
Abstract
α-Synuclein (α-syn) amyloid fibrils are the major component of Lewy bodies, which are the pathological hallmark of Parkinson’s disease (PD) and other synucleinopathies. High-resolution structure of α-syn fibril is important for understanding its assembly and pathological mechanism. Here, we determined a fibril structure of full-length α-syn (1–140) at the resolution of 3.07 Å by cryo-electron microscopy (cryo-EM). The fibrils are cytotoxic, and transmissible to induce endogenous α-syn aggregation in primary neurons. Based on the reconstructed cryo-EM density map, we were able to unambiguously build the fibril structure comprising residues 37–99. The α-syn amyloid fibril structure shows two protofilaments intertwining along an approximate 21 screw axis into a left-handed helix. Each protofilament features a Greek key-like topology. Remarkably, five out of the six early-onset PD familial mutations are located at the dimer interface of the fibril (H50Q, G51D, and A53T/E) or involved in the stabilization of the protofilament (E46K). Furthermore, these PD mutations lead to the formation of fibrils with polymorphic structures distinct from that of the wild-type. Our study provides molecular insight into the fibrillar assembly of α-syn at the atomic level and sheds light on the molecular pathogenesis caused by familial PD mutations of α-syn.
Similar content being viewed by others


Introduction
The formation of amyloid fibrils is a common pathological hallmark of human neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD).1,2,3 The insoluble, non-crystalline and heterogeneous nature of amyloid fibrils had made it difficult to decipher their atomic structures by X-ray crystallography or NMR spectrometry.4,5,6,7 The application of cryo-electron microscopy (cryo-EM) technology has recently succeeded in obtaining the atomic structures of the amyloid fibrils formed by Aβ and Tau,8,9 two key amyloid-forming proteins involved in AD.
α-Syn fibrils play a central role in PD, dementia with Lewy bodies, multiple system atrophy, and other synucleinopathies.10,11 Deposition of α-syn amyloid fibrils in Lewy bodies is a diagnostic histological hallmark of these diseases.10,12,13 Duplication and triplication of SNCA, the gene encoding α-syn, as well as multiple α-syn missense mutations have been identified in association with early onset of PD,14,15,16,17,18,19,20,21,22 suggesting a causal role of α-syn dysfunction and aggregation in mediating neurodegeneration. However, the fibril structure of α-syn has not yet been determined, nor do we understand the molecular mechanism of missense α-syn mutations involved in promoting PD pathogenesis.
α-Syn fibrils may present different morphology and pathology under different conditions.23,24 Recently, different endogenous α-syn strains from glial cytoplasmic inclusions and Lewy bodies derived from the brains of patients with multiple system atrophy and Lewy body disease have been reported to exhibit distinctive conformation and pathology.25 In this work, we determined an amyloid fibril structure of full-length α-syn at an overall resolution of 3.07 Å by cryo-EM. Together with atomic-force microscopy (AFM), our study demonstrates that PD familial mutations disrupt the wild-type amyloid fibril and induce the formation of distinctive fibril structures, which may be related to the early onset of PD.
Results
Pathological amyloid fibrils formed by recombinant human α-syn
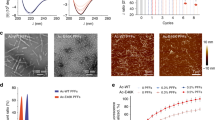
The fibril was formed by recombinant full-length α-syn (1–140) with N-terminal acetylation. N-acetylation has been reported to be a general post-translational modification of native α-syn.26 The recombinant α-syn was incubated in a buffer containing 50 mM Tris, pH 7.5, 150 mM KCl and pre-formed fibril seeds at 37 °C with agitation for 3 days. Under this condition, α-syn formed homogeneous and unbranched fibrils as shown by negative-staining transmission electron microscopy (TEM) (Fig. 1a). The X-ray diffraction of aligned fibrils showed a typical cross-β diffraction pattern with the strongest peaks at 4.8 Å and near 10 Å (Supplementary information, Fig. S1). These fibrils induced cytotoxicity of HEK 293T cells in a dose-dependent manner (Fig. 1b). Addition of these fibrils in the culture of rat primary neurons seeded the aggregation of endogenous S129-phosphorylated α-syn, indicating the capability of transmission and propagation of these fibrils (Fig. 1c).

Pathological fibrils of recombinant full-length α-syn. a A negative-staining TEM image of the α-syn fibrils. b Cytotoxicity of the fibrils to HEK 293T cells assessed by the MTT assay. Cells were treated with indicated concentration of α-syn fibrils for 24 h. The fibril concentrations were calculated from the monomer concentration before fibrillation minus the monomer concentration in the supernatant after fibrillation. Data were normalized to cells treated with PBS buffer (gray bar). Values are means ± SD of 6 replicates. Statistical significance was measured by t-test. ** represents P < 0.01, *** represents P < 0.001. c Extracellular α-syn fibrils induce endogenous α-syn aggregation in rat primary cortical neurons. Primary cortical neurons were treated with 0, 10, 100 nM pre-formed α-syn fibril seeds (PFF) at 8 day in vitro (DIV) for 12 days, respectively. Fixed neurons were stained for DAPI (blue), the dendritic marker MAP2 (green) and S129-phosphorylated α-syn (red) at 20 DIV
Cryo-EM structure of the α-syn fibril
We reconstructed the 3D density map of the α-syn fibril by cryo-EM (Supplementary information, Table S1 and Fig. S2). The density map showed a left-handed helix with a width of ~ 10 nm and a helical pitch of 239 nm (Fig. 2a). The fibril is composed of two protofilaments intertwining along an approximate 21 screw axis. α-Syn subunits in each protofilament stack along the fibril axis with a helical twist of −0.72° and rise of 4.78 Å in good agreement with the X-ray fibril diffraction at ~4.8 Å (Fig. 2b, c; Supplementary information, Fig. S1). Two opposing subunits are dimerized with an approximate 21 screw symmetry (Fig. 2c). Thus, there are 500 layers of dimers per helical pitch. The overall resolution of the density map is 3.07 Å and most side chains are well visible (Fig. 2c; Supplementary information, Fig. S3). Local resolution estimation shows that the cryo-EM density in the dimer interface has the highest resolution of 2.90 Å, which indicates that the dimer interface is the most stable region of the fibril (Fig. 3).

Cryo-EM reconstruction density map of the α-syn fibril. a Calculated density of a single fibril. Fibril width and twisting pitch are indicated. “Pitch” here refers to a 360° helical turn. The two intertwining protofilaments are colored in purple and yellow, respectively. b A side view of the density map. The density map was calculated by applying the helical symmetry to eight subunits in the central region of the 3D reconstruction. The helical rise is indicated. c A top view of the EM density map. The twist angle is graphically illustrated

Local resolution estimation. EM reconstruction map colored based on local resolutions. The central region of the map exhibits the highest resolution, which was used to build the structure model. The colored map demonstrates the local resolution ranging from 2.9 to 6.0 Å
According to the cryo-EM density map, we were able to unambiguously build the atomic model for residues 37–99 (Fig. 4a). The model showed that α-syn folds into a β-strand-rich architecture with a Greek key-like topology consistent with the previously reported solid-state NMR structure of an α-syn protofilament27 (Fig. 4b; Supplementary information, Fig. S4). The hydrophobic residues fold inside to form a stable core with the hydrophilic residues at the periphery (Fig. 4c). The dimer interface is composed of residues 50–57 (50HGVATVAE57) from opposing α-syn subunits and contains a typical parallel in-register steric-zipper configuration (Fig. 5a, b). The steric-zipper interface is stabilized by the hydrophobic interactions between the side chains of A53 and V55 (Fig. 5b). E57 and H50 at the edge of the steric zipper core, together with K45, form electrostatic interactions that block the access to the hydrophobic zipper interface and further stabilize the dimer interface (Fig. 5b, c).

Atomic structure of the α-syn fibril. a The cryo-EM density map with structural model. b The structure model of multiple subunits shown in different views. Two protofilaments are colored in purple and yellow, respectively. The fibril axis is indicated. The distance between neighboring subunits in one protofilament is indicated. The β strands are numbered and labeled. c The surface of two opposing subunits is shown according to the hydrophobicity of the residues. The structure is shown in the same view as a

Important interactions in the α-syn fibril structure. a A top view of the fibril structure with two dimeric subunits colored in purple and yellow, respectively. The zoom-in views of boxes i–iv are shown in b–e. b The dimer interface comprises residues 50–57. The side chains involved in the interface are shown as spheres in the top view. The side view shows the parallel in-register steric zipper in the dimer interface. Symbol ’ represents residues from opposing subunits. c Electrostatic interactions among H50, K45 and E57’. The distances between side chains are indicated. d, e Salt bridges formed by K58 and E61, and E46 and K80, respectively. Residues identified as familial PD mutation sites are colored in red
Familial PD mutation sites are critical for the fibril structure
The fibril structure remarkably showed that four out of six PD familial mutations identified to date, i.e. H50Q, G51D and A53T/E,18,19,20,21 led to changes in amino-acid residues localized at the dimer interface (Fig. 5b). Based on the fibril structure, the G51D and A53T/E mutations introduce hydrophilic or even charged residues into the hydrophobic steric-zipper interface, which would disrupt the dimer interface; while the H50Q mutation may break the electrostatic interactions among H50, K45 and E57 from the opposing subunit (Fig. 5c). Note that residues 50–57, that form the dimer interface and contain these mutation sites, are highly flexible in the protofilament structure indicated by solid-state NMR27 (Supplementary information, Fig. S4). Thus, the formation of dimer leads to the conformation stabilization of residues 50–57, which also suggests the importance of the dimer interface in the over-all fibril assembly.
In addition, there are another two pairs of electrostatic interactions, i.e., K58-E61 and E46-K80, that form intra-molecular salt bridges, important for the folding of the Greek-key topology (Fig. 5d, e). The salt bridge of K58 and E61 constrains the turn between β3 and β4, whereas that of E46 and K80 fixes β1 and β6. Notably, E46K is also a PD familial mutation.17 According to the structure, the E46K mutation would break the salt bridge between E46 and K80 to destabilize the fibril structure (Fig. 5e). The prevalent ability of familial PD mutations to interfere with the formation of this structure argues the pathophysiological significance of the α-syn fibril.
Familial PD mutations alter the structure of α-syn fibrils
Since these five familial PD mutations (H50Q, G51D, A53T/E, and E46K) may disrupt the wild-type fibril assembly, yet these mutants can still form amyloid fibrils, we considered the possibility that these mutant fibrils might exhibit features distinguishable from that of the wild-type, as previously suggested regarding the molecular differences of mutant fibrils.28,29 To further structurally characterize and compare the morphology of the mutant fibrils, we conducted AFM to measure the helical parameters of the mutant fibrils. The result showed that under the same conditions, these five disease mutations resulted in the formation of fibrils with distinctive morphologies characterized by different fibril width, ranging from ~7 to 10 nm, and different helical periodicity (Fig. 6; Supplementary information, Fig. S5). The wild-type fibril features a left-handed helix with a width of 10.5 ± 0.8 nm and a periodicity of 120 ± 14 nm (half pitch), in a good agreement with the cryo-EM structure (Figs. 1a, 6b, c). The fibrils formed by A53T and E46K mutants deviated from that of the wide-type with a reduced helical periodicity of 60–80 nm. In addition, the E46K fibril presents as a right-handed fibril distinct from the left-handed chirality of the wild-type and A53T fibrils (Fig. 6c; Supplementary information, Fig. S5). In contrast to the twisted fibrils, the fibrils formed by A53E, G51D and H50Q mutants exhibited no visible twist. Thus, the fibrils formed by these mutants might be straight or their helical twists are too large to be measured by AFM. Finally, the fibrils formed by A30P mutant showed no difference from that of the wild-type measured by AFM (Fig. 6c; Supplementary information, Fig. S5), which is in agreement with the atomic fibril structure, as A30 is not involved in the formation of fibril core (Fig. 4a), as well as a previous study which suggests that A30 might be involved in membrane binding.30 Thus, our results suggest that PD familial mutations, except for A30P, lead to polymorphic fibril structures with features distinct from that of the wild type, which may render different aggregation kinetics and fibril dynamics, and underlie neuropathology of α-syn.29,31,32,33,34

Different structures of α-syn fibrils formed by various familial PD mutants. a Graphic illustration of fibril helical parameters. AFM measures the periodic spacing that for fibrils composed of two protofilaments, equals to half of the helical pitch. b Characterization of the helical parameters of the WT α-syn fibril by AFM. 2D and 3D AFM images are shown. Analyses of the cross section and along the fibril are indicated with black and red lines, respectively. Data for the mutant fibrils are shown in Supplementary information, Fig. S5. c Table on the left shows the helical parameters of WT and mutant fibrils. Values are means ± SD of 8 replicates. ND represents “not detectable”. A graphic presentation of the values is shown on the right. PD mutation sites involved in the formation of the fibril core are shown in orange. A30 that is not involved in the structure is in light gray
Discussion
In this study, we report the atomic structure of a full-length α-syn fibril determined by cryo-EM. The advancement of cryo-EM technology has made it possible to determine the atomic and complete structure of amyloid fibrils, such as the successes in determining the fibril structures of Aβ, Tau8,9 and α-syn (this work). However, the flexibility including the curvature of the fibrils and the variation of the helical periodicity, represented by the unevenly distributed resolution in the 3D reconstruction (Fig. 3), is a challenge for determining the fibril structure at the atomic resolution. Another challenge is that there is only a single pair of visible layer lines in the power spectrum due to the long periodicity (several hundreds of nanometers) and very small subunit (~4.8 Å along the helical axis), which leads to an extreme difficulty in determining the helical symmetry of the amyloid fibrils. We introduced a method to solve this problem, which was based on the repeated segments identified by the 2D classification (see Materials and methods), and succeeded in revealing the near-atomic resolution structure of the α-syn fibril at 3.07 Å. To determine the chirality, we used AFM as an independent tool which is shown to be very useful (Fig. 6). We noticed that during the review process of our work, Stahlberg and colleagues published the cryo-EM structure of a fibril formed by C-terminally truncated α-syn (1–121).35 The structure showed an opposite chirality–a right-handed helix rather than a left-handed helix as we determined in our work. The helical rise and twist angle of the two structures are also different. It is possible in consideration of fibril polymorphism, that the differences between the two structures come from the different α-syn constructs (i.e., a truncated α-syn versus a full-length α-syn).
The amyloid fibril of full-length human α-syn studied in this work, although was formed in vitro, is pathological with high cytotoxicity and capability of transmission and propagation in neurons. Importantly, the key residues that maintain this structure, include four of the five PD familial mutation sites, highlighting the pathophysiological significance of this fibril structure. We further describe the polymorphous structures of familial PD mutant fibrils and demonstrate that the mutations result in distinctive fibril structures that might be involved in the early onset of PD. Our study sheds light on the understanding of α-syn fibril assembly and the relationship between fibril structure and PD pathology.
Materials and methods
Preparation of WT and mutant α-syn with N-terminal acetylation
The gene encoding human wild-type α-syn (1–140) was inserted into pET22 vector. The six familial PD mutations of α-syn including A30P, E46K, H50Q, G51D, A53E and A53T were generated by a Quick Change site-directed mutagenesis kit. The α-syn constructs were transformed into E. coli expression cell line BL21 (DE3). Co-expression of fission yeast N-acetyltransferase complex B with α-syn was used to generate N-terminally acetylated α-syn.36 Protein expression was induced by 1 mM isopropyl-1-thio-D-galactopyranoside (IPTG) at 37 °C for 4 h. Protein purification was carried out at 4 °C. Bacteria were firstly harvested and lysed in 100 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride. The cell debris was discarded by centrifugation at 16,000× g for 30 min. The supernatant was heated for 10 min at 100 °C. After 30 min centrifugation at 16,000× g, streptomycin (20 mg/mL) was added to remove nucleic acid, followed by centrifugation at 16,000× g for 30 min. 2 M HCl was added to the supernatant dropwise until the pH reached 3.5, followed by another centrifugation of 16,000× g for 30 min. The protein supernatant was then dialyzed into the buffer (25 mM Tris–HCl, pH 8.0) overnight. The dialyzed sample was purified by Q column (GE Healthcare, 17-5156-01), followed by Superdex 75 (GE Healthcare, 28-9893-33).
α-Syn fibril formation
The acetylated α-syn including WT and mutations was concentrated to 500 µM in fibril growth buffer (50 mM Tris, pH 7.5, 150 mM KCl, 0.05% NaN3). Protein sample was incubated at 37 °C with agitation (900 rpm) in ThermoMixer (Eppendorf). After 3 days, the fibrillar sample was diluted to 25 µM, sonicated for 30 s (1 s on, 1 s off) on ice. The α-syn fibril seeds obtained by sonication (0.5%, v/v) were added to 25 µM acetylated α-syn monomer. The mixture was further incubated at 37 °C with agitation (900 rpm) for 3 days to obtain the mature fibrils for cryoEM, X-ray fibril diffraction, and AFM studies. To prepare α-syn fibril seed for 293T cell viability assay and primary neuron cell transmission assay, the mature fibrils were concentrated by centrifugation, washed with PBS and sonicated for 30 s (1 s on, 1 s off) on ice.
Cell viability assay
Cell Titer 96 non-radioactive cell proliferation kit (Promega, 4100) was used to measure cell viability. Trypsin-digested HEK 293T cells (6,000 cells/well) were plated to a 96-well plate with total volume of 90 µL. HEK 293T cells were cultured in DMEM media containing 10% FBS and 1% penicillin-streptomycin at 37 °C, 5% CO2 for 18 h. Then, 10 µL of fibril seeds were added and incubated for 24 h at the final concentrations of 0, 0.01, 0.1, 1 and 10 µM. Next, dye solution and Solution/Stop Mix were sequentially added into each well and mixed following the manufacturer’s instructions. Finally, the absorbance was recorded at 570 nm and 650 nm (background) with a Varioskan Flash multimode reader (Thermo Scientific). Each experiment contains six replicates. Statistical significance was determined by Student’s t-test using GraphPad Prism.
Primary neuronal cultures and cell transmission assay
Pregnant Sprague-Dawley (SD) rats were purchased from Shanghai SIPPR BK Laboratory Animals Ltd. (Shanghai, China). All experiments on mice were performed according to the protocols approved by Animal Care Committee of the Interdisciplinary Research Center on Biology and Chemistry (IRCBC), Chinese Academy of Sciences (CAS). Primary cortical neurons were prepared from E16–E18 embryos. Dissociated cortical neurons were plated onto coverslips with poly-D-lysine coated in 24-well plate at a density of 150,000 cell/well. Primary cortical neurons were maintained in Neurobasal media replenished with B-27 supplement, 0.5 mM L-glutamine and 1% penicillin-streptomycin at 37 °C, 5% CO2. The α-syn seeds or PBS were added into culture medium at 8 day in vitro (DIV). Neurons were treated with PBS or fibril seeds for 12 days with the final α-syn concentrations of 0, 10, and 100 nM as indicated. Treated neurons were harvested for immunocytochemistry at 20 DIV. Each experiment was performed in triplicates and repeated three times.
Immunofluorescence imaging
Primary cortical neurons cultured on coverslips were fixed with 4% paraformaldehyde and 4% sucrose in PBS at 20 DIV, followed by permeabilized with 0.15% Triton X-100. The sample was blocked in blocking buffer (5% bovine serum albumin in PBS) for 1 h at room temperature. Neurons were incubated with anti-p129-α-syn (Abcam, ab51253, 1:1,000 dilution), anti-MAP2 (Abcam, ab5392, 1:1,000 dilution) antibodies in blocking buffer at 4 °C overnight. After washing with PBS for three times, neurons were stained with appropriate Alexa Fluor 488- or Alexa Fluor 568-conjugated secondary antibodies (Life Technologies, A11039 and A11036, respectively, 1:1,000 dilution) for 1 h at room temperature. After washing with PBS for another three times, coverslips were then mounted onto glass slides with ProLong Gold antifade reagent with 4′,6-Diamidino-2-Phenylindole (DAPI). The fluorescent images were acquired by an SP8 confocal microscope.
Electron microscopy
WT and mutant α-syn fibrils were examined by negative-staining electron microscopy. A drop of 5 μL of the fibril solution was loaded onto a glow discharged grid with continuous carbon film (Beijing Zhongjingkeyi Technology Co., Ltd.). The grid was washed by 5 μL distilled water and followed by 5 μL 3% w/v uranyl acetate, and then stained with 5 μL 3% w/v uranyl acetate for 45 s. The excess buffer was removed by filter paper. Finally, the grid was dried in air. Tecnai T12 microscope (FEI Company) operated at 120 kV was used for imaging.
An aliquot of 4 μL of 25 μM α-syn fibril solution was applied to a glow-discharged holey carbon grid (Quantifoil R1.2/1.3, 300 mesh), blotted for 6 s, and plunge-frozen in liquid ethane using FEI Vitrobot Mark IV. The cryo-EM micrographs were acquired on a FEI Titan Krios microscope operated at 300 kV and equipped with a Gatan K2 Summit camera. 32 frames in super-resolution mode of the K2 camera were recorded for each exposure of 8 s. The electron dose rate was set to ~8.2 counts/physical pixel/s. The total dose is ~50 e/Å2. The pixel size of the super-resolution images is 0.66 Å. The defocus ranging from −1.5 μm to −2.0 μm was used. The automated data collection software, eTas (developed by Bo Shen at Xueming Li Lab), was used for all data collection.
Image pre-processing
MotionCorr237 was used for beam-induced motion correction and weighting the radiation damage. The aligned and integrated images were binned by two folds, yielding a pixel size of 1.32 Å, and used for further processing. CTFFIND 4.1.538 was used to estimate defocus parameters. 2D and 3D classifications, 3D refinement were performed using RELION 2.1.39
Helical parameters determination
The helical parameters, twist angle and rise, were first estimated based on the following image analysis, and then refined during 3D reconstruction using Relion 2.1.
The rise was measured based on the position of a strong layer line (pointed in Supplementary information, Fig. S2) in the power spectrum of a selected filament, corresponding to ~4.8 Å. The rise is identical to the distance of adjacent β-strands in β-sheet.
The twist angle was estimated based on the measurement to the length of a helical pitch. Because the length of a pitch is much longer than the size (equal to the rise, 4.8 Å) of a subunit along the fibril, we can ignore the characters of each subunit in the low-contrast cryo-EM image and approximately analyze the fibril as a continuous thread. Considering the image is a projection of the helix, the image features along the filament are approximately repeated at a distance of half pitch. By 2D classification of the fibril segments, we were able to find the repeated segments (belong to the same 2D class) on the same fiber. Measuring the distance between two adjacent segments gave the length of half pitch. Statistics of all fibrils were performed, which gave the average length of half pitch of ~115 nm corresponding to ~480 asymmetric units per pitch. Accordingly, the twist angle was calculated as ~0.75°. The examination by AFM was performed and confirmed the length of a pitch (Fig. 6). And AFM analysis to the texture and brightness change on the surface of the fibrils indicated that the fibril took a left-handed helical structure. Therefore, the twist angle should be −0.75°.
By 2D class averages of the filament segments, we observed the subunits on two sides of the filament took a staggered arrangement. Therefore, each asymmetric unit in our fibrils may also contain two α-syn subunits with an approximate 21 screw symmetry. Accordingly, the helical parameters are changed to the rise of 4.8/2 = 2.4 Å and the twist angle of (360° – 0.75°)/2 = 179.63°.
Helical reconstruction
In total 2,556 fibrils were picked manually using RELION from 148 micrographs, and then were segmented with a step of 28.8 Å into 119,703 particles with a box size of 264 Å. 2D classification were performed by RELION. 102,673 segments in the classes showing clear structure features were selected for further processing.
A cylinder model was created using relion_helix_toolbox subroutine in RELION and low-pass filtered to 60 Å as the initial model of the 3D classification. 3D classification to a single class (K = 1 in RELION) was carried out with helical symmetry optimization enabled. The newly generated density map was used for further 3D classifications with 3–5 classes. 43,677 segments were selected by the 3D classifications, and subjected to the final 3D refinement with helical symmetry optimization enabled. The final helical parameters were optimized to the rise of 2.39 Å and the twist angle of 179.64°, which is corresponding to the rise of 4.78 Å and −0.72° for subunits in one protofilament. The reconstruction map was sharpened with a B-factor of −91 Å2 using post-processing subroutine in RELION. And the overall resolution was reported at 3.07 Å based on the gold-standard FSC = 0.143 criteria. The local resolution was estimated by ResMap.40
Model building and refinement
A homology model based on the NMR structure (PDB entry code: 2N0A)27 was built and modified by COOT.41 The model with 6 adjacent layers (12 promoters) was refined using the real-space refinement in PHENIX.42,43 The subunit dimers in the middle of 6 layers was extracted and used as the final model.
Atomic force microscopy (AFM)
All of the α-syn fibril samples were prepared in the fibril growth buffer to a final concentration of 25 μM. 7 μL fibril solution was incubated on a freshly cleaved mica surface for 3 min at room temperature, followed by washing by Milli-Q water to remove unbound fibrils. The sample was then dried with nitrogen flow and probed in air by using Multimode 8 scanning probe microscope (Bruker) on scanAsyst mode. Measurements were carried out using SNL-10 probe with a spring constant of 0.35 N/m and a resonance frequency of 65 kHz (Bruker). Images were acquired at a fixed resolution (512 × 512 data points) with scan rate at 1 Hz. Images and height were processed with supplied software (NanoScope Analysis 1.5, Bruker).
Accession codes
Density maps of α-syn are available through EMDB with entry code: EMD-6988. Model of α-syn is deposited in the Protein Data Bank with entry code: 6A6B. The data that support the findings of this study are available from the corresponding authors upon request.
References
Goedert, M., Clavaguera, F. & Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325 (2010).
Jucker, M. & Walker, L. C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51 (2013).
Knowles, T. P., Vendruscolo, M. & Dobson, C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014).
Tycko, R. Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 23, 1528–1539 (2014).
Riek, R. & Eisenberg, D. S. The activities of amyloids from a structural perspective. Nature 539, 227–235 (2016).
Qiang, W., Yau, W. M., Lu, J. X., Collinge, J. & Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 541, 217–221 (2017).
Rodriguez, J. A. et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490 (2015).
Gremer, L. et al. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 358, 116–119 (2017).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Spillantini, M. G. et al. alpha-Synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Goedert, M. & Spillantini, M. G. Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol. Psychiatry 3, 462–465 (1998).
Goedert, M., Spillantini, M. G., Del Tredici, K. & Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24 (2013).
Moore, D. J., West, A. B., Dawson, V. L. & Dawson, T. M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 28, 57–87 (2005).
Chartier-Harlin, M. C. et al. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
Singleton, A. B. et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841 (2003).
Krüger, R. et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18 106–108 (1998).
Zarranz, JuanJ. et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neuro. 55, 164–173 (2004).
Appel-Cresswell, S. et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813 (2013).
Lesage, S. et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neuro. 73, 459–471 (2013).
Pasanen, P. et al. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180.e2181–2185 (2014).
Polymeropoulos, M. H. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997).
Goedert, M., Jakes, R. & Spillantini, M. G. The synucleinopathies: twenty years on. J. Park. Dis. 7, S53–S71 (2017).
Peelaerts, W. et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 (2015).
Guo, J. L. et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117 (2013).
Peng, C. et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563 (2018).
Theillet, F. X. et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530, 45–50 (2016).
Tuttle, M. D. et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat. Struct. Mol. Bio. 23, 409–415 (2016).
Choi, W. et al. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 576, 363–368 (2004).
Khalaf, O. et al. The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 289, 21856–21876 (2014).
Jo, E., Fuller, N., Rand, R. P., St George-Hyslop, P. & Fraser, P. E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 315, 799–807 (2002).
Flagmeier, P. et al. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of alpha-synuclein aggregation. Proc. Natl Acad. Sci. USA 113, 10328–10333 (2016).
Reynolds, N. P. et al. Mechanism of membrane interaction and disruption by alpha-synuclein. J. Am. Chem. Soc. 133, 19366–19375 (2011).
Fares, M. B. et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509 (2014).
Lazaro, D. F. et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS ONE 10, e1004741 (2014).
Guerrero-Ferreira, R. et al. Cryo-EM structure of alpha-synuclein fibrils. eLife 7, e36402 (2018).
Johnson, M., Coulton, A. T., Geeves, M. A. & Mulvihill, D. P. Targeted amino-terminal acetylation of recombinant proteins in E. coli. PLoS ONE 5, e15801 (2010).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63 (2014).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Afonine, P. V., Headd, J. J., Terwilliger, T. C. & Adams, P. D. New tool: phenix.real_space_refine. Comp. Cryst. Newsl. 4, 43–44 (2013).
Acknowledgements
This work was supported by the Major State Basic Research Development Program (2016YFA0501902 to C.L., X.L. and 2016YFA0501102 to X.L.), the National Natural Science Foundation (NSF) of China (31470748 to C.L. and 31570730 and 31722015 to X.L.), the Chinese Academy of Sciences (to C.L.), The “1000 Talents Plan” of China to C.L. and X.L., the Advanced Innovation Center for Structural Biology (to X.L.), Tsinghua-Peking Joint Center for Life Sciences (to X.L.), Shanghai Pujiang Program (to D.L.). We acknowledge Tsinghua University Branch of China National Center for Protein Sciences Beijing for providing facility supports in cryo-EM.
Author information
Authors and Affiliations
Contributions
X.L., C.L., D.L., Y.L. and C.Z. designed the project. Y.L. and F.L. performed the cryo-electron microscopy experiments. Y.L. and Z.Luo built and refined the structure model. C.Z., X.G. and X.Z. performed the biochemical and cellular assays. Z.Liu performed the AFM experiments. Y.L., C.Z., D.L., X.L. and C.L. analyzed the data and contributed to manuscript discussion and editing. D.L. and X.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Li, Y., Zhao, C., Luo, F. et al. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res 28, 897–903 (2018). https://doi.org/10.1038/s41422-018-0075-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-018-0075-x
Keywords
This article is cited by
-
Overlaps and divergences between tauopathies and synucleinopathies: a duet of neurodegeneration
Translational Neurodegeneration (2024)
-
Misfolded protein oligomers: mechanisms of formation, cytotoxic effects, and pharmacological approaches against protein misfolding diseases
Molecular Neurodegeneration (2024)
-
Phosphorylation and O-GlcNAcylation at the same α-synuclein site generate distinct fibril structures
Nature Communications (2024)
-
Structure of alpha-synuclein fibrils derived from human Lewy body dementia tissue
Nature Communications (2024)
-
Peptide-based approaches to directly target alpha-synuclein in Parkinson’s disease
Molecular Neurodegeneration (2023)
