrated that the disruption of LKB1 or AMPK signaling promotes aerobic glycolysis, in part, through HIF-1α, which results in increased transcription of glycolytic enzymes.222,223,224,225 LKB1–AMPK-dependent regulation of HIF-1α may also partly depend on suppression of mTORC1.226 LKB1–AMPK signaling may indirectly orchestrate the differentiation of Th17 and Treg cell lineages through HIF-1α- or ACC1-mediated changes in glycolysis and mitochondrial oxidative metabolism.118,119,146 Additionally, recent work demonstrated that LKB1 promotes stable Foxp3 expression,215,227 as well as Th2-like Treg cell development independently of AMPK215,228 and mTORC1–HIF-1α signaling but dependent on β-catenin signaling.228 LKB1 signaling is required for mitochondrial function and mitochondria-dependent metabolic programs upon TCR-mediated Treg cell activation,228 including FAO or purine and pyrimidine metabolism.229 These findings highlight that LKB1 and AMPK orchestrate metabolic reprogramming to regulate T cell differentiation and Treg cell function.
AMPK signaling and adaptation to metabolic stress in T cells
Adaptive immune responses are metabolically demanding and require adaptation to nutrient and metabolic alterations to support their survival and proliferative expansion at sites of activation and infection. As noted above, mTORC1 signaling, combined with mTORC2 activity, coordinates many of the initiating events that are necessary to meet these metabolic demands.31,66 AMPK also enables T cell metabolic adaptation, which can occur independently of TCR signaling. For instance, T cells in glucose-depleted conditions have impaired cellular proliferation, survival and function,60,230 and the absence of AMPKα1 further enhances cell death.190 AMPK promotes T cell survival by supporting glutaminolysis and mitochondrial OXPHOS to maintain intracellular ATP levels in the absence of glucose by promoting the expression of genes involved in glutamine uptake and metabolism.190 Further, AMPK regulates mitochondrial homeostasis through PGC-1α-mediated mitochondrial biogenesis and by phosphorylating mitochondrial fission factor to initiate mitochondrial fission,231,232 which may allow for sustained glycolysis and anti-tumor function of T cells.126,233 In addition, AMPK mediates recycling of damaged mitochondria through ULK1,234 a process that can be induced by elevated production of mitochondria-derived reactive oxygen species.235 AMPKα1 deficiency consequently impairs primary effector CD8+ T cell responses to viral and bacterial infections in vivo, or the expansion of CD4+ Th1 and Th17 cells in lymphopenic environments.190 Thus, AMPK controls metabolic reprogramming during nutrient starvation and mitochondrial homeostasis to promote effector T cell responses.
Emerging perspectives
Understanding the regulation of the LKB1–AMPK signaling axis is an important area of immunological research for several reasons. T cells must adapt to inflammatory and nutrient-depleted conditions, such those that occur in the tumor microenvironment;60,236 therefore, pathways such as AMPK that mediate metabolic flexibility are likely to have critical implications in adoptive T cell therapy. While LKB1 phosphorylates many kinases, AMPK has been the primary target examined in most studies. However, there is emerging evidence that LKB1 has AMPK-independent roles in T cell biology, including Treg cell-dependent suppression of autoimmunity and T cell-dependent inhibition of intestinal polyp formation.215,228,237 Thus, the contribution of LKB1 downstream targets in T cell biology and metabolism is an exciting area that requires more exploration.
An emerging field in immunology is the regulation of T cell biology by nutrient and metabolite signaling. The LKB1–AMPK axis serves as a critical signaling nexus to integrate metabolic cues for T cell function and fate. For instance, several targets of LKB1 and AMPK are implicated in epigenetic regulation of chromatin accessibility, such as by the protein deacetylase sirtuin 1 (Sirt1). AMPK enhances Sirt1 activity by increasing intracellular NAD+ levels,238 which may lead to altered chromatin structure through histone deacetylation, as well as protein activity through post-translational modifications. In T cells, AMPK may promote memory CD8+ T cell development, in part, through Sirt1, which can regulate human memory CD8+ T cell metabolism and cytotoxic function through FoxO1 that is implicated in T cell differentiation and memory CD8+ T cell formation.238,239,240,241 However, the role of Sirt1 for epigenetic reprogramming in T cells remains to be explored. Thus, the contribution of downstream mediators of LKB1–AMPK signaling to impart metabolic regulation of T cell fate is an exciting area for future studies.
Immunometabolic signaling networks
The major signaling pathways described above are not functionally exclusive; rather, they are intertwined and reciprocal, together culminating in the proper metabolic regulation to meet context-specific needs for cellular function. As such, many of these regulatory inputs converge at common nodes, which include both individual molecules and entire cellular processes as discussed below.
Integration of signaling pathways to regulate key cellular and metabolic nodes
In general, elevated activity of PI3K–AGC kinases and mTOR, together with decreased LKB1–AMPK activity, is associated with cell growth, proliferation and effector function, which is associated with changes in metabolic programming as summarized in Fig. 4. The crosstalk between these signaling pathways can occur through the direct reciprocal antagonism, such as between mTORC1 and AMPK190,207,242 or Akt and LKB1,187 or via indirect inhibition of downstream signaling effectors. For example, the ULK1 complex, which promotes autophagy, is inhibited and activated by mTORC1 and AMPK signaling, respectively.172,173,234,235 Further, autophagy may act as an ‘off-switch’ for mTORC1 activation and glycolysis under nutrient-deficient conditions.26 This inhibition likely favors cell survival and maintains cellular quiescence at the expense of cell growth and proliferation.243 Understanding the mechanisms for the interplay between autophagy and mTORC1 may be therapeutically relevant for immune-related diseases, such as autoimmune disorders and cancer.

Under nutrient-deprived conditions, LKB1–AMPK signaling inhibits anabolism-associated programs, such as glycolysis and fatty acid synthesis, while promoting mitochondrial homeostasis and autophagy; however, these mechanisms require additional investigation in primary T cells. AMPK directly inhibits mTORC1 through phosphorylation of its obligate adapter protein Raptor (not depicted). During activation and/or replete nutrient conditions, PI3K–Akt and mTORC1 signaling promotes glycolysis, mitochondrial biogenesis and fatty acid synthesis, while inhibiting autophagy. Akt can reportedly phosphorylate LKB1 to suppress its functional localization, but this regulation is not yet reported to occur in T cells.
The integration of signaling by upstream complexes is ultimately linked to regulation of downstream metabolic programming. For instance, LKB1–AMPK signaling inhibits fatty acid synthesis and mevalonate metabolism in certain contexts through the direct regulation of ACC1 or ACC2 and HMGCR,212,213 while mTORC1 signaling promotes the function of these molecules in T cells by upregulating gene expression downstream of the SREBP transcription factors.31,153 However, it is important to note that although AMPK can promote FAO in T cells,216 it is unclear whether AMPK negatively regulates fatty acid synthesis in T cells. AMPK signaling can also oppose glycolysis through negative regulation of mTORC1–HIF-1α signaling.222,223,224,225,226 However, when glucose is limiting, AMPK signaling can also promote glutamine metabolism and flux via the mitochondrial TCA cycle in T cells.190 Also, LKB1 and AMPK signaling can influence mitochondrial fitness and biogenesis in a potentially mTORC1-independent manner.228,229,231,232 Thus, the crosstalk between PI3K–Akt, mTOR, LKB1 and AMPK signaling (along with other immunometabolic signaling networks, such as those mediated by Regnase-1244), likely promotes metabolic adaptations that allow cells to meet energetic and biosynthetic demands to function in unique microenvironments.
The signaling pathways that dictate metabolic programs in immune cells are also reciprocally influenced by metabolites and nutrients. This ‘bidirectional metabolic signaling’ provides another layer of regulation on kinase-dependent signaling and gene transcription to control immune cell function in different contexts.2 As described above, mTORC1 signaling is dynamically regulated by nutrients and metabolites, with amino acids promoting mTORC1 activation through modulation of upstream proteins such as the Rag complex.63,112,116 The subcellular localization of nutrients can also influence signaling and metabolic programming, as shown by recent observations that TCR stimulation inhibits pyruvate translocation into the mitochondria, leading to the promotion of aerobic glycolysis.32 Conversely, diminished nutrient conditions can regulate signaling pathways to control metabolism. For example, low cellular glucose and glutamine can activate AMPK.190 In addition, elevated AMP or ADP concentrations promote AMPK activation, which limits anabolism and promotes mitochondrial homeostasis to replenish ATP stores.188 Finally, several key metabolites derived from mitochondria-associated metabolism, including α-KG, 2-hydroxyglutarate and acetyl-CoA, serve as co-factors for epigenome-modifying enzymes to influence transcriptional activity.2 Thus, the metabolites generated as a consequence of kinase-dependent metabolic signaling also have signaling roles to impart effects on cellular function.
Concluding remarks
The role of immune signaling networks in immunometabolism is an exciting area of research, with broad implications for therapy and human health. In this review, we highlighted the critical roles for PI3K–AGC, mTOR and LKB1–AMPK signaling in T cell function and fate. While the field has made much progress in understanding how these signaling molecules are regulated and how they reprogram T cell metabolism, many outstanding questions still remain, including: (1) Do altered receptor signal strength and duration modulate T cell responses through different downstream targets? (2) Do specific upstream regulators and downstream targets of AGC kinases, mTOR and LKB1 transmit nutrient and immunological signals to define T cell subset-specific metabolic programs? (3) How do upstream signaling networks (e.g., LKB1–AMPK) integrate metabolite sensing with T cell metabolic and epigenetic programming? In addition, the roles of other signaling networks, including the conserved Hippo family kinases and mitogen activated-protein kinase pathways, in immunometabolism are only beginning to be understood.245,246 Technical advances and systems immunology approaches should also accelerate our understanding of how these signaling networks crosstalk with metabolic programs to control T cell outcomes. Finally, determining how relatively well-known upstream immune signals (e.g., TCR, cytokine receptors) impart different functional programs on metabolism may provide new therapeutic targets for improving adoptive T cell therapy, such as anti-tumor CAR-T cell therapy.
References
Saravia, J., Chapman, N. M. & Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 16, 634–643 (2019).
Chapman, N. M., Boothby, M. R. & Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 20, 55–70 (2020).
Bilanges, B., Posor, Y. & Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 20, 515–534 (2019).
Sauer, K. & Cooke, M. P. Regulation of immune cell development through soluble inositol-1,3,4,5-tetrakisphosphate. Nat. Rev. Immunol. 10, 257–271 (2010).
Kane, L. P. & Weiss, A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol. Rev. 192, 7–20 (2003).
Man, K. & Kallies, A. Synchronizing transcriptional control of T cell metabolism and function. Nat. Rev. Immunol. 15, 574–584 (2015).
Tarasenko, T. et al. T cell-specific deletion of the inositol phosphatase SHIP reveals its role in regulating Th1/Th2 and cytotoxic responses. Proc. Natl. Acad. Sci. USA 104, 11382–11387 (2007).
Buckler, J. L., Walsh, P. T., Porrett, P. M., Choi, Y. & Turka, L. A. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J. Immunol. 177, 4262–4266 (2006).
Gomez-Rodriguez, J. et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J. Exp. Med. 211, 529–543 (2014).
Bensinger, S. J. et al. Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J. Immunol. 172, 5287–5296 (2004).
Walsh, P. T. et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J. Clin. Investig. 116, 2521–2531 (2006).
Hu, Z. et al. Acylglycerol kinase maintains metabolic state and immune responses of CD8(+) T cells. Cell Metab. 30, 290–302 (2019).
Liu, X. et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J. Clin. Investig. 120, 2497–2507 (2010).
Huynh, A. et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat. Immunol. 16, 188–196 (2015).
Shrestha, S. et al. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 16, 178–187 (2015).
Uche, U. U. et al. PIK3IP1/TrIP restricts activation of T cells through inhibition of PI3K/Akt. J. Exp. Med. 215, 3165–3179 (2018).
Johnson, M. O. et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795 (2018).
Vinuesa, C. G. et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435, 452–458 (2005).
Essig, K. et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity 47, 1067–1082 (2017).
McLeod, I. X., Zhou, X., Li, Q. J., Wang, F. & He, Y. W. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J. Immunol. 187, 5051–5061 (2011).
Parekh, V. V. et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J. Immunol. 190, 5086–5101 (2013).
Willinger, T. & Flavell, R. A. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc. Natl. Acad. Sci. USA 109, 8670–8675 (2012).
Park, S. G. et al. The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nat. Immunol. 10, 158–166 (2009).
Macintyre, A. N. et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34, 224–236 (2011).
Finlay, D. K. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 (2012).
Wei, J. et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 17, 277–285 (2016).
Park, S. G. et al. T regulatory cells maintain intestinal homeostasis by suppressing gammadelta T cells. Immunity 33, 791–803 (2010).
Biondi, R. M. et al. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 19, 979–988 (2000).
Lee, K. et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010).
Delgoffe, G. M. et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 12, 295–303 (2011).
Yang, K. et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 39, 1043–1056 (2013).
Menk, A. V. et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 22, 1509–1521 (2018).
Jones, N. et al. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat. Commun. 10, 2042 (2019).
Jacobs, S. R. et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 180, 4476–4486 (2008).
Wieman, H. L., Wofford, J. A. & Rathmell, J. C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 18, 1437–1446 (2007).
Gubser, P. M. et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 14, 1064–1072 (2013).
Kerdiles, Y. M. et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat. Immunol. 10, 176–184 (2009).
Ouyang, W., Beckett, O., Flavell, R. A. & Li, M. O. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity 30, 358–371 (2009).
Carlson, C. M. et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442, 299–302 (2006).
Ouyang, W. et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491, 554–559 (2012).
Newton, R. H. et al. Maintenance of CD4 T cell fitness through regulation of Foxo1. Nat. Immunol. 19, 838–848 (2018).
Luo, C. T., Liao, W., Dadi, S., Toure, A. & Li, M. O. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature 529, 532–536 (2016).
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M. & Hemmings, B. A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 (1995).
Ohteki, T. et al. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J. Exp. Med. 192, 99–104 (2000).
Jellusova, J. et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 18, 303–312 (2017).
Vodnala, S. K. et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, eaau0135 (2019).
Eil, R. et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537, 539–543 (2016).
Karmaus, P. W. F. et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 565, 101–105 (2019).
Wu, T. et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1, eaai8593 (2016).
Im, S. J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016).
Nish, S. A. et al. CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J. Exp. Med. 214, 39–47 (2017).
Klebanoff, C. A. et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight 2, 95103 (2017).
Abu Eid, R. et al. Enhanced therapeutic efficacy and memory of tumor-specific CD8 T cells by ex vivo PI3K-delta inhibition. Cancer Res. 77, 4135–4145 (2017).
Chellappa, S. et al. The PI3K p110delta isoform inhibitor idelalisib preferentially inhibits human regulatory T cell function. J. Immunol. 202, 1397–1405 (2019).
Kaneda, M. M. et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016).
De Henau, O. et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447 (2016).
Hawse, W. F., Boggess, W. C. & Morel, P. A. TCR signal strength regulates Akt substrate specificity to induce alternate murine Th and T regulatory cell differentiation programs. J. Immunol. 199, 589–597 (2017).
Luo, W. et al. The AKT kinase signaling network is rewired by PTEN to control proximal BCR signaling in germinal center B cells. Nat. Immunol. 20, 736–746 (2019).
Vaeth, M. et al. Store-operated Ca(2+) entry controls clonal expansion of T cells through metabolic reprogramming. Immunity 47, 664–679 (2017).
Ho, P. C. et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 (2015).
Gorentla, B. K., Wan, C. K. & Zhong, X. P. Negative regulation of mTOR activation by diacylglycerol kinases. Blood 117, 4022–4031 (2011).
Shin, J., O’Brien, T. F., Grayson, J. M. & Zhong, X. P. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J. Immunol. 188, 2111–2117 (2012).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017).
Sarbassov, D. D. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168 (2006).
Yang, K. et al. Metabolic signaling directs the reciprocal lineage decisions of alphabeta and gammadelta T cells. Sci. Immunol. 3, eaas9818 (2018).
Tan, H. et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503 (2017).
Delgoffe, G. M. et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 (2009).
Yang, K., Neale, G., Green, D. R., He, W. & Chi, H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 12, 888–897 (2011).
Chornoguz, O. et al. mTORC1 promotes T-bet phosphorylation to regulate Th1 differentiation. J. Immunol. 198, 3939–3948 (2017).
Zeng, H. et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 45, 540–554 (2016).
Kishore, M. et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity 48, 831–832 (2018).
Zeng, H. et al. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490 (2013).
Chapman, N. M. et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 9, 2095 (2018).
Gerriets, V. A. et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 17, 1459–1466 (2016).
Apostolidis, S. A. et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat. Immunol. 17, 556–564 (2016).
Pollizzi, K. N. et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J. Clin. Investig. 125, 2090–2108 (2015).
Shrestha, S. et al. Tsc1 promotes the differentiation of memory CD8+ T cells via orchestrating the transcriptional and metabolic programs. Proc. Natl. Acad. Sci. USA 111, 14858–14863 (2014).
Araki, K. et al. mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112 (2009).
Rao, R. R., Li, Q., Odunsi, K. & Shrikant, P. A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32, 67–78 (2010).
Sowell, R. T., Rogozinska, M., Nelson, C. E., Vezys, V. & Marzo, A. L. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J. Immunol. 193, 2067–2071 (2014).
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P. & Guan, K. L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945 (2008).
Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008).
Sancak, Y. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010).
Ma, L., Chen, Z., Erdjument-Bromage, H., Tempst, P. & Pandolfi, P. P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 (2005).
Roux, P. P., Ballif, B. A., Anjum, R., Gygi, S. P. & Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 101, 13489–13494 (2004).
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J. & Cantley, L. C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162 (2002).
Inoki, K., Li, Y., Zhu, T., Wu, J. & Guan, K. L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 (2002).
Inoki, K., Li, Y., Xu, T. & Guan, K. L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 (2003).
Angarola, B. & Ferguson, S. M. Weak membrane interactions allow Rheb to activate mTORC1 signaling without major lysosome enrichment. Mol. Biol. Cell 30, 2750–2760 (2019).
Tsai, S. et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab. 28, 922–934 (2018).
Gattinoni, L. et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 15, 808–813 (2009).
Liu, P. et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 5, 1194–1209 (2015).
Gan, X., Wang, J., Su, B. & Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 286, 10998–11002 (2011).
Abu-Remaileh, M. et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017).
Wang, S. et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194 (2015).
Efeyan, A. et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683 (2013).
Chantranupong, L. et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164 (2016).
Saxton, R. A., Chantranupong, L., Knockenhauer, K. E., Schwartz, T. U. & Sabatini, D. M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 536, 229–233 (2016).
Saxton, R. A. et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 351, 53–58 (2016).
Wolfson, R. L. et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48 (2016).
Peng, M., Yin, N. & Li, M. O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 159, 122–133 (2014).
Bar-Peled, L. et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106 (2013).
Han, J. M. et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149, 410–424 (2012).
Tsun, Z. Y. et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 52, 495–505 (2013).
Yoon, M. S. et al. Leucyl-tRNA synthetase activates Vps34 in amino acid-sensing mTORC1 signaling. Cell Rep. 16, 1510–1517 (2016).
Bonfils, G. et al. Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell 46, 105–110 (2012).
Duran, R. V. et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 47, 349–358 (2012).
Castellano, B. M. et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311 (2017).
Jewell, J. L. et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198 (2015).
Son, S. M. et al. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 29, 192–201 (2019).
Nakaya, M. et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705 (2014).
Shi, H. et al. Amino acids license kinase mTORC1 activity and Treg cell function via small G proteins Rag and Rheb. Immunity 51, 1–16 (2019).
Howden, A. J. M. et al. Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat. Immunol. 20, 1542–1554 (2019).
Sinclair, L. V. et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14, 500–508 (2013).
Ikeda, K. et al. Slc3a2 mediates branched-chain amino-acid-dependent maintenance of regulatory T cells. Cell Rep. 21, 1824–1838 (2017).
Do, M. H. et al. Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J. Exp. Med. 217, e20190848 (2020).
Wang, R. N. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Shi, L. Z. et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011).
Dang, E. V. et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011).
Bantug, G. R. et al. Mitochondria-endoplasmic reticulum contact sites function as immunometabolic hubs that orchestrate the rapid recall response of memory CD8(+) T cells. Immunity 48, 542–555 (2018).
Wu, C. et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517 (2013).
Kleinewietfeld, M. et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–522 (2013).
Heikamp, E. B. et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat. Immunol. 15, 457–464 (2014).
Ron-Harel, N. et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 24, 104–117 (2016).
Morita, M. et al. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14, 473–480 (2015).
Buck, M. D. et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016).
van der Windt, G. J. et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 (2012).
Klein Geltink, R. I. et al. Mitochondrial priming by CD28. Cell 171, 385–397 (2017).
Tarasenko, T. N. et al. Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab. 25, 1254–1268 (2017).
Sena, L. A. et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 (2013).
Chisolm, D. A. et al. CCCTC-binding factor translates interleukin 2- and alpha-ketoglutarate-sensitive metabolic changes in T cells into context-dependent gene programs. Immunity 47, 251–267 (2017).
Tyrakis, P. A. et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236–241 (2016).
Weinberg, S. E. et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499 (2019).
Peng, M. et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484 (2016).
Baixauli, F. et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498 (2015).
Ma, E. H. et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357 (2017).
Yang, M. & Vousden, K. H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 16, 650–662 (2016).
Lian, G. et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. Elife 7, e36158 (2018).
Mak, T. W. et al. Glutathione primes T cell metabolism for inflammation. Immunity 46, 1089–1090 (2017).
Ma, E. H. et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 1–15 (2019).
Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J. H., Asara, J. M. & Manning, B. D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733 (2016).
Aronova, S. et al. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 7, 148–158 (2008).
Sofi, M. H. et al. Ceramide synthesis regulates T cell activity and GVHD development. JCI Insight 2, 91701 (2017).
Rotolo, J. A. et al. Cytolytic T cells induce ceramide-rich platforms in target cell membranes to initiate graft-versus-host disease. Blood 114, 3693–3706 (2009).
Bai, A., Kokkotou, E., Zheng, Y. & Robson, S. C. Role of acid sphingomyelinase bioactivity in human CD4+ T-cell activation and immune responses. Cell Death Dis. 6, e1828 (2015).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333 (2014).
Lee, J. et al. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. 192, 3190–3199 (2014).
Abu-Elheiga, L., Matzuk, M. M., Abo-Hashema, K. A. & Wakil, S. J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291, 2613–2616 (2001).
Lee, J. E. et al. Acetyl CoA carboxylase 2 is dispensable for CD8+ T cell responses. PLoS ONE 10, e0137776 (2015).
Peterson, T. R. et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011).
Nickels, J. T. Jr New links between lipid accumulation and cancer progression. J. Biol. Chem. 293, 6635–6636 (2018).
Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 109, 1125–1131 (2002).
Kidani, Y. et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 14, 489–499 (2013).
Bensinger, S. J. et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134, 97–111 (2008).
Venkateswaran, A. et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 97, 12097–12102 (2000).
Cheng, H. Y. et al. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J. Clin. Investig. 126, 3236–3246 (2016).
Wang, F. et al. S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat. Immunol. 17, 514–522 (2016).
Yang, W. et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655 (2016).
Angela, M. et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat. Commun. 7, 13683 (2016).
Klotz, L. et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 206, 2079–2089 (2009).
Alvarez-Curto, E. & Milligan, G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem. Pharmacol. 114, 3–13 (2016).
Bachem, A. et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8(+) T cells. Immunity 51, 285–297 (2019).
Balmer, M. L. et al. Memory CD8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity 44, 1312–1324 (2016).
Sun, M. et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 9, 3555 (2018).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Miyamoto, J. et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 10, 4007 (2019).
Haghikia, A. et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 43, 817–829 (2015).
O’Sullivan, D. et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88 (2014).
Cui, G. et al. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 161, 750–761 (2015).
Kim, Y. C. & Guan, K. L. mTOR: a pharmacologic target for autophagy regulation. J. Clin. Investig. 125, 25–32 (2015).
Hosokawa, N. et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009).
Ganley, I. G. et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305 (2009).
Kim, J. & Guan, K. L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 21, 63–71 (2019).
Puleston, D. J. et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 3. https://doi.org/10.7554/eLife.03706 (2014).
Xu, X. et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 15, 1152–1161 (2014).
DeVorkin, L. et al. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep. 27, 502–513 (2019).
Kabat, A. M. et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife 5, e12444 (2016).
Gu, X. et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818 (2017).
So, L. et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci. Signal. 9, ra57 (2016).
Rollings, C. M., Sinclair, L. V., Brady, H. J. M., Cantrell, D. A. & Ross, S. H. Interleukin-2 shapes the cytotoxic T cell proteome and immune environment-sensing programs. Sci. Signal. 11, eaap8112 (2018).
Hsu, P. P. et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322 (2011).
Lizcano, J. M. et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 (2004).
Jaleel, M. et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 579, 1417–1423 (2005).
Kullmann, L. & Krahn, M. P. Controlling the master-upstream regulation of the tumor suppressor LKB1. Oncogene 37, 3045–3057 (2018).
Boudeau, J. et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 22, 5102–5114 (2003).
Liu, L., Siu, F. M., Che, C. M., Xu, A. & Wang, Y. Akt blocks the tumor suppressor activity of LKB1 by promoting phosphorylation-dependent nuclear retention through 14-3-3 proteins. Am. J. Transl. Res. 4, 175–186 (2012).
Hardie, D. G., Ross, F. A. & Hawley, S. A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012).
Xiao, B. et al. Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 (2011).
Blagih, J. et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54 (2015).
Zhang, C. S. et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 (2017).
Hawley, S. A. et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2, 28 (2003).
Woods, A. et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 (2003).
Shaw, R. J. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 101, 3329–3335 (2004).
Hurley, R. L. et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 280, 29060–29066 (2005).
Hawley, S. A. et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2, 9–19 (2005).
Woods, A. et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 (2005).
Tamas, P. et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J. Exp. Med. 203, 1665–1670 (2006).
Zheng, Y., Delgoffe, G. M., Meyer, C. F., Chan, W. & Powell, J. D. Anergic T cells are metabolically anergic. J. Immunol. 183, 6095–6101 (2009).
MacIver, N. J. et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J. Immunol. 187, 4187–4198 (2011).
Siggs, O. M. et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc. Natl. Acad. Sci. USA 113, E3706–E3715 (2016).
Park, H. et al. Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity 36, 769–781 (2012).
Park, H., Tsang, M., Iritani, B. M. & Bevan, M. J. Metabolic regulator Fnip1 is crucial for iNKT lymphocyte development. Proc. Natl. Acad. Sci. USA 111, 7066–7071 (2014).
Possik, E. et al. Folliculin regulates ampk-dependent autophagy and metabolic stress survival. PLoS Genet. 10, e1004273 (2014).
Baba, M. et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. USA 103, 15552–15557 (2006).
Ramiscal, R. R. et al. Attenuation of AMPK signaling by ROQUIN promotes T follicular helper cell formation. Elife 4, e08698 (2015).
Gwinn, D. M. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008).
Corradetti, M. N., Inoki, K., Bardeesy, N., DePinho, R. A. & Guan, K. L. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 18, 1533–1538 (2004).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Watanabe, R. et al. Autophagy plays a protective role as an anti-oxidant system in human T cells and represents a novel strategy for induction of T-cell apoptosis. Eur. J. Immunol. 44, 2508–2520 (2014).
Jones, R. G. et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 (2005).
Shackelford, D. B. & Shaw, R. J. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009).
Fullerton, M. D. et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 19, 1649–1654 (2013).
Lin, R. et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat. Cell Biol. 17, 1484–1496 (2015).
Timilshina, M. et al. Activation of mevalonate pathway via LKB1 is essential for stability of Treg cells. Cell Rep. 27, 2948–2961 (2019).
Pearce, E. L. et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107 (2009).
Rolf, J. et al. AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 43, 889–896 (2013).
Raud, B. et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 28, 504–515 (2018).
Saravia, J. et al. Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci. Adv. 6, eaaw6443 (2020).
Divakaruni, A. S. et al. Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis. Cell Metab. 28, 490–503 (2018).
Phan, A. T. et al. Constitutive glycolytic metabolism supports CD8(+) T cell effector memory differentiation during viral infection. Immunity 45, 1024–1037 (2016).
Faubert, B. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 17, 113–124 (2013).
Faubert, B. et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc. Natl. Acad. Sci. USA 111, 2554–2559 (2014).
Kishton, R. J. et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 23, 649–662 (2016).
Shackelford, D. B. et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. USA 106, 11137–11142 (2009).
Duvel, K. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010).
Wu, D. et al. Lkb1 maintains Treg cell lineage identity. Nat. Commun. 8, 15876 (2017).
Yang, K. et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature 548, 602–606 (2017).
He, N. et al. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc. Natl. Acad. Sci. USA 114, 12542–12547 (2017).
Chang, C. H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Toyama, E. Q. et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281 (2016).
Jager, S., Handschin, C., St-Pierre, J. & Spiegelman, B. M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 104, 12017–12022 (2007).
Scharping, N. E. et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 701–703 (2016).
Egan, D. F. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011).
Rabinovitch, R. C. et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 21, 1–9 (2017).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015).
Poffenberger, M. C. et al. LKB1 deficiency in T cells promotes the development of gastrointestinal polyposis. Science 361, 406–411 (2018).
Canto, C. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 (2009).
Motta, M. C. et al. Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551–563 (2004).
Jeng, M. Y. et al. Metabolic reprogramming of human CD8(+) memory T cells through loss of SIRT1. J. Exp. Med. 215, 51–62 (2018).
Luo, C. T. & Li, M. O. Foxo transcription factors in T cell biology and tumor immunity. Semin. Cancer Biol. 50, 13–20 (2018).
Ling, N. X. Y. et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2, 41–49 (2020).
Garcia-Prat, L. et al. Autophagy maintains stemness by preventing senescence. Nature 529, 37–42 (2016).
Wei, J. et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476 (2019).
Carr, E. L. et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 185, 1037–1044 (2010).
Du, X. et al. Hippo/Mst signalling couples metabolic state and immune function of CD8alpha(+) dendritic cells. Nature 558, 141–145 (2018).
Acknowledgements
The authors apologize to those whose key contributions could not be cited due to space limitations. This work was supported by NIH AI105887, AI131703, AI140761, AI150241, AI150514 and CA221290 (to H. C.).
Author information
Authors and Affiliations
Contributions
All authors contributed to the content discussion for the review article and to the writing and editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saravia, J., Raynor, J.L., Chapman, N.M. et al. Signaling networks in immunometabolism. Cell Res 30, 328–342 (2020). https://doi.org/10.1038/s41422-020-0301-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-020-0301-1
