
Abstract
Metastasis, the development of secondary malignant growths at a distance from a primary tumor, is the cause of death for 90% of cancer patients, but little is known about how metastatic cancer cells adapt to and colonize new tissue environments. Here, using clinical samples, patient-derived xenograft (PDX) samples, PDX cells, and primary/metastatic cell lines, we discovered that liver metastatic colorectal cancer (CRC) cells lose their colon-specific gene transcription program yet gain a liver-specific gene transcription program. We showed that this transcription reprogramming is driven by a reshaped epigenetic landscape of both typical enhancers and super-enhancers. Further, we identified that the liver-specific transcription factors FOXA2 and HNF1A can bind to the gained enhancers and activate the liver-specific gene transcription, thereby driving CRC liver metastasis. Importantly, similar transcription reprogramming can be observed in multiple cancer types. Our data suggest that reprogrammed tissue-specific transcription promotes metastasis and should be targeted therapeutically.
Similar content being viewed by others


Introduction
Tumor metastasis is the cause of death for 90% of cancer patients, and no currently available therapies target this multi-step process in which cancer cells spread from the local tissue of a primary tumor to distant organs where they establish secondary tumors.1 Although epithelial-to-mesenchymal transition,2 tumor-secreted exosomes,3 metabolic reprogramming,4 and epigenetic regulators, as well as other genes5,6,7,8,9 have been implicated in metastasis, little is known about how primary cancer cells adapt to and colonize new tissue environments.
Tumor metastasis refers to the movement of tumor cells from a primary site to distant organs that they progressively colonize.10 More than 100 years ago, Paget suggested the idea of metastasis as the interaction of “seeds” and “soil”,11 but subsequent research has yielded only a limited understanding of the mechanism(s) through which metastatic cancer cells (“seeds”) adapt to and colonize a new tissue environment (“soil”), the crucial steps of the metastasis process.12
It has been reported that the expression of tissue-specific or cell lineage genes, which are regulated by general and cell type-specific transcription factors through regulatory genomic elements, such as enhancers, determines cell identity13,14,15,16 and may also promote macrophages adapting to a particular environment in innate immune system.17
Here, by comparing gene expression profiles between primary CRC tumors/cell lines and liver metastatic tumors/cell lines, we interestingly found that liver metastatic tumors gain a liver-specific gene program and lose a colon-specific gene program. Further, we demonstrate that this directional transcription reprogramming is driven by epigenetic reprogramming and we show that the liver-specific transcriptional factors (TFs) FOXA2 and HNF1A bind to the gained epigenetic enhancers of liver metastatic cells and induce liver-specific transcription, thereby promoting CRC liver metastasis. Collectively, our findings establish that the tissue-specific transcription reprogramming promotes CRC liver metastasis.
Results
Tissue-specific transcription reprogramming in liver metastatic colon tumors
To explore the importance of tissue-specific genes during metastasis, we initially evaluated the tissue-specific transcriptome profiles of primary and liver-metastatic CRC tumors (Fig. 1a) in three publicly available CRC datasets.18,19,20 We used the Genotype-Tissue Expression (GTEx, https://gtexportal.org/home/) classification to define tissue-specific gene signatures in this analysis. We performed an unbiased Gene Set Enrichment Analysis (GSEA)21 of these published datasets to identify the statistically significant differences in global gene expression between primary and liver-metastatic CRC tumors. The GSEA showed that the set of significantly upregulated genes in the liver-metastatic CRC tumors was significantly enriched for a liver-specific signature (FDR < 0.00001), and the set of genes with down-regulated transcription was significantly enriched for a colon-specific gene signature (FDR < 0.00001) in all these three datasets (Fig. 1b). We confirmed that these enrichments were still significant even after removing up-regulated genes that could possibly have been introduced by contamination from normal liver tissues by utilizing a rational biological algorithm (Fig. S1a–c).

Reprogrammed tissue-specific transcription in liver metastatic CRC tumors. a Schematic diagram demonstrating primary and liver-metastatic tumors for our CRC experiments. b GSEA of liver-specific gene set, as defined by using the expression data from GTEx project database, in human liver metastasis CRC tumors from the GSE41258, GSE49355, and GSE50760 datasets. Genes are ranked by log2 fold changes of averaged expression values of multiple primary and liver metastasis CRC samples. The normalized enrichment scores (NES) and tests of statistical significance (FDR) are shown. met., metastasis. c GSEA of liver-specific gene set, as defined using the expression data from the GTEx project database, for human liver metastasis CRC tumors growing in mice (PDX model). Genes are ranked by log2 fold change of expression values in liver metastasis CRC sample versus primary CRC sample. The normalized enrichment scores (NES) and tests of statistical significance (FDR) are shown. met., metastasis. d Plots showing the IHC scores for LIPC, INHBE, and CYP27A1 in human primary CRC tumors and in liver metastatic tumors. Each dot represents one primary or liver metastatic CRC tumor, and the red bar represents the mean value. Statistically-significant P values are indicated with asterisks (*P < 0.05, **P < 0.01, ***P < 0.001, by t-test). e Representative IHC images of LIPC, INHBE and CYP27A1 expression in human primary CRC tumor and liver metastatic CRC tumor. f Liver-specific genes with significantly up-regulated transcription in liver metastatic CRC tumor samples. The expression data are from the GSE41258 dataset. Each dot represents one primary or liver metastatic CRC tumor; red bars represent mean values. Statistically significant P values are indicated with asterisks (***P < 0.001, by t-test). g Proportion surviving analyses were performed using published clinical data in the cancer genome atlas (TCGA). The subset of CRC patients from the GSE41258 dataset with high expression levels for liver-specific genes in their primary tumors had significantly worse overall survival (OS) outcomes than did the subset of patients with low expression levels for these genes. Liver specific genes were obtained by overlapping highly expressed genes in human liver metastatic samples compared with primary tumors (log2FC > 0.6) and a tissue-specific gene set defined by GTEx (see Methods) (Left panel). The subset of CRC patients with high expression levels for LIPC and CYP27A1 in their primary tumors had significantly worse overall survival (OS) outcomes than did the subset of patients with low expression levels for these genes (Right two panels). High, Top 20% patients; Low, bottom 20% patients; Med, the rest; FC: fold change of expression. P values were calculated by a log-rank (Mantel-Cox) test
Patient-derived xenograft (PDX) samples are human tumors growing in mice; only the cancer cells in a PDX sample are from human. By using a published method22 that can distinguish human mRNAs from mouse mRNAs in PDX samples, we again found that the liver-specific gene signature is gained and the colon-specific genes signature is lost in a liver metastatic tumor model (Fig. 1c). It bears emphasis that this result clearly indicates that the observed reprogrammed tissue-specific transcription occurs in the cancer cells (human cells) but does not occur in the stromal cells (mouse cells) within the examined PDX tumors.
To further confirm that liver-specific genes are highly expressed in CRC cells within liver metastatic tumors, immunohistochemistry (IHC) assays were performed on colon cancer tissue arrays. We used a previously described scoring system23 to quantify the data from IHC assays of tissue arrays, and found that the levels of liver-specific proteins such as LIPC, INHBE, and CYP27A1 are significantly higher in CRC cells within liver metastatic tumors than in CRC cells within primary CRC tumors (Fig. 1d, e and Tables S1–S3), findings consistent with the high mRNA levels that we had earlier observed for these three genes in liver metastatic tumors (Figs. 1f, S2a, b). Further, the IHC images clearly supported that these liver-specific proteins were highly expressed in cancer cells but not in the other cells of the examined tumors (Fig. 1e). Extending these findings towards clinical relevance, analysis using the TCGA database data set for primary CRC tumors revealed that high expression of the liver-specific gene set, as well as individual liver-specific genes such as LIPC and CYP27A1, was invariably significantly associated with poor prognosis for CRC patients (Fig. 1g).
To investigate whether this intriguing tissue-specific transcription reprogramming is a general feature of metastasis for multiple target organs and/or cancer types, we next analyzed a large number of published transcriptome datasets. Using the same analysis and classification criteria as above, these analyses revealed that such transcription reprogramming also occurs in colon-to-lung metastasis, prostate-to-liver metastasis, prostate-to-bone metastasis, kidney-to-lung metastasis, breast-to-brain metastasis, and pancreas-to-liver metastasis (Fig. S3a). Consistently, analysis of patient data also revealed that once the distant tissue-specific gene set is highly expressed in primary tumors, at least for the liver-specific gene set in pancreas tumors, bone-specific gene set in prostate tumors, lung-specific gene set in CRC tumors, brain-specific gene set in breast tumors, patients featuring such transcription reprogramming had significantly worse overall survival (OS) outcomes than patients with low expression levels for these genes (Fig. S3b). Thus, this analysis suggested that metastatic cells lose their primary tissue-specific gene program before colonization of the secondary site and distant tissue-like genes may be already expressed in primary tumors but the expression levels are lower than those in distant metastasis tumors. Viewed together, these extensive experimental and in silico findings collectively indicate that many types of metastatic cells can lose the tissue-specific transcription program of the organ from which they originate and can gain the tissue-specific transcription program of a distant organ.
Reprogrammed tissue-specific transcription in liver metastatic CRC cells
We further checked this tissue-specific transcription reprogramming in paired CRC cancer cell lines (Fig. 2a). SW620 cells were originally derived from a lymph node metastasis and can easily metastasize to liver in xenografts.24,25 SW480 cells were derived from primary CRC tumor and show no ability to metastasize.24,25 Notably, SW620 and SW480 cells were derived from the same patient24 and thus share the same genetic background, providing an appropriate model to study reprogrammed transcription, especially that regulated by epigenetic mechanisms. Gene expression profiling by RNA-seq revealed striking differences between non-metastatic SW480 and liver-metastatic SW620 cells (Fig. S4a) and the expression differences of some example genes were validated by qRT-PCR (Fig. S4b). GSEA on these two cell lines showed that a liver-specific gene signature was enriched in the significantly up-regulated genes in SW620 cells (FDR = 0.02), while a colon-specific gene signature was enriched in the significantly down-regulated genes (FDR = 0.04) (Fig. 2b). Specifically, SW620 cells had up-regulated expression of 112 liver-specific genes and down-regulated expression of 77 colon-specific genes (Fig. S4c). Encouragingly, about half of the up-regulated liver-specific genes (50/112) and one third of the down-regulated colon-specific genes (24/77) from the analysis of these two cell lines had similar expression trends in primary and liver-metastatic CRC tumor samples (Fig. 2c and Table S4).

Reprogrammed tissue-specific transcription in liver metastatic CRC cells. a Schematic diagram demonstrating paired primary and liver metastasis cells for our CRC experiments. b GSEA of liver-specific gene set (left) and colon-sigmoid-specific gene set (right) as defined from the GTEx project database in SW620 and SW480 cells. The genes are ranked by the log2 fold change of the FPKM values in SW620 and SW480 cells. The NES and FDR are shown. met., Metastasis. c Heatmap showing expression levels of 50 out of 112 liver- and 24 out of 77 colon-specific genes in SW480 and SW620 cells are consistent with them in human primary and liver metastatic CRC tumors. Gene expression values for tumor samples are from GSE41258 dataset. met., metastasis. d Tissue-specific transcription reprogramming occurs in metastatic cancer cells (CCLE data) of 11 types of cancer metastases. The red and blue bars in the chart show the % of cell pairs gain gene signature of distant tissue and original tissue, respectively. We integrated multiple GSEA for representative examples into a bubble map. Each enrichment is summarized as a bubble in a color matching the population in which the gene set was enriched. Bubble area and color intensity indicate NES and FDR, respectively. met., Metastasis. e GSEA of original- and distant-tissue gene sets in prostate-to-bone metastasis, colon-to-lung metastasis, lung-to-liver metastasis, prostate-to-brain metastasis and pancreas-to-liver metastasis. The NES and FDR are shown. The number of samples used for the GSEA are listed in brackets for primary and metastatic tumors, respectively. met., metastasis
We next re-analyzed the data from a study of paired metastatic breast cancer cell lines, in which murine breast cancer 4T1 cells that are known to metastasize to various sites were injected in vivo followed by analysis of the metastatic subpopulations that colonized and grew in the lungs, bones, and liver.4 These metastatic subpopulations were isolated and purified, thus having no contamination of stromal cells or normal tissue cells.26 Consistent with our experimental and other in silico findings, this re-analysis revealed tissue-specific transcription reprogramming. Specifically, compared with the parental 4T1 cells, the subpopulation of liver-metastatic 4T1 cells showed high expression of a liver-specific gene set but not any lung- or bone-specific gene sets. Likewise, the lung-metastatic 4T1 subpopulation showed high expression of a lung-specific gene set but not any liver- or bone-specific gene sets (Fig. S5a). These consistent findings from an independent data set again strongly support that tissue-specific transcription reprogramming occurs in metastatic cancer cells.
To investigate whether this transcription reprogramming also occurs in cell lines of other cancer types, we analyzed gene expression datasets from Cancer Cell Line Encyclopedia (CCLE) containing 165 primary and 14 metastatic cell lines for 8 different cancer types and 6 distant organs (Table S5). Using the same analysis and classification criteria, we found that tissue-specific transcription reprogramming occurs in many metastatic cancer cells associated with a variety of distant organs (Fig. 2d). Some GSEA examples are shown (Figs. 2e and S5b). These results from CCLE are consistent with those from clinical tumor samples, which again confirmed that the tissue-specific transcription reprogramming may represent a general feature of metastatic cancers.
Collectively, besides demonstrating that liver metastatic CRC tumors, PDX samples, and CRC cell lines gain a liver-specific transcription program and lose a colon-specific transcription program as these tumor cells undergo metastasis, these results show that such tissue-specific transcription reprogramming actually occurs in multiple types of metastatic human tumors and cell lines.
The reprogrammed transcription is associated with a reshaped enhancer landscape
To investigate how the reprogrammed transcription occurred, we checked the landscapes of enhancers. Enhancers have been implicated in the regulation of tissue-specific or cell lineage gene expression.13 We therefore used chromatin immunoprecipitation sequencing (ChIP-seq) to investigate variations in the enhancer landscapes between SW620 and SW480 cells. Analysis using two antibodies against enhancer-specific histone modifications (H3K27ac and H3K4me2) showed that the deposition patterns of H3K27ac and H3K4me2 differed significantly between SW620 and SW480 cells (Figs. 3a and S6a). An integrated analysis of our RNA-seq and ChIP-seq datasets showed that genes with up-regulated mRNA expression tended to be associated with H3K27ac and H3K4me2 deposition near their genomic loci in SW620 cells (Figs. 3b and S6b). Analysis of liver-specific gene expression in SW620 cells also showed that the genome regions around loci encoding up-regulated liver-specific genes had enriched H3K27ac (P = 2.2e-16, by t-test) and H3K4me2 (P = 2.2e-16, by t-test) deposition (Fig. 3c). In order to examine whether this tissue-specific transcription reprogramming in other cancer types is also associated with reshaped enhancer landscape, we analyzed published H3K27ac ChIP-seq datasets from multiple primary and metastatic cancer cells (see METHODS). We found indeed that this epigenetics-driven transcription reprogramming also occurs in prostate-to-bone metastasis, colon-to-lung metastasis and pancreas-to-liver metastasis (Fig. 3d).

Variations in enhancer and super-enhancer landscape between primary and liver metastatic CRC cells. a Venn diagram indicating overlap and specificity of enhancers marked by H3K4me2 and H3K27ac in SW480 and SW620 cells. b Heatmap showing the densities of enhancers marked by H3K4me2 and H3K27ac at the nearest differentially expressed genes between non-metastatic SW480 and liver metastatic CRC SW620 cells. Rows are ordered the same for all plots. RNA-seq and ChIP-seq signals are shown as log2 of tag counts normalized to 1 × 107 uniquely mapped tags. c Boxplots showing log2 ratios of SW620 to SW480 tag densities for genomic regions marked by H3K4me2 (left) and H3K27ac (right) around genes that are colon-specific genes with high expression in SW480 cells and liver-specific genes with high expression in SW620 cells. Statistically significant P value is indicated with asterisks (***P < 0.005, by t-test). spec., specific; reg., regulation. d Boxplots showing log2 ratios of PC-3 (bone metastatic prostate cancer cells) to 22Rv1 (primary prostate cancer cells) (left), T84 (lung metastatic CRC cells) to HCT116 (primary CRC cells) (middle) and Capan-1 (liver metastatic pancreas cancer cells) to Capan-2 (primary pancreas cancer cells) (right) tag densities for genomic regions marked by H3K27ac around tissue-specific genes that are relatively highly expressed in corresponding cells. Statistically significant P value is indicated with asterisks (***P < 0.005, by t-test). spec., specific; reg., regulation. e Enhancers are ranked by increasing H3K27ac ChIP-seq signal in primary (SW480) and liver-prone metastasis CRC cells (SW620). Points in red indicated super-enhancers, which are past the point where the slope is greater than 1. f Venn diagram indicating overlap and specificity of super-enhancers in SW480 and SW620 cells. g Scatterplot of the relationship between the ratio of SW480 to SW620 H3K27ac tag density at super-enhancers (x axis) and the ratio of nearest gene expression (y axis). The RNA-seq and ChIP-seq signals are shown as log2 of tag counts normalized to 1 × 107 uniquely mapped tags. Red dots represent some liver-specific genes (FOXA2, SARDH, ENO3, SLC9A3R2, ABHD2, LRP5 and PROX1) associated with super-enhancers. The Pearson correlation coefficient is 0.807 and P < 2.2e-16. h UCSC genome browser images of super-enhancers around a liver-specific gene SARDH and a colon-specific gene GLI3 in SW620 and SW480 covered genomic regions marked by H3K27ac. Bars labeled with SE indicate super-enhancers
The reprogrammed transcription is associated with a reshaped super-enhancer landscape
Super-enhancers are a small fraction of total enhancers and encompass broad chromatin domains with H3K27ac deposition near genes essential for defining cell identity.27,28 By identifying super-enhancers in SW480 and SW620 cells based on our H3K27ac ChIP-seq datasets (Fig. 3e), we found that in addition to the 264 super-enhancers common to both cell lines, there are 280 and 215 unique super-enhancers in SW620 and SW480 cells, respectively (Fig. 3f). Comparison between our ChIP-seq and RNA-seq data revealed a high Pearson correlation coefficient (R = 0.807, P value < 2.2e-16) between the genome-wide distribution of super-enhancers and the expression levels of the nearest genes in these two cell lines (Fig. 3g). Notably, a number of liver-specific genes were found near SW620-unique super-enhancers in SW620 cells (Fig. S6c). Also, some liver-, bone- and lung-specific genes were close to unique super-enhancers in liver metastatic pancreas cancer cells, bone metastatic prostate cancer cells and lung metastatic CRC cells, respectively (Fig. S6d). Some representative examples of super-enhancers were shown (Figs. 3h and S6e).
A small molecule bromodomain inhibitor JQ1 can disrupt the binding of bromodomain containing 4 (BRD4) to enhancers29 and thus inhibits enhancer activity. We used JQ1 to disrupt the influence of enhancers on gene transcription in SW620 cells to explore the role of reshaped enhancer landscapes in the reprogramming of tissue-specific transcription in metastatic CRC cells. JQ1 treatment of SW620 cells indeed resulted in the down-regulation of a set of liver-specific genes (Fig. S7a, b). GSEA further confirmed that the liver-specific gene signature was significantly enriched in the set of down-regulated genes from JQ1-treated SW620 cells (FDR = 0.005) (Fig. S7c). Further, we noted that the SW620-unique enhancers are significantly more enriched near the genes that were down-regulated by JQ1 treatment than common enhancers (P = 4.148e-06 and < 2.2e-16, Fisher’s exact test, for H3K27ac and H3K4me2 marked enhancers, respectively) (Fig. S7d). Our JQ1 treatment results thus indicate that reprogramming of transcription in metastatic CRC cells is driven by reshaped enhancer landscapes. BRD4 inhibitors have been reported to suppress CRC cells metastasizing to the liver in mice, but very little is known about any related mechanisms of action.30 Our findings suggest that the anti-metastasis effect of BRD4 inhibition may be conferred by preventing metastatic CRC cells from executing the liver-specific transcription program that is specified and driven by an altered enhancer landscape.
Unique enhancers in liver metastatic CRC cells are bound by liver-specific transcription factor FOXA2
To determine whether some key transcription factors were involved in enhancer landscape-oriented reprogramming of tissue-specific transcription, we conducted a de novo binding motif analysis to examine all of the gained enhancers in metastatic cancer cells, such as SW620 (4635 gained enhancers) (Figs. 4a and S8a), CAPAN1 (13,484), PC-3 (27,041) and T84 (5297) cells (Fig. S8b). Interestingly, motifs for many distant-organ specific-TFs are highly enriched in the gained enhancers in metastatic cancer cells (Figs. 4a and S8b). For example, we found that the liver-specific TF FOXA binding motif is the most highly-enriched one in liver metastatic CRC SW620 cells (Fig. 4a). The FOXA1 binding motif is highly enriched in liver metastatic pancreas cancer cells (Capan-1) (Fig. S8b). FOXA1 was reported to activate the expression of liver-specific genes such as albumin and transthyretin.31 It was also recently reported that FOXA1 can occupy and activate metastasis-associated enhancers and drive pancreas cancer metastasis.32 The binding motif for RUNX proteins is highly enriched in bone metastatic PC-3 prostate cancer cells (Fig. S8b), and RUNX proteins like RUNX2 have been reported to regulate bone-related genes and play important roles in bone development.33 Moreover, it is well documented that prostate tumors with high expression of RUNX2 aggressively metastasize to the bone.34,35 Motif of ELF TFs is highly enriched in lung metastatic CRC cell line T84 (Fig. S8b), where ELF3 was reported to be frequently activated in lung adenocarcinoma.36 By contrast, DNA motifs for many primary-tissue-specific TFs are significantly enriched in the primary-cancer-cell-unique enhancers (Fig. S8a, c). Together, these results from DNA motif analysis indicated that tissue-specific TFs might play important roles in the regulation of tissue-specific gene expression in metastatic cancer cells.

FOXA2 is required for the activation of liver-specific genes and liver colonization of CRC cells. a Top enriched DNA binding motifs with significant P values, identified in a de novo motif analysis of SW620-unique enhancers (non-promoter regions marked by H3K4me2, far from 3 kb up- or downstream of TSS). b The blue and red bars in the chart show the expression of FOXA2 in normal colon and liver tissues, respectively. The Y axis shows the mean RPKM value. RNA-seq data were obtained from the GTEx project database. c UCSC genome browser images of a super-enhancer around the liver-specific gene FOXA2 in SW620 and SW480 covered genomic regions marked by H3K27ac and H3K4me2, with corresponding RNA-seq data for SW480 and SW620 cells. Bars labeled with SE indicate super-enhancers. d Immunoblot analysis was used to assess the efficiency of FOXA2 knockdown in SW620 cells. GAPDH served as a loading control (Left panel). GSEA of liver-specific signatures as defined from the GTEx project database is shown. Genes are ranked by the log2 fold change of the FPKM values in SW620 FOXA2-knockdown versus shNC cells. The NES and FDR are shown (Right two panels). e Immunoblot analysis was used to assess efficiency of FOXA2 knockdown in Colo205 cells. GAPDH served as a loading control (Left panel). GSEA of liver-specific signatures as defined from the GTEx project database is shown. Genes are ranked by the log2 fold change of the FPKM values in Colo205 FOXA2-knockdown versus shNC cells. The NES and FDR are shown (Right two panels). f Immunoblot analysis was used to assess efficiency of FOXA2 overexpression in SW480 cells. GAPDH served as a loading control (Left panel). GSEA of liver-specific signatures as defined from the GTEx project database is shown. Genes are ranked by the log2 fold change of the FPKM values in SW480 FOXA2-overexpression versus Control cells. The NES and FDR are shown (Right panel). g Immunoblot analysis was used to assess efficiency of FOXA2 overexpression in HCT116 cells. GAPDH served as a loading control (Left panel). GSEA of liver-specific signatures as defined from the GTEx project database is shown. Genes are ranked by the log2 fold change of the FPKM values in HCT116 FOXA2-overexpression versus Control cells. The NES and FDR are shown (Right panel). h FOXA2 is highly expressed in human liver metastatic CRC tumors as compared with primary tumors. The expression data are from the GSE41258 dataset. Each dot represents one primary or liver metastatic CRC tumor, and the red bar represents the mean value. Statistically significant P values are indicated with asterisks (***P < 0.001, by t-test). i Plots showing the IHC scores for the nuclear FOXA2 in human primary CRC tumors and liver metastatic CRC tumors. Each dot represents one primary or liver metastatic CRC tumor, and the red bar represents the mean value. Statistically-significant P values are indicated with asterisks (**P < 0.005, by t-test). j Representative IHC images of FOXA2 expression in paired CRC primary tumors and liver metastatic tumors. Scale bars, 500 µm. k Representative images of the liver colonies upon intrahepatic injection of SW620 shNC and shFOXA2 cells. l The stacked bars indicate the percentage of mice with liver colonies or with no liver colonies relative to the total number of mice in each section. Data were analyzed using Pearson’s Chi-square test. *P < 0.05, ***P < 0.001. The hepatic colonization rate is indicated at the bottom. m Representative images of the liver colonies (Top panel) upon intrahepatic injection of SW480 Control and FOXA2-overexpressing cells. The stacked bars indicate the percentage of mice with liver colonies and with no liver colonies relative to the total number of mice in each section. Data were analyzed using Pearson’s Chi-square test. n.s., not significant. The hepatic colonization rate is indicated at the bottom (Bottom panel). n Representative images of the liver colonies (Top panel) upon intracecal injection of HCT116 Control and FOXA2-overexpressing cells. The stacked bars indicate the percentage of mice with liver colonies and with no liver colonies relative to the total number of mice in each section. Data were analyzed using Pearson’s Chi-square test. n.s., not significant. The hepatic metastasis rate is indicated at the bottom (Bottom panel)
The FOXA binding motif was among the top three most enriched motifs in the gained enhancers in liver metastatic CRC cells (Fig. 4a). There are three members of the FOXA gene family, and FOXA2 is a well-known liver lineage-determining transcription factor,37 which is also highly expressed in liver metastatic SW620 cells (Fig. S9a) and in normal liver tissues (Figs. 4b and S9b). Notably, the FOXA2 locus is covered by a gained super-enhancer in SW620 cells (Fig. 4c). To check the DNA binding sites of these distant-tissue-specific TFs in the metastatic cancer cells, we performed ChIP-seq to examine the FOXA bindings in CRC cells as an example. In accordance with our motif analysis, ChIP-seq analysis with an antibody against FOXA2 identified 5247 and 12,412 FOXA2-bound sites in SW480 and SW620 cells, respectively (Fig. S9c), and showed that in SW620 cells FOXA2 occupied 35% of the SW620-unique enhancers but only occupied 0.01% of the SW480-unique enhancers (Fig. S9d). Moreover, the deposition of H3K4me2 around FOXA2 binding sites was also significantly stronger in SW620 cells than that in SW480 cells (Fig. S9e).
FOXA2 is required for the expression of liver-specific genes in CRC cells and up-regulated in liver-metastatic CRC tumors
To investigate the role of FOXA2 in transcription reprogramming and CRC liver metastasis, we knocked down FOXA2 in CRC cells using two shRNAs (Fig. 4d). GSEA results showed that the liver-specific gene signature was significantly repressed in shFOXA2 cells compared to shNC SW620 cells (Fig. 4d). We also used qPCR to validate that the downregulation of a number of liver-specific genes did occur upon FOXA2 knockdown (Fig. S10a). The promoter of some of these liver-specific gene loci, including LIPC, which was reported to be highly expressed in human CRC liver metastatic tumors and to be required for CRC liver metastasis,38 can be bound and activated by FOXA2 (Fig. S10b).
We also knocked down FOXA2 in another CRC cell line (colo205; Fig. 4e) and performed RNA-seq. Consistently, the GSEA results showed that the liver-specific gene signature was significantly repressed in shFOXA2 cells as compared with shNC cells (Fig. 4e). Interestingly, the overexpression of FOXA2 in two additional CRC cell lines that are derived from primary tumors (SW480 and HCT116) could not activate the expression of the liver-specific gene set (Fig. 4f, g). Considering the primary functions of the liver, we were encouraged to find that a gene ontology analysis showed the top-ranked enriched term for the down-regulated genes upon FOXA2 knocking down was ‘metabolic process’ (Fig. S10c). These results indicated that FOXA2 is required but not sufficient for the activation of liver-specific gene expression in CRC cells and supported that the liver-specific TF FOXA2 binds to the unique enhancers present in liver-metastatic CRC cells to activate the transcription of a set of liver-specific genes.
To explore the clinical relevance of our findings, we analyzed published gene-expression datasets for CRC tumor samples. We found that FOXA2 is more highly expressed in human CRC liver metastatic tumors than in primary CRC tumors (Fig. 4h). In addition, the IHC results in tissue arrays clearly showed that the FOXA2 protein levels were significantly higher in liver metastatic CRC tumors than those in primary CRC tumors (P = 0.004, by t-test) (Figs. 4i, j, S10d and Table S6). When we analyzed published clinical data, we found that the subset of CRC patients with high expression levels for FOXA2-target genes (91 genes, Table S7) in their primary tumors had significantly worse overall survival (P = 0.006, by log-rank test) and recurrence free survival (P = 0.007, by log-rank test) outcomes than did a subset of patients with low expression levels for these genes (Fig. S10e), strongly supporting a functional role for FOXA2 in promoting CRC metastasis.
FOXA2 is required for the colonization of CRC cells in the liver
We used three xenograft models to examine the effect of FOXA2 knockdown and over-expression on liver colonization of CRC cells in mice. First, 6 weeks after intrahepatic-injection-based implantation39 of shFOXA2 or shNC SW620 cells into the livers of nude mice, many large (macroscopically visible) metastases were found in livers of mice implanted with shControl SW620 cells, whereas significantly fewer (and smaller) metastases were found in livers of mice implanted with shFOX2 SW620 cells (Fig. S10f). The FOXA2 shRNA mice also had a significantly lower colonization rate (Fig. 4k, l). Importantly, knockdown of FOXA2 in SW620 cells did not inhibit cell proliferation in vitro (Fig. S10g) and did not inhibit the growth of subcutaneous tumors in nude mice (Fig. S10h, i), indicating that FOXA2’s effects on promoting CRC tumor growth are context-dependent and that this promotion function apparently occurred in the livers of these animals. Thus, our results clearly demonstrate that knockdown of FOXA2 inhibits liver-specific gene transcription, thereby interrupting the colonization of CRC cells in the liver. Second, when we overexpressed FOXA2 in SW480 cells with less liver metastatic capacity and injected them into mouse livers, we found almost no difference from controls in the extent of liver colonization about 2 months later (Fig. 4m). Third, we intracecally injected another primary CRC cell line HCT116 that over-expressed FOXA2 and again found no difference in the extent of liver metastasis between mice overexpressing FOXA2 and control mice (Fig. 4n).
Notably, our experiments with the highly metastatic SW620 cells demonstrate that the knockdown of FOXA2 clearly does repress the expression of liver-specific genes in metastatic CRC cells (Fig. 4d, e); we also found that overexpression of FOXA2 in cell lines with relatively less metastatic capacity (SW480 and HCT116) did not activate the expression of liver-specific gene signature (Fig. 4f, g). So, the impacts of FOXA2 knockdown and overexpression on the regulation of liver-specific gene transcription in CRC cells are consistent with the liver colonization and metastasis capability of these CRC cells, again indicating that the reprogramming of tissue-specific transcription is integral to the progression of CRC liver metastasis. Further, these in vivo results also support that FOXA2 is required but not sufficient for promoting CRC liver colonization and metastasis.
The liver-specific TF HNF1A regulates the expression of liver-specific genes in CRC cells
In addition to the FOXA binding motif, we noticed that liver-specific TF HNF1A binding motif is also significantly enriched in the unique enhancers in SW620 cells (Fig. 4a). HNF1A, originally described in the liver,40 is a well-known essential transcription factor that controls the expression of many hepatic genes and is critical for diverse metabolic functions in the liver.41 HNF1A is highly expressed in normal liver tissue compared with colon tissue (Fig. 5a). Our RNA-seq data indicated that HNF1A is highly expressed in liver metastatic SW620 cells compared with primary SW480 cells (Fig. S4c) and our ChIP-seq data also revealed the gain of enhancers around the HNF1A locus in liver metastatic cells (Fig. 5b).

HNF1A is required and sufficient for the expression of liver specific genes in CRC cells and liver colonization and metastasis. a The blue and red bars in the chart show HNF1A gene expression in normal colon and liver tissues, respectively. The Y axis shows the mean RPKM value. The RNA-seq data were obtained from the GTEx project database. b UCSC genome browser images of an enhancer around the liver-specific gene HNF1A in SW620 and SW480 covered genomic regions marked by H3K27ac and H3K4me2. c Immunoblot analysis was used to assess efficiency of HNF1A knockdown in SW620 cells. GAPDH served as a loading control (Top panel). GSEA of liver-specific signatures as defined from the GTEx project database is shown. Genes are ranked by the log2 fold change of the FPKM values in HNF1A-knockdown versus shNC cells. The NES and FDR are shown (Bottom two panels). d Gene ontology analysis of down-regulated genes by HNF1A knockdown. e Immunoblot analysis was used to assess efficiency of HNF1A overexpression in HCT116 cells. GAPDH served as a loading control (Top panel). GSEA of liver-specific signatures as defined from the GTEx project database is shown. Genes are ranked by the log2 fold change of the FPKM values in HCT116 HNF1A-overexpression versus Control cells. The NES and FDR are shown (Bottom panel). f HNF1A is highly expressed in human liver metastatic CRC tumors as compared with primary tumors. The expression data are from the GSE41258 dataset. Each dot represents one primary or liver metastatic CRC tumor, and the red bar represents the mean value. Statistically-significant P values are indicated with asterisks (***P < 0.001, by t-test). g Luciferase measurements from different time points upon intracecal injection of HCC022 shNC and shHNF1A cells. An shHNF1A virus pool (3 HNF1A shRNA sequences) was expressed in HCC022 cells and shNC and shHNF1A cells were injected into mice using intracecal injection (n = 4 shNC mice and n = 4 shHNF1A mice). Then a time-course analysis of luciferase measurements in mice was performed; Black lines represent shNC control group and red lines represent shHNF1A mice group. Error bars represent standard deviation of the mean. Student’s t test, P < 0.01, n.s., not significant. h The stacked bars indicate the percentage of mice with liver metastases or with no liver metastases relative to the total number of mice in each section. The hepatic metastasis rate of HCC022 cells is indicated at the bottom. Data were analyzed using Pearson’s Chi-square test. **P < 0.01. i Representative images of the metastases upon intracecal injection of HCT116 control and HNF1A-overexpressing cells. j The stacked bars indicate the percentage of mice with liver metastases or with no liver metastases relative to the total number of mice in each section. The hepatic metastasis rate is indicated at the bottom. Data were analyzed using Pearson’s Chi-square test. *P < 0.05. k Representative images of the metastases upon intrahepatic injection of control and HNF1A-overexpressing HCT116 cells. l The stacked bars indicate the percentage of mice with liver colonies and without liver colonies relative to the total number of mice in each section. The hepatic colonization rate is indicated at the bottom. Data were analyzed using Pearson’s Chi-square test. **P < 0.01
To investigate the potential role of HNF1A in tissue-specific transcription reprogramming, we knocked down or overexpressed HNF1A in CRC cells. GSEA analysis showed that the liver-specific gene signature was enriched among the down-regulated genes in the shHNF1A SW620 cells as compared with shNC SW620 cells (Fig. 5c), suggesting this liver-specific TF also regulates a subset of liver-specific genes in SW620 cells. Interestingly, we found that the liver-specific gene LIPC, which is a CRC liver metastasis-associated gene, is also regulated by HNF1A (Fig. S11a). Moreover, considering the primary metabolic functions of the liver, we were encouraged to find that the top-ranked enriched terms for the down-regulated genes upon HNF1A knockdown were related to metabolic process and included “arachidonic acid metabolism” (Fig. 5d). In addition, we overexpressed HNF1A in CRC HCT116 cells and GSEA analysis revealed that, compared to controls, the liver-specific gene set was significantly upregulated in the HNF1A-overexpressing HCT116 cells (Fig. 5e). These results clearly establish that the liver-specific TF HNF1A is required and sufficient to activate the expression of liver-specific genes in CRC cells.
HNF1A is required and sufficient for the colonization of metastatic CRC cells in liver
To evaluate the clinical relevance of HNF1A with CRC liver metastasis, we examined the expression of HNF1A in CRC tumors. By analyzing a published transcriptome dataset, we found that HNF1A is highly expressed in liver metastatic tumors compared with primary CRC tumors (Fig. 5f). We further analyzed published clinical data and observed that the subset of CRC patients with high expression levels for HNF1A-target genes in their primary tumors had significantly worse overall survival (P = 0.021, by log-rank test) and recurrence free survival (P = 0.000, by log-rank test) outcomes than a subset of patients with low expression levels for these genes (Fig. S11b). Together, these results from clinical samples strongly suggest that HNF1A promotes CRC liver metastasis.
We next used two different mouse xenograft models to evaluate the functional role of HNF1A in CRC liver metastasis. The HCC022 human cell line is derived from a PDX sample and exhibits strong liver metastatic capability. We stably knocked down HNF1A with pooled shRNAs targeting HNF1A in luciferase-expressing HCC022 cells and performed intracecal injection to generate an orthotopic xenograft mouse model. At 4 weeks post-injection, 4 out of 4 mice in the shNC group presented strong luciferase activity in the liver, while 0 out of 4 mice in the shHNF1A group exhibited luciferase activity in the liver (Fig. 5g, h). We also anatomized the mice and extracted the liver. Consistently, shNC mice developed aggressive and serious liver metastases in the liver, while shHNF1A mice had barely any liver metastases (Fig. S11c).
To investigate whether HNF1A is sufficient to promote liver metastasis of CRC cells, we also used an orthotopic mouse model to examine the metastasis potential with HNF1A-overexpressing HCT116 cells. At ~6 weeks post-intracecal injection, we observed that 8 of 8 mice in the HNF1A overexpression group developed liver metastases, while only half of the mice (6 of 12) in the control group developed liver metastases (Fig. 5i, j). Metastasis is certainly a complex process consisting of multiple steps, and we focused on the last step, which is liver colonization. Therefore, we overexpressed HNF1A in HCT116 cells and performed intrahepatic injection in mice to test tumor formation. This experiment revealed that the number of mice which developed tumors was significantly higher in these mice relative to control mice, in agreement with our model of liver colonization of CRC cells mediated by liver TFs (Fig. 5k, l). Moreover, we found knockdown of HNF1A in SW620 cells and HCC022 cells and overexpression of HNF1A in HCT116 cells did not inhibit cell proliferation in vitro (Fig. S11d), indicating that the effects of HNF1A on promoting CRC tumor growth are context-dependent and that this promotion function apparently occurs in the livers of these animals. Collectively, these in vivo results suggest that the liver-specific TF HNF1A is not only required but also sufficient to promote liver colonization and metastasis of CRC cells, a finding that is also consistent with our observations about the reprogrammed tissue-specific transcription that occurs in CRC cells.
Discussion
Here we investigated how metastatic cancer cells, such as CRC cells, with original-tissue specificity adapt to the environment of and colonize a distant tissue, like liver. First, we discovered reprogrammed tissue-specific transcription, simultaneous gain of liver-specific and loss of colon-specific transcription programs, in liver metastatic CRC tumors. This observation was supported by transcriptome datasets from multiple cohorts of clinical CRC tumors and PDX samples, as well as by RNA-seq analysis of paired CRC cell lines. Second, we provided evidence that these transcriptional changes likely result from reprogrammed enhancer landscapes, including both typical and super-enhancers. Third, we showed that FOXA2 and HNF1A, two well-known liver lineage-determining transcription factors, mediated the unique enhancer changes to activate a set of liver-specific genes in liver metastatic CRC cells. Finally, we demonstrated that inactivation of FOXA2 or HNF1A inhibited liver-specific transcription and thus impaired the colonization and metastasis of CRC cells in the liver; overexpression of HNF1A activated liver-specific transcription and enhanced CRC liver colonization and metastasis.
Our findings indicate the direct contribution of a reprogrammed tissue-specific transcription program to the adaption and colonization of metastatic CRC cells in the liver. Notably, in addition to CRC, this transcription reprogramming is also observed for multiple metastatic cancer types in distant organs. Further studies are needed to understand how the epigenetic landscapes in metastatic cancer cells are reshaped and explore the potential original-to-distant tissue transition in other cancer types. A very recent published paper illustrated that combination of forkhead box proteins and hepatocyte nuclear factors could induce expression of liver-specific genes and reprogram hepatocellular carcinoma cells to hepatocyte-like cells.42
Our results showed that knockdown of FOXA2 could inhibit the liver-specific gene program (Fig. 4d, e) and mitigate liver colonization (Fig. 4k, l), while overexpression of FOXA2 could not activate the liver-specific gene program (Fig. 4f, g) and could not enhance the liver colonization potential of primary colon cancer cells (Fig. 4m, n). These results indicate that FOXA2 is required but not sufficient to drive CRC liver metastasis. Consistently, our results showed that knockdown of HNF1A inhibited the liver-specific gene program (Fig. 5c) and mitigated CRC liver metastasis (Fig. 5g, h). Overexpression of HNF1A activated the liver-specific gene program (Fig. 5e) and promoted CRC liver colonization and metastasis (Fig. 5i–l). These results indicate that HNF1A is required and sufficient to activate liver-specific gene program in CRC cells, thereby affecting CRC liver metastasis in vivo. All these in vitro and in vivo results for FOXA2 and HNF1A strongly support our hypothesis that the liver-specific transcription reprogramming is required and sufficient to drive CRC liver colonization and metastasis. Since many but not all genes regulated by FOXA2 and HNF1A in metastatic colon cancer cells are liver-specific genes, non-liver-specific genes, alone or in combination with liver-specific genes, may also contribute to liver metastasis of colon cancer cells, which need to be further studied in the future.
Our study demonstrated that both FOXA2 and HNF1A can bind to the unique enhancers present in metastatic CRC cells and thereby regulate the expression of liver-specific genes. Although our study did not directly investigate any connections between the functions of FOXA2 and HNF1A, an emerging body of evidence implicates the functional cooperation of FOXA2 and HNF1A in normal liver development. Specifically, it has been reported that combination of FOXA2 and HNF1A can activate the expression of hepatocyte-related genes to promote efficient hepatic differentiation from human embryotic stem cells (hESCs) and from induced pluripotent stem cells (hiPSCs).43 It has also been reported that approximately 50% of HNF1 binding sites are also occupied by FOXA2 and that HNF1A regulates intestinal epithelial cell fate both directly and via interactions with FOXA2. A very recent study also showed that HNF1A binding was not significant when it was expressed alone but that such binding was significantly enhanced when HNF1A was co-expressed alongside FOXA2,44 suggesting that FOXA2 may function to enhance HNF1A binding.
Intriguingly, a previous study reported that liver-specific gene LIPC is required for CRC liver metastasis,38 and our study revealed that the expression of this liver metastasis-associated liver-specific gene is positively regulated by FOXA2 and HNF1A. Knockdown of FOXA2 or HNF1A could reduce gene expression of LIPC (Figs. S10a and S11a). However, overexpression of FOXA2 could not upregulate LIPC (Fig. S11e), while overexpression of HNF1A significantly enhanced LIPC expression (Fig. S11f). These findings demonstrate that FOXA2 is required but not sufficient for liver-specific gene expression, while HNF1A is required and sufficient for liver-specific gene expression. Viewed together, this information suggests that FOXA2 and HNF1A cooperate to some degree in the activation of downstream liver-specific gene expression.
Our findings establish that the binding of liver-specific TFs to the enhancers of a reprogrammed epigenetic landscape promotes CRC liver colonization and metastasis. Distinct usage of enhancers is the driver for reprogrammed gene transcription and promotion of metastasis. Usage and function of enhancer elements are affected by various factors. Tissue environment could drive the selection and function of enhancers, which control tissue-specific transcription of macrophages.17 Roe et al. reported that the pioneer factor FOXA1 could alter enhancer landscape, which drives pancreas cancer liver metastasis.32 Also, Mutation of DNA sequences in enhancer element could alter enhancer activity, affect the binding of transcription factors or promote aberrant gain of enhancer function.17,45,46
We showed that liver-specific gene program could be used to predict overall survival in primary colon cancers (Figs. 1g, S10e and S11b), indicating that some cancer cells already acquire liver-specific characteristics while still in colons. We also found distant organ-specific gene expression program pre-exists in some primary tumors of other cancer types, including liver metastatic pancreas cancer, bone metastatic prostate cancer, lung metastatic CRC and brain metastatic breast cancer (Fig. S3b). The survival analysis suggested that distant tissue-like gene program may already exist in primary tumors but the gene expression levels are low. However, the expression levels of these genes are enhanced in distant metastatic tumors. This observation generally exists in other cancer types and organs as well. This idea actually is consistent with a previous report about how breast cancer clones selected by CAF (cancer associated factors)-derived factors are primed for metastasis in the bone microenvironment.47 It has been reported that once prostate and breast cancer cells metastasize to the bone, they try to resemble osteoblasts by expressing bone cell-related genes. The ability of this osteomimicry acquired by cancer cells give them more chances to proliferate in and colonize the bone tissue.48
Here, our results indicate the feature that the metastatic cancer cells mimic the cells in distant organs is not limited to bone metastatic prostate or breast cancer cells, and is commonly observed for various other cancer types, which we deem as “original to distant transition” (ODT). Importantly, our mechanistic study further discloses that ODT is driven by reshaped epigenetic landscapes that are subsequently accessed by distant tissue-specific transcription factors in metastatic cancer cells.
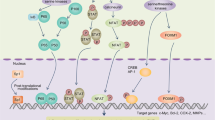
The model we proposed can explain well our observations about the epigenetic reprogramming of tissue-specific transcription that occurs in liver metastatic CRC cells (Fig. 6). Importantly, this model predicts that multiple stages of the reprogramming process could be targeted therapeutically and provides us potential selective targets, including the enzymes responsible for reshaping epigenetic landscapes and/or the transcription factors or other regulators that function in the altered transcription programs. Targeting these potential targets can reverse the reprogrammed enhancer and transcription landscapes for metastasis therapy. Here, JQ1 treatment demonstrated that inhibition of the function of gained enhancers in SW620 cells led to suppression of liver-specific genes. However, we need to find more selective targetings to explore the therapeutics of CRC liver metastasis in the future. The ODT process likely occurs in multiple cancer types, and similar therapeutic strategies in theory should also work in other cancers. Recalling that there are at present no therapies to treat metastasis, and considering that this process causes the death of 90% of cancer patients, the mechanistic insights from our study suggest a timely new conceptual framework for scientists and clinicians to use as they seek to detect, treat, and perhaps even prevent metastasis.

Model for tissue-specific transcription reprogramming that promotes liver metastasis of colorectal cancer
Materials and methods
Cell culture
SW480, SW620, HCT116 cell lines were obtained from China Infrastruture of Cell Line Resources and cultured as described.49 PDX cell line HCC022 was given by Prof. Wei Guo from Tsinghua University. Briefly, SW480, SW620, HCT116 and HCC022 cells were cultured in DMEM (Gibco Cat. No. C11995500BT) supplemented with 10% fetal bovine serum (Gemini Cat. No. 900–108) and 1% penicillin/streptomycin (Gibco Cat. No. 15140–122). Cells were cultured at 37 °C with 5% CO2. Cells used to inject mice were stably transfected with luciferase.
Plasmids and shRNA infections
A lentiviral U6-based expression vector containing PuroR-T2A-mCherry was used to express shRNAs. The lentiviral vector was digested by BsmBI, followed by annealed shRNA oligos insertion, to clone shRNA expression plasmids. We used two shRNA targeting sites against FOXA2 as follows: FOXA2-shRNA1, 5′ GAACGGCATGAACACGTACAT 3′ (from Sigma-Aldrich Corporation, TRCN0000014915); FOXA2-shRNA2, 5′ GGAACACCACTACGCCTTCAA 3′. We used a pool of three shRNAs targeting sites against HNF1A as follows: HNF1A-shRNA1, 5′ CACTCCCATGAAGACGCAGAA 3′; HNF1A-shRNA2, 5′ AGACTGCAGAAGTACCCTCAA 3′; HNF1A-shRNA3, 5′ GCTCCCGCAGACTATGCTCAT 3′. shControl, 5′ CAACAAGATGAAGAGCACCAA 3′.
FOXA2 overexpression plasmid was purchased from Nanjing Jingbai biotechnology company. Briefly, FOXA2 cDNA was subcloned into pLenti-CMV-RFP-Puro. HNF1A cDNA was subcloned into CMV-T2A-eGFP. Briefly, the lentiviral vector was digested by EcoRI and XbaI, followed by cDNA insertion to clone HNF1A overexpression plasmids.
Lentiviral vector particles were produced by tri-transfection of plasmids harboring the packaging construct, the transfer vector and the envelope-expressing construct into 293T cells using DNAfect reagents (Cwbio Cat. No. CW0806). Viral supernatants were harvested and used for infections or stored at −80 °C. Stable FOXA2 knockdown cell lines were generated by using lentiviral U6-based expression vectors. Stable populations were selected with 2 μg/ml puromycin (Sigma-Aldrich Cat. No. P9620). Knockdown was confirmed by RT-qPCR and western blotting.
Western blotting
Cells were lysed in RIPA buffer. Proteins were separated by 10% polyacrylamide gel and transferred to polyvinylidene membranes (Bio-Rad Cat. No. 170–4159), which were blocked for 1 h at room temperature in TBS with Tween 20 (TBST) containing 5% BSA and subsequently probed with primary antibodies overnight at 4 °C. After incubating the membrane with anti-rabbit peroxidase-conjugated secondary antibody (Cell Signaling Technology Cat. No. 7074), protein levels were detected with SuperSignal West Pico reagents (ThermoFisher Scientific Cat. No. 34095). Primary antibodies were prepared in 5% BSA in TBST. The following primary antibodies were used: anti-FOXA2 (Abcam Cat. No. ab108422), anti-HNF1A (proteintech Cat. No. 22426–1-AP), and anti-GAPDH (Cell Signaling Technology Cat. No. 2118) antibodies.
SYBR-Green real-time PCR
Total RNA was extracted from cells by using TRIzol® Reagent (Ambion Cat. No. 15596018), and reverse-transcribed to cDNA by using the Thermo Scientific RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific Cat. No. K1622). Quantitative PCR (qPCR) involved use of the KAPA SYBR FAST Universal qPCR Kit (KAPA Cat. No. KK4601) in triplicate, with normalization to GAPDH. Primer sequences (from 5′ to 3′) are listed as follows: FOXA2 forward primer, TTGCTGGTCGTTTGTTGTGG; FOXA2 reverse primer, GTTCATGTTGCTCACGGAGG; HNF1A forward primer, TGGTCAAGTCCTACCTGCAG; HNF1A reverse primer, TCTTCAATCAGCCCTCCCTG; LIPC forward primer, GTTCATGTTGCTCACGGAGG; LIPC reverse primer, GGCTGAAGCTGTTCATGTCA; STARD10 forward primer, GGCTGAAGCTGTTCATGTCA; STARD10 reverse primer, TTCCACTCGGGGTACTTGAG; SARDH forward primer, TTCCACTCGGGGTACTTGAG; SARDH reverse primer, AGCCCCACCAGGTAGAACTT; GAPDH forward primer, AGCCCCACCAGGTAGAACTT and GAPDH reverse primer, AGCCTTCTCCATGGTGGTGAAGAC.
Proliferation assay
The effect of FOXA2 knockdown on SW620 cell proliferation was monitored in real-time by using the Incucyte Live-Cell Imaging System (Essen BioScience, USA). SW620 (shFOXA2) and SW620 (shControl) cells (8000 per well) were seeded in 96-well plates. Automated phase contrast images were acquired by use of an Incucyte microscope. The Incucyte Live-Cell Imaging System provides an imbedded contrast-based confluence algorithm to compute monolayer confluence for each image and at each time point. Multiple images are collected per well and averaged to provide a representative statistical measure of confluence, thus allowing quantification of cell growth inside the cell culture incubator.
Tissue microarray and immunohistochemistry (IHC)
The human CRC tissue microarray (Shanghai Outdo Biotech Cat. No. HLin-Ade075Met-01) containing 75 cases of normal colon tissues, primary CRC tumors and metastases was purchased for IHC to determine FOXA2 expression. Of the 75 cases, there were 27 primary tumors and 18 liver metastases. The number of matched pairs of primary tumors and liver metastases is 18. IHC analysis was performed by Outdo Biotech (Shanghai, China) using standard techniques as described.23 Briefly, all specimens on the tissue microarray were evaluated by H&E (hematoxylin-eosin) staining to ensure the pathological types before IHC staining. The tissue microarray was probed using the primary antibody (1:4000 dilution) against FOXA2 (Abcam Cat. No. ab108422), primary antibody (1:200) against LIPC (proteintech, Cat. No. 21133–1-AP), primary antibody (1:100) against CYP27A1 (proteintech, Cat. No. 14739-1-AP), and primary antibody (1:75) against INHBE (ATLAS, Cat. No. HPA016843). The degree of immunostaining was scored by two independent investigators without prior knowledge of the clinical data. The IHC scores were calculated as previously described.23 In brief, more than 1000 tumor cells for each sample were analyzed under microscope. The percentage of positively nuclear stained tumor cells was recorded and varied from 0–100%. Designated protein expression was quantified using a grading system based on the percentage of protein-positive cells and the intensity of nuclear staining. The percentage of protein-positive cells was scored as follows: 0, <5%; 1, 5–24%; 2, 25–49%; 3, 50–75%; 4, >75%. The intensity of nuclear staining was scored as follows: 0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining. A final IHC score was calculated by multiplying the scores of “percentage of protein-positive cells” and “intensity of nuclear staining”. All primary data are shown (Tables S1–S3, S6). HE images of these tumor tissues have been examined by four clinicians and experts. There are no normal liver tissues in the tissue microarray as they can be distinguished from liver metastases by their distinct morphology and organization.
Mouse model
Mice were housed in facilities managed by the Tsinghua University Animal Resources Center and all experiments were performed in accordance with Tsinghua University’s Animal Care and Use Committee guidelines. SW620 cells were under domestication in liver for three times. Briefly, we used 6-week-old female Balb/c nude mice to domesticate SW620 cells. About 3 weeks later after intrahepatic injection, the mice burdened with liver tumors were sacrificed. Liver tumors were then resected and minced under sterile conditions. The minced tissues were placed in DMEM medium with 100 U/ml collagenase and hyaluronidase for 1.5 h at 37 °C and then filtered by 200-mesh filter followed by centrifugation. Next, cells were re-suspended and grown in DMEM medium with 10% FBS. Intrahepatic injection was performed as described.39 Briefly, female Balb/c nude mice, 5–8 weeks old, were used for surgery. Mice were anesthetized with avertin (From Tsinghua University Animal Resources Center). The skin was incised and tumor cells (5 × 106) with shFOXA2 or shControl in 40 μl PBS were injected into the right liver lobe under the capsule. Mice were killed ~6 weeks later, and the number of liver metastases and the metastatic area were quantified. Subcutaneous injection of tumor cells was performed as described.39 Briefly, tumor cells (2 × 106) were re-suspended in 40 μl PBS and were injected subcutaneously into the right flank of nude mice for 1 injection site per mouse. Six days after injection and every 2 days thereafter, the length and width of tumors were measured. Volume was calculated as length × (width2)/2.50 Intracecal injection was performed using a similar method as described previously.51 Briefly, mice were anesthetized with avertin (From Tsinghua University Animal Resources Center). A small laparotomy incision was performed on the lower right abdomen. The cecum was located and carefully placed onto a sterile field. Tumor cells (5 × 106) were injected into the outer wall tissue layer of the cecum.
RNA-seq library preparation and sequencing
RNA-sequencing (RNA-seq) libraries were prepared by using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB Cat. No. E7420), according to the manufacturer’s instructions. The sequencing was performed by Hiseq1500 (Illumina). The RNA-seq data produced in this study were listed (Table S8).
RNA-seq and microarray data analysis
The fastq files from RNA-seq experiments were mapped to the human genome (hg19) by using STAR52 with parameters --outFilterMismatchNoverLmax 0.05. To measure expression, we calculated the raw counts for each gene by using the analyzeRepeats command from HOMER53 (http://homer.salk.edu/homer/) with the option “rna” and the default parameters. We identified genes with differential expression between SW480 and SW620 cells by using edgeR54 with several criteria (|log2fc| ≥ 1, logCPM ≥ 1 and false discovery rate [FDR] ≤ 0.05).
For published microarray datasets, GSE41258,18 GSE49355,19 GSE6919,55 GSE32269,56 GSE85258,57 GSE52604,58 GSE4295259 and GSE1928160 and RNA-seq dataset, GSE50760,20 we downloaded the normalized expression values directly from Gene Expression Omnibus (GEO) database. We also downloaded the gene expression datasets (i.e. Gene-centric RMA-normalized mRNA expression data) of primary and metastatic cancer cell lines from Cancer Cell Line Encyclopedia (CCLE) website (https://portals.broadinstitute.org/ccle/home).
ChIP-seq library preparation and sequencing
About 5–10 × 106 crosslinked cells were used for ChIP-seq, as described.61 After crosslinking, chromatin was fragmented by sonication, and the mixture was purified with magnetic beads (Millipore Cat. No. 16–157) conjugated with 1 ng of the antibodies against H3K4me1 (Abcam Cat. No. ab8895), H3K4me2 (Millipore Cat. No. 07–030), H3K4me3 (Millipore Cat. No. 07–473), H3K27ac (Abcam Cat. No. ab4729) or FOXA2 (Proteintech Cat. No. 22474–1-AP). ChIP-sequencing libraries were prepared by using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB Cat. No. E7370) according to the manufacturer’s instructions. After barcoding, pooled DNA was sequenced (HiSeq 1500, Illumina) to achieve a minimum of 1 × 107 aligned reads per sample. The ChIP-seq data produced in this study were listed (Table S8).
ChIP-seq data analysis
Fastq files from ChIP-seq experiments were mapped to the human genomes (hg19) by using STAR52 with parameters --outFilterMismatchNoverLmax 0.05. For the ChIP-seq of histone modification, enriched loci were identified by using the findPeaks command from HOMER53 (http://homer.salk.edu/homer/) with the option “-style histone”, 4-fold enrichment over the input sample, 4-fold enrichment over local background, at FDR = 0.001, and the ChIP-seq signals are normalized to 10 million mapped reads per experiment.
Peaks of H3K4me2 3 kb away from gene’s TSS (either upstream or downstream) were defined as enhancers. Super-enhancers were identified following the original strategy used by the Young lab27 using our H3K27ac data. First, peaks are found using similar method applied in analysis of any other ChIP-Seq data set. The peaks found in ChIP-seq of histone modification within a given distance are ‘stitched’ together into larger regions (by default over 12.5 kb). The super-enhancer signal of each of these regions is then determined by the number of total normalized reads minus the number of normalized reads in the input. Next, these scores were normalized to the highest score and the number of putative enhancer regions, then these regions are sorted by their normalized scores. The super-enhancers are identified as regions past the point where the slope is greater than 1.
Those potential enhancer regions marked with histone markers are assigned to closest TSS, using script ‘annotatePeaks.pl’ with default parameters in the homer software (http://homer.ucsd.edu/homer/index.html). By using this strategy, we link a peak/region to its related gene.
Genomic binding peaks for transcription factor FOXA2 were identified with option “-style factor”, 2-fold enrichment over the input sample, 2-fold enrichment over local background, at the false discovery rate of 0.001.
All the peaks of histone and transcription factor were annotated to the nearest TSS (transcription start site), TTS (transcription termination site), Exon (Coding), 5′ UTR Exon, 3′ UTR Exon, Intronic, or Intergenic of genes using annotatePeaks.pl command.
All peaks between the different cell types per comparison were merged into one peak set by using mergePeaks –size given. To obtain differentially bound peaks, tags were counted from each experiment by using getDifferentialPeaks and were considered significant with default parameters (4-fold difference and P = 0.0001). These enhancer regions with peak signals in SW620 cells increased more than 4-fold when compared with those in SW480 cells were named as “SW620-unique” regions; the regions with opposite situation were named as “SW480-unique” regions. The similarly bound peaks were determined by using the “-same” option. Peaks which were not part of either the differentially bound or similarly bound peaks were filtered out.
Besides, we downloaded the raw H3K27ac ChIP-seq data for PC-3, HCT116 and 22Rv1 cell lines from ENCODE project (https://www.encodeproject.org/), and Capan-1 and Capan-2 cell lines from GSE64560.62 The H3K27ac ChIP-seq data for T84 cell line was generated by ourselves. The analyses on these data were performed as described above.
De novo motif finding and known motif enrichment
We used HOMER53 for de novo motif discovery and known motif enrichment. Motif finding were performed on 1000 bp length sequences related to the histone modification (H3K4me2, H3K27ac) peak center, which located >3 kb up- and down-stream of TSS, whereas motif finding for transcript factors (FOXA2) was performed on sequence of given peak length. Briefly, sequences were divided into target and background sets. Background sequences were then selectively weighted to equalize the distributions of G/C content in target and background sequences to avoid comparing sequences of different general sequence content. Firstly, motifs of 8, 10 and 12 bp were identified separately by exhaustively screening all oligonucleotides for enrichment in the target set compared with the background set using the cumulative hypergeometric distribution to score enrichment. Up to two mismatches were allowed in each oligonucleotide sequence to increase the sensitivity of the method. Top oligonucleotides for each length with the lowest P values were then converted into probability matrices and heuristically optimized to maximize hypergeometric enrichment of each motif in the given data set. As optimized motifs were found, they were removed from the dataset to facilitate the identification of additional motifs in subsequent rounds. HOMER also screens the enrichment of previously identified motifs through analysis of published ChIP-ChIP and ChIP-Seq data sets by calculating the known motifs’ hypergeometric enrichment in the same set of G/C normalized sequences used for de novo analysis. Sequence logos were generated using WebLOGO (http://weblogo.berkeley.edu).
The de novo motif finding and known motif enrichment analysis for 22Rv1 versus PC-3, HCT116 versus T84, and Capan-2 versus Capan-1 were performed as described above.
Gene Set Enrichment Analysis
We defined tissue-specific gene sets by using the expression values yielded by the Genotype-Tissue Expression (GTEx) project and Tissue-specific Gene Expression and Regulation (TiGER) datasets (http://bioinfo.wilmer.jhu.edu/tiger/). We downloaded the file that contained the median RPKM of each gene by tissues and cell types from the latest release version, V6p. For each gene, we ranked the median expression value for each tissue or cell type in decreasing order. Genes defined as tissue-specific needed to meet two criteria: (1) gene expression ranked in the top 5 among all tissue and cell types and (2) also highly expressed (>90th percentile of all genes) in particular tissues and cell types. If tissue-specific gene sets for some tissues are not included in GTEx, we downloaded them from TiGER database (http://bioinfo.wilmer.jhu.edu/tiger/). The log2 ratios were computed for several datasets with normalized expression data in different conditions. The log2 ratios of expressions of tissue-specific genes in the primary versus liver metastatic tumor samples were computed for GSE41258,18 GSE49355,19 GSE50760,20 GSE6919,55 GSE32269,56 GSE85258,57 GSE52604,58 GSE4295259 and GSE1928160 datasets. The log2 ratios of expressions of tissue-specific genes in the primary versus metastatic cell lines were computed for CCLE (Table S5). The log2 ratios of expressions of tissue-specific genes in the DMSO versus JQ1 treatment on SW620 cells were also calculated. Then, the pre-ranked Gene Set Enrichment Analysis21 was performed with tissue-associated gene sets and dataset with default parameters. We show the scatter plot of FDR (q-values) versus normalized enrichment score (NES) for each analysis. Since stromal cells exhibit no organ specificity based on transcriptomic analysis, they should not interfere with our observation of tissue-specific transcription reprogramming.63
Removing expression noises caused by contamination of mouse genome
Sequencing reads are assigned simultaneously to human and mouse genome using Xenome software.22 Ambiguous reads and reads mapped to neither or both of the two genomes are filtered out. The remaining reads are mapped to human genome by STAR.
Removing expression noises caused by contamination of host organ
In order to further confirm the tissue-specific gene signatures we found in the patient samples, we removed differentially expressed probes/genes that could be introduced by the contamination of distant organ (i.e. normal liver tissue) in the metastasis samples. We adapted a published method19 to remove ambiguous probes/genes. Because hepatic metastasis (HM) sample may be contaminated by certain normal hepatic tissue (HN), the measured expression value of HM for each probe/gene, mHM, is different from the real expression value:
where λ is the ratio of HN contamination in HM. Thus, we removed the ambiguous up-regulated probes/genes between hepatic metastasis sample (HM) and primary colon tumor (CT) using two criteria:
where \(\widetilde {HM}\) and \(\widetilde {CT}\) are the median expression values of multiple samples in the same data set. We assume the maximum contamination ratio is 20% (\(\lambda _{max} = 0.2\)). The results based on different values of λmax are shown in Supplementary Fig. 1 as well. For datasets (GSE49355, GSE50760) without normal liver tissue (HN), the expression values of probes/genes in HN were calculated using corresponding samples in GSE41258 (array) or GTEx database (RNA-seq).
We examined the enrichment of tissue-specific gene signatures with odds ratios calculated from four numbers (Supplementary Fig. 1): (1) total number of genes; (2) number of tissue-specific gene signatures (i.e. liver specific genes defined by GTEx database); (3) number of up-regulated genes filtered by the above two criteria; (4) number of liver specific genes filtered by the above two criteria. P value of each odds ratio is calculated using Fisher’s exact test.
We used the same strategy to examine the colon-sigmoid specific gene signatures down-regulated in the metastasis samples (Supplementary Fig. 1).
Kaplan-Meier survival analysis
We downloaded RNA-seq data for COAD patients with clinical data in the TCGA from the NCI Cancer Genomics Hub (CGHub).64 For overall survival (OS) analysis, the event call was derived from the “vital status” parameter. The “time_to_event” is in days, equal to “days_to_death” if the patient died; if the patient is still alive, the time variable is the maximum (“days_to_last_known_alive”, “days_to_last_followup”). For recurrence-free survival (RFS), the event call was derived from the “new_tumor_event_after_initial_treatment” parameter. The “time_to_event” is in days, equals to maximum (“days_to_new_tumor_event_after_initial_treatment”, “days_to_tumor_recurrence”) if there is an event. In the Kaplan-Meier survival analysis, 433 and 203 patients were used for OS and RFS analysis, respectively. We first divided patients into two groups (bottom 20% and top 20%) by median expression level of signature genes, then used Kaplan-Meier survival analysis65 to analyze OS and RFS via the survival package (https://cran.r-project.org/web/packages/survival) in the R environment for statistical computing and computed significance with the log-rank test.
References
Steeg, P. S. Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016).
Yang, J. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 (2004).
Hoshino, A. et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015).
Dupuy, F. et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015).
Bos, P. D. et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–U1137 (2009).
Chen, Q. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–+ (2016).
Wang, H. et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 27, 193–210 (2015).
van der Weyden, L. et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 541, 233–236 (2017).
Chakrabarti, R. et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol. 14, 1212–+ (2012).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1, 571–573 (1889).
Fidler, I. J. Timeline - the pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 3, 453–458 (2003).
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009).
Attanasio, C. et al. Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342, 1241006 (2013).
Nord, A. S. et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 (2013).
Heinz, S., Romanoski, C. E., Benner, C. & Glass, C. K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154 (2015).
Gosselin, D. et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014).
Sheffer, M. et al. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc. Natl Acad. Sci. USA 106, 7131–7136 (2009).
Del Rio, M. et al. Specific extracellular matrix remodeling signature of colon hepatic metastases. PLoS ONE 8, e74599 (2013).
Kim, S. K. et al. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol. Oncol. 8, 1653–1666 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Conway, T. et al. Xenome-a tool for classifying reads from xenograft samples. Bioinformatics 28, i172–i178 (2012).
Bai, D. M. et al. The ATPase hCINAP regulates 18S rRNA processing and is essential for embryogenesis and tumour growth. Nat. Commun. 7, 12310 (2016).
Leibovitz, A. et al. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 36, 4562–4569 (1976).
Flatmark, K., Maelandsmo, G. M., Martinsen, M., Rasmussen, H. & Fodstad, O. Twelve colorectal cancer cell lines exhibit highly variable growth and metastatic capacities in an orthotopic model in nude mice. Eur. J. Cancer 40, 1593–1598 (2004).
Rose, A. A. et al. Osteoactivin promotes breast cancer metastasis to bone. Mol. Cancer Res 5, 1001–1014 (2007).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Hu, Y. et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int. J. Mol. Sci. 16, 1928–1948 (2015).
Cirillo, L. A. et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289 (2002).
Roe, J. S. et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 170, 875–888.e20 (2017).
Yamashiro, T., Aberg, T., Levanon, D., Groner, Y. & Thesleff, I. Expression of Runx1, -2 and -3 during tooth, palate and craniofacial bone development. Mech. Dev. 119, S107–S110 (2002).
Brubaker, K. D., Vessella, R. L., Brown, L. G. & Corey, E. Prostate cancer expression of runt-domain transcription factor Runx2, a key regulator of osteoblast differentiation and function. Prostate 56, 13–22 (2003).
Barnes, G. L. et al. Fidelity of Runx2 activity in breast cancer cells is required for the generation of metastases-associated osteolytic disease. Cancer Res. 64, 4506–4513 (2004).
Enfield, K. S. et al. Elf3 is a novel oncogene frequently activated by genetic and epigenetic mechanisms in lung adenocarcinoma. J. Thorac. Oncol. 8, S351–S352 (2013).
Lee, C. S., Friedman, J. R., Fulmer, J. T. & Kaestner, K. H. The initiation of liver development is dependent on Foxa transcription factors. Nature 435, 944–947 (2005).
Ding, Q. et al. APOBEC3G promotes liver metastasis in an orthotopic mouse model of colorectal cancer and predicts human hepatic metastasis. J. Clin. Invest. 121, 4526–4536 (2011).
Herynk, M. H. et al. Down-regulation of c-Met inhibits growth in the liver of human colorectal carcinoma cells. Cancer Res. 63, 2990–2996 (2003).
Hayashi, Y. et al. Liver enriched transcription factors and differentiation of hepatocellular carcinoma. Mol. Pathol. 52, 19–24 (1999).
Shih, D. Q. et al. Hepatocyte nuclear factor-1 alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 27, 375–382 (2001).
Cheng, Z. et al. Conversion of hepatoma cells to hepatocyte-like cells by defined hepatocyte nuclear factors. Cell Res. https://doi.org/10.1038/s41422-018-0111-x (2018).
Takayama, K. et al. Generation of metabolically functioning hepatocytes from human pluripotent stem cells by FOXA2 and HNF1 alpha transduction. J. Hepatol. 57, 628–636 (2012).
Donaghey, J. et al. Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nat. Genet. 50, 250-+ (2018).
Mansour, M. R. et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377 (2014).
Cohen, A. J. et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun. 8, 14400–14400 (2017).
Zhang, X. H. F. et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154, 1060–1073 (2013).
Huang, W. C. et al. Human osteocalcin and bone sialoprotein mediating osteomimicry of prostate cancer cells: Role of cAMP-dependent protein kinase a signaling pathway. Cancer Res. 65, 2303–2313 (2005).
Hewitt, R. E. et al. Validation of a model of colon cancer progression. J. Pathol. 192, 446–454 (2000).
Naito, S., Voneschenbach, A. C., Giavazzi, R. & Fidler, I. J. Growth and Metastasis of tumor-cells isolated from a human renal-cell carcinoma implanted into different organs of nude-mice. Cancer Res. 46, 4109–4115 (1986).
Cespedes, M. V. et al. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am. J. Pathol. 170, 1077–1085 (2007).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Chandran, U. R. et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7, 64 (2007).
Cai, C. M. et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Invest. 123, 1109–1122 (2013).
Ho, T. H. et al. Differential gene expression profiling of matched primary renal cell carcinoma and metastases reveals upregulation of extracellular matrix genes. Ann. Oncol. https://doi.org/10.1093/annonc/mdw652 (2016).
Salhia, B. et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS ONE 9, e85448 (2014).
Van den Broeck, A., Vankelecom, H., Van Eijsden, R., Govaere, O. & Topal, B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. J. Exp. Clin. Cancer Res. 31, 68 (2012).
Barry, S. et al. S100P is a metastasis-associated gene that facilitates transendothelial migration of pancreatic cancer cells. Clin. Exp. Metastasis 30, 251–264 (2013).
Wang, D. et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474, 390–394 (2011).
Diaferia, G. R. et al. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 35, 595–617 (2016).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Wilks, C. et al. The Cancer Genomics Hub (CGHub): overcoming cancer through the power of torrential data. Database (Oxford) 2014, bau093 (2014).
Efron, B. Logistic-regression, survival analysis, and the Kaplan-Meier curve. J. Am. Stat. Assoc. 83, 414–425 (1988).
Acknowledgements
We thank Yusheng Wu and Prof. Xuerui Yang (Tsinghua University, China) for help with cell proliferation assay. We thank Qian Gao for help with HiSeq 1500 operation. We thank members of the Tsinghua University Animal Resources Center for their care of the mice. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov. This work was supported by a grant from the National Natural Science Foundation of China (81673460, 31771461, 81972798) and funding from Tsinghua-Peking Joint Center for Life Sciences, Beijing Municipal Science & Technology Commission and Fok Ying-Tong Education Foundation. This study was also supported by Beijing Advanced Innovation Center for Structural Biology and Bio-Computing Platform of Tsinghua University Branch of China National Center for Protein Sciences (Beijing). I.G.B. is supported by the U.S. Department of Defense (W81XWH-14-1-0089 and W81XWH-12-1-0358), and the Ovarian Cancer Research Fund Alliance (Liz Tilberis Award) bequest from the Estate of Agatha Pepper Fort.
Author information
Authors and Affiliations
Contributions
D.W. and Z.J.L. conceived and supervised the project with assistance from I.G.B. S.S.T. performed the experiments with the help of M.Y., Q.Y.W., Y.M.Z., S.W.C., Y.C., Q.S.J., Q.Y.G., Z.C. and Y.J.C. Y.E.L. performed the computational analyses with the help from R.Q., Y.M.H., S.S.L., Q.K.L., Q.S.J. and Q.Y.G. D.W., Z.J.L. and S.S.T. wrote the manuscript with contributions from I.G.B., G.W., P.Y.W. and from all the rest of authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Teng, S., Li, Y.E., Yang, M. et al. Tissue-specific transcription reprogramming promotes liver metastasis of colorectal cancer. Cell Res 30, 34–49 (2020). https://doi.org/10.1038/s41422-019-0259-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-019-0259-z
This article is cited by
-
Single-cell transcriptomics reveals multiple chemoresistant properties in leukemic stem and progenitor cells in pediatric AML
Genome Biology (2023)
-
H3K27me3-H3K4me1 transition at bivalent promoters instructs lineage specification in development
Cell & Bioscience (2023)
-
Fe-doped carbon dots: a novel biocompatible nanoplatform for multi-level cancer therapy
Journal of Nanobiotechnology (2023)
-
Identification of multiple organ metastasis-associated hub mRNA/miRNA signatures in non-small cell lung cancer
Cell Death & Disease (2023)
-
Multilevel proteomic analyses reveal molecular diversity between diffuse-type and intestinal-type gastric cancer
Nature Communications (2023)
