
Abstract
In the human body, 50–70 billion cells die every day, resulting in the generation of a large number of apoptotic bodies. However, the detailed biological role of apoptotic bodies in regulating tissue homeostasis remains unclear. In this study, we used Fas-deficient MRL/lpr and Caspase 3−/− mice to show that reduction of apoptotic body formation significantly impaired the self-renewal and osteo-/adipo-genic differentiation of bone marrow mesenchymal stem cells (MSCs). Systemic infusion of exogenous apoptotic bodies rescued the MSC impairment and also ameliorated the osteopenia phenotype in MRL/lpr, Caspase 3−/− and ovariectomized (OVX) mice. Mechanistically, we showed that MSCs were able to engulf apoptotic bodies via integrin αvβ3 and reuse apoptotic body-derived ubiquitin ligase RNF146 and miR-328-3p to inhibit Axin1 and thereby activate the Wnt/β-catenin pathway. Moreover, we used a parabiosis mouse model to reveal that apoptotic bodies participated in the circulation to regulate distant MSCs. This study identifies a previously unknown role of apoptotic bodies in maintaining MSC and bone homeostasis in both physiological and pathological contexts and implies the potential use of apoptotic bodies to treat osteoporosis.
Similar content being viewed by others


Introduction
Apoptosis is programmed cell death involving distinct cell shrinkage, chromatin condensation, and plasma blebbing.1 It is necessary for over 50 billion cells to undergo apoptosis each day in the human body to maintain tissue homeostasis.2,3,4,5,6,7 Excessive apoptosis causes the progression of several neurological disorders such as Alzheimer’s, Parkinson’s, and Huntington’s diseases.8 Conversely, absence of apoptosis may be associated with autoimmune diseases, such as systemic lupus erythematosus (SLE),9,10 also known as lupus, in which the body’s immune system mistakenly attacks healthy tissue in many parts of the body. Apoptosis generates a large number of apoptotic bodies containing a variety of cellular components including microRNAs, mRNAs, DNAs, proteins, and lipids.11,12,13 Apoptotic bodies are engulfed by macrophages, dendritic cells, epithelial cells, endothelial cells, and fibroblasts, and corpses are subsequently internalized, ingested, and degraded in the lysosomes.14,15,16,17,18 Since engulfment of apoptotic cells may prime macrophages to generate molecular memory, it is speculated that apoptotic bodies may facilitate intercellular communication through transfer of cellular factors.13,19 However, whether apoptotic processes and apoptotic body production regulate stem cell function is largely unknown.
Bone marrow mesenchymal stem cells (MSCs) are non-hematopoietic stem cells with the capacity for self-renewal and multipotent differentiation that maintain bone marrow homeostasis.20,21 MSCs can differentiate into osteoblasts, adipocytes, fibroblasts, chondrocytes and non-mesenchymal cell types.22,23 In addition, MSCs can inhibit proliferation and function of several major immune cells, such as T and B lymphocytes, dendritic cells and natural killer cells, to regulate immune responses. Therefore, MSCs have been identified as a promising cell source for tissue regeneration and immune therapies.24,25,26,27,28 In this study, we show that MSCs phagocytose apoptotic bodies and reutilize multiple cellular factors from apoptotic bodies to maintain their stem cell properties.
Results
Staurosporine-induced acceleration of apoptotic body production rescued impaired MSCs in apoptosis-deficient mouse models
Apoptosis can be initiated by extrinsic death receptor or intrinsic mitochondrial pathways, both resulting in the same execution procedure.29 The FasL/Fas pathway is a well-characterized extrinsic apoptosis pathway, and caspase 3 is critical for the final execution phase of apoptosis. MRL/lpr mice are homozygous for Fas mutation, which show functional deficient of Fas.30,31,32 In this study, we used Fas-deficient MRL/lpr and caspase 3 knockout (Casp3−/−) mice as apoptosis-deficient models to examine how apoptosis affects bone marrow MSCs. First, we developed an in vivo apoptosis-tracing assay to assess apoptosis rates in the bone marrow. After FITC-labeled Annexin V was intravenously injected, the number of FITC-Annexin V-positive cells in the femoral bone marrow was calculated under a fluorescent microscope at two hours post-injection. The apoptosis rates were significantly reduced in MRL/lpr and Casp3−/− mice when compared with wild-type controls (Fig. 1a; Supplementary information, Figure S1a). During the apoptotic process, cells release a large number of 1–5 µm extracellular vesicles, named apoptotic bodies.33,34,35,36 We next asked whether the number of apoptotic bodies was reduced in apoptosis-deficient mouse models. After sequential centrifugation of whole bone marrow,37,38 we found that there were significantly fewer 1–5 µm extracellular vesicles in the resulting pellet than in the control groups (Fig. 1b; Supplementary information, Figure S1b). These extracellular vesicles were positive for Annexin V and negative for CD62P antibody staining, as assessed by flow cytometric analysis. 1 and 10 μm beads were used to define gate size, and 4.5 μm counting beads were used to calculate the number of extracellular vesicles. In order to exclude potential platelet contamination, apoptotic bodies were defined as 1–5 μm in size as well as Annexin V positive and platelet marker CD62P negative (Fig. 1c). The number of apoptotic bodies was calculated according to the number of Annexin V-positive and CD62P-negative cells versus the number of counting beads by the absolute cell counting method (Supplementary information, Figure S1c).39 Flow cytometric analysis confirmed that the number of apoptotic bodies was decreased in the bone marrow of MRL/lpr and Casp3−/− mice when compared to the control groups (Fig. 1c; Supplementary information, Figure S1d).

Staurosporine-induced elevated levels of apoptotic bodies rescued impaired MSCs in MRL/lpr Mice. a 3 ng of Staurosporine (STS) was intraperitoneally administered to MRL/lpr mice twice a week for 4 weeks (total of 8 injections). Annexin V was injected via the tail vein 2 h prior to sample collection to assess the apoptosis rate in the bone marrow. Immunostaining showed that STS injection rescued the reduced number of apoptotic cells in the bone marrow of MRL/lpr mice (n = 5). b Fewer apoptotic body-sized extracellular vesicles were observed in pellets derived from MRL/lpr mice (n = 5) than in those collected from the control mice. After 4 weeks of STS treatment, the amount of apoptotic body-sized extracellular vesicles was increased in MRL/lpr mice. c The number of apoptotic bodies was counted by flow cytometry. 1 and 10 μm diameter beads and 4.5 μm counting beads were used to gate 1–5 μm-sized microvesicles. Annexin V+ and CD62P− events were counted as apoptotic bodies. The flow cytometric calculation showed that the number of apoptotic bodies from the bone marrow of MRL/lpr mice (n = 5) was reduced compared to the wild-type control group. After 4 weeks of STS treatment, the number of apoptotic bodies from the bone marrow of MRL/lpr mice (n = 5) was increased. d BrdU labeling and continuous passage assay showed that MRL/lpr MSCs (n = 5) had reduced proliferation and population doubling rates when compared to the wild-type control group. After 4 weeks of STS treatment, proliferation and population doubling rates were increased in MRL/lpr MSCs (n = 5). e Compared to wild-type MSCs, MRL/lpr MSCs showed reduced capacities to form mineralized nodules when cultured under the osteogenic inductive conditions, assessed by alizarin red staining (n = 5), and reduced expression of osteogenic markers Runx2 and ALP, assessed by Western blot. After 4 weeks of STS treatment, reduced mineralized nodule formation and expression of Runx2 and ALP were rescued in MRL/lpr MSCs (n = 5). f MRL/lpr MSCs showed reduced capacities to form new bone when implanted into immunocompromised mice subcutaneously using HA/TCP as a carrier. After 4 weeks of STS administration, reduced bone formation capacity was rescued in MRL/lpr MSCs (n = 5). H&E staining showed new bone (B) and HA/TCP (HA) carrier. g Compared to wild-type MSCs, MRL/lpr MSCs showed a reduced capacity to differentiate into adipocytes when cultured under the adipogenic inductive conditions, as assessed by Oil red O staining (n = 5), along with reduced expression of adipogenic markers PPARγ and LPL, as assessed by Western blot. After 4 weeks of STS administration, reduced adipocyte formation and expression of PPARγ and LPL were rescued in MRL/lpr MSCs (n = 5). All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. ***P < 0.001; **P < 0.01; *P < 0.05. Scale bar, 5 μm (a), 50 μm (f)
Since previous studies indicated that MSCs from MRL/lpr and Casp3−/− mice showed impaired osteogenic differentiation,40,41 here we showed that MSCs from MRL/lpr mice and Casp3−/− mice had decreased proliferation and population doubling rates, as assessed by BrdU labeling and continuous passage assay, respectively (Fig. 1d; Supplementary information, Figure S1e). Additionally, MSCs from MRL/lpr and Casp3−/− mice showed reduced capacities for osteogenic differentiation, as indicated by decreased calcium nodule formation and expression of osteogenic markers runt-related transcription factor 2 (Runx2) and alkaline phosphatase (ALP) when cultured under the osteogenic inductive conditions (Fig. 1e; Supplementary information, Figure S1f). Moreover, MSCs from MRL/lpr and Casp3−/− mice showed significantly reduced capacities to generate new bone when implanted into immunocompromised mice subcutaneously using hydroxyapatite tricalcium phosphate (HA/TCP) as a carrier (Fig. 1f; Supplementary information, Figure S1g). When compared with the control group, MSCs from MRL/lpr and Casp3−/− mice also showed significantly reduced capacities to differentiate into adipocytes under the adipogenic inductive conditions, as indicated by a reduced number of Oil red O-positive adipocytes and downregulated expression of adipogenic markers peroxisome proliferator-activated receptor γ (PPARγ) and lipoprotein lipase (LPL) (Fig. 1g; Supplementary information, Figure S1h). These data suggest that deficiency of apoptosis may impair MSC self-renewal and multipotent differentiation.
We next asked whether accelerated apoptosis could rescue impaired MSCs in MRL/lpr and Casp3−/− mice. Since staurosporine (STS) induces cell apoptosis via both caspase-dependent and -independent pathways,42,43 we administrated 3 ng of STS per mouse twice a week for 4 weeks and found that STS treatment induced a significantly increased apoptosis rate and the production of apoptotic bodies in the bone marrow of MRL/lpr and Casp3−/− mice (Fig. 1a–c; Supplementary information, Figure S1a, b, d). Interestingly, we found that STS treatment rescued impaired MSCs in MRL/lpr and Casp3−/− mice, increasing their proliferation and population doubling rates, as assessed by BrdU labeling assay and continuous passage assay, respectively (Fig. 1d; Supplementary information, Figure S1e). Osteogenic and adipogenic differentiation of these cells was also upregulated, as assessed by mineralized nodule formation, expression of Runx2 and ALP, in vivo bone formation when implanted into immunocompromised mice, ratio of Oil Red O-positive adipocytes and expression of adipogenic markers PPARγ and LPL (Fig. 1e–g; Supplementary information, Figure S1f–h). These data imply that STS-induced elevation of apoptotic body production can rescue impaired MSCs in MRL/lpr and Casp3−/− mice.
Apoptotic bodies rescued impaired MSCs in MRL/lpr and Casp3 −/− mice
In order to examine whether exogenous apoptotic bodies could rescue impaired MSCs in MRL/lpr and Casp3−/− mice, we used STS to induce apoptosis in cultured allogenic MSCs and subsequently isolated apoptotic bodies using a sequential filter system (Supplementary information, Figure S2a). We confirmed that these apoptotic bodies expressed apoptotic body-specific surface markers Annexin V, C1q and TSP-1, as assayed by immunofluorescent staining and flow cytometric analysis, respectively (Supplementary information, Figure S2b). We further identified that these apoptotic bodies were 1–5 μm in size and contained cellular components (Supplementary information, Figure S2c–f). When 4 × 106 apoptotic bodies were intravenously infused once a week for 4 weeks, impaired MSCs from MRL/lpr and Casp3−/− mice were rescued, as indicated by increased BrdU labeling and population doubling rates (Fig. 2a; Supplementary information, Figure S2g), improved mineralized nodule formation, elevated expression of Runx2 and ALP (Fig. 2b; Supplementary information, Figure S2h), increased in vivo bone formation (Fig. 2c; Supplementary information, Figure S2i), increased adipocyte formation and elevated expression of PPARγ and LPL (Fig. 2d; Supplementary information, Figure S2j). Since bone disorders are common symptoms of SLE, we further showed that apoptotic body infusion was able to ameliorate the osteopenia phenotype in MRL/lpr and Casp3−/− mice, as assessed by microCT analysis (Fig. 2e; Supplementary information, Figure S2k). Since MRL/lpr mice represent a disease model for SLE, these data suggest that exogenous apoptotic body treatment may offer a therapeutic effect to rescue impaired MSCs and osteopenia in SLE.

Systemic infusion of exogenous apoptotic bodies rescued impaired MSCs in MRL/lpr mice. a After inducing culture-expanded MSCs to undergo apoptosis via STS for 15 h, apoptotic bodies (AB) were isolated and purified. 4 × 106 apoptotic bodies were injected into MRL/lpr mice via the tail vein once a week. After 4 weeks of apoptotic body infusion, BrdU labeling and continuous passage assay showed that the decreased proliferation and population doubling rates were rescued in MRL/lpr MSCs. b After 4 weeks of apoptotic body infusion, MSCs from MRL/lpr mice showed significantly increased capacities to form mineralized nodules, as assessed by alizarin red staining (n = 5), and upregulated expression of osteogenic markers Runx2 and ALP compared to MRL/lpr mice, as assessed by Western blot. c MSCs from apoptotic body-treated MRL/lpr mice showed increased capacities to generate new bone when implanted into immunocompromised mice subcutaneously using HA/TCP as a carrier (n = 5). H&E staining showed newly formed bone (B) and HA/TCP (HA) carrier. d MSCs from apoptotic body-treated MRL/lpr mice showed significantly increased capacities to differentiate into adipocytes under the adipogenic inductive culture conditions, as assessed by Oil red O staining (n = 5), and upregulated expression of adipogenic markers PPARγ and LPL, as assessed by Western blot. e After 4 weeks of apoptotic body treatment, the femurs of MRL/lpr mice showed significantly increased bone mineral density (BMD) and bone volume/total volume (BV/TV), as assessed by microCT (n = 5). f After intravenous infusion of PKH67-labeled apoptotic bodies for 24 h, immunofluorescent staining showed PKH67 co-localized with CD105-, CD73- and CD44-positive cells in the femurs of MRL/lpr mice. All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. ***P < 0.001; **P < 0.01; *P < 0.05. Scale bar, 50 μm (c), 200 μm (e), 10 μm (f)
Our previous study showed that recipient MSCs could engulf exosomes from systemically infused MSCs.41 Here we examined whether recipient MSCs could engulf apoptotic bodies after apoptotic body infusion. Immunofluorescent staining showed that PKH67-labeled apoptotic bodies could be detected in CD105-, CD73- and CD44-positive MSCs in the bone marrow of MRL/lpr and Casp3−/− mice at 24 h post-infusion (Fig. 2f; Supplementary information, Figure S2l). Next, we found that about 44.6% of the PKH67-labeled apoptotic bodies in the bone marrow were taken up by CD105-positive cells and 19.7% PKH67-labeled apoptotic bodies were taken up by CD11b-positive monocytes (Supplementary information, Figure S2m). These data suggest that impaired MSCs are indeed able to phagocytose exogenous apoptotic bodies in vivo.
To further confirm that apoptotic bodies are able to rescue impaired MRL/lpr and Casp3−/− MSCs, we used an in vitro co-culture system to show that treatment with 4 × 106 apoptotic bodies, assessed after 24 h, rescued 1 × 106 MRL/lpr and Casp3−/− MSCs, as indicated by increased BrdU labeling and population doubling rates (Supplementary information, Figure S3a and j), improved mineralized nodule formation and elevated expression of Runx2 and ALP (Supplementary information, Figure S3b and k), elevated bone formation when transplanted into immunocompromised mice (Supplementary information, Figure S3c and l), increased adipocyte formation and elevated expression of PPARγ and LPL (Supplementary information, Figure S3d and m) when compared to the control groups. These in vitro experimental data confirmed that apoptotic bodies play a direct role in regulating stem cell properties of MSCs. Since uptake of apoptotic bodies was detected in MRL/lpr and Casp3−/− MSCs, we used PKH26-labeled apoptotic bodies to treat cultured MSCs and confirmed that the MSCs could engulf apoptotic bodies in vitro (Supplementary information, Figure S3e). Apoptotic bodies expose phosphatidylserine (PtdSer) in membrane surface to serve as an “eat me” signal to be recognized by phagocytes for engulfment process.44 We showed that MSCs expressed αvβ3 and αvβ5, but not MerTK; they also expressed MFGE8, which is capable of connecting αvβ3 and αvβ5 with PtdSer in apoptotic bodies (Supplementary information, Figure S3f). To determine whether αvβ3 or αvβ5 contribute to the engulfment of apoptotic bodies, we used siRNA to knock down αvβ3 and αvβ5 expression, respectively (Supplementary information, Figure S3g and h) and found that knockdown of αvβ3, but not αvβ5, blocked the engulfment of apoptotic bodies by MSC (Supplementary information, Figure S3i). These data suggest that αvβ3 may play an essential role in MSC-mediated engulfment of apoptotic bodies.
Apoptotic body treatment upregulated Wnt/β-catenin pathway in MRL/lpr and Casp3 −/− MSCs via reusing RNF146
Next, we sought to determine how apoptotic bodies rescue impaired MSCs in apoptosis-deficient mice. We used mouse stem cell signaling RT2 Profiler PCR array to show that systemic apoptotic body infusion markedly altered the gene expression profiles of MRL/lpr and Casp3−/− MSCs (Supplementary information, Figure S4a). Then we used Ingenuity Pathway Analysis (IPA) “Canonical Pathway” analysis to evaluate activation z-scores to predict the most highly activated relevant pathway. This analysis showed that the Wnt/β-catenin pathway had the highest activation score in MRL/lpr and Casp3−/− MSCs at 4 weeks after apoptotic body infusion (Supplementary information, Figure S4b). These data suggested that apoptotic body treatment may activate the Wnt/β-catenin pathway to rescue impaired MRL/lpr and Casp3−/− MSCs. We used a Western blot assay to confi6rm that apoptotic body infusion upregulated the expression levels of active β-catenin and reduced the expression of Axin1 at 4 weeks post-infusion in MRL/lpr and Casp3−/− MSCs (Fig. 3a; Supplementary information, Figure S4c). However, the expression levels of other Wnt/β-catenin components, including Fzd, APC, did not show significant changes. Axin2 only showed slight upregulation compared to that in MSCs from un-treated MRL/lpr and Casp3−/− mice (Fig. 3a; Supplementary information, Figure S4c). Therefore, we focused our study on the role of Axin1 in apoptotic body-regulated Wnt/β-catenin signaling. Recently it has been reported that Axin protein undergoes Poly-ADP-ribosylation (PARsylation) via Tankyrases 1 and 2 (Tnks1/2),45 leading to the promotion of Axin ubiquitination by RNF146,46,47 an E3 ubiquitin ligase, and subsequently its proteasomal degradation. Additionally, Axin can be destabilized by Smurf2 ubiquitin ligase- or ubiquitin-regulated protease USP34-mediated ubiquitination.48,49 A Western blot showed that apoptotic body infusion upregulated the levels of RNF146, but not UPS34 or Smurf2, in MRL/lpr MSCs (Fig. 3b; Supplementary information, Figure S4d). To confirm that apoptotic body treatment upregulates the expression levels of RNF146 and active-β-catenin, we used allogenic apoptotic bodies at low doses (4 × 106 apoptotic bodies) and high doses (8 × 106 apoptotic bodies), respectively, to treat 1 × 106 culture-expanded MRL/lpr MSCs and found that apoptotic body treatment induced a dose-dependent upregulation of RNF146 and active-β-catenin, along with marked downregulation of Axin1 (Fig. 3c). However, real-time PCR analysis showed that apoptotic body treatment failed to affect rnf146 expression at the mRNA level (Fig. 3d), suggesting that apoptotic bodies may directly elevate the levels of RNF146 without genetic regulation. After confirming by Western blot that apoptotic bodies contain RNF146 (Fig. 3e), we transfected MSCs with EGFP-RNF146 plasmids and used STS treatment to generate EGFP-RNF146-positive apoptotic bodies (Supplementary information, Figure S4e). These apoptotic bodies were systemically infused into MRL/lpr and Casp3−/− mice, and we detected CD105 and GFP double-positive cells in the bone marrow at 24 h post-infusion, as assessed by immunofluorescent staining (Fig. 3f; Supplementary information, Figure S4f). This suggested that MSCs are able to engulf RNF146 derived from systemically infused apoptotic bodies. As expected, culture-expanded MRL/lpr and Casp3−/− MSCs were also able to engulf EGFP-RNF146-positive apoptotic bodies in the culture (Fig. 3g; Supplementary information, Figure S4g).

Systemically infused apoptotic bodies rescued impaired MRL/lpr MSCs through reuse of RNF146 to upregulate Wnt/β-catenin pathway. a Western blot analysis showed that apoptotic body infusion upregulated active Wnt/β-catenin expression and downregulated Axin1 expression in MRL/lpr MSCs. There was no significant change in β-catenin, Axin2, APC, or Fzd expression. β-Actin was used as a protein loading control. b Western blot showed that apoptotic body infusion upregulated the level of RNF146, but not Smurf2 or USP34, in MRL/lpr MSCs. β-Actin was used as a protein loading control. c When a low dose (4 × 106) and a high dose (8 × 106) of apoptotic bodies were used to treat 1 × 106 culture-expanded MRL/lpr MSCs, upregulation of active-β-catenin and RNF146, along with downregulation of Axin 1, were observed in a dose-dependent manner, as assessed by Western blot. d Real-time PCR analysis showed that apoptotic body treatment failed to affect mRNA expression levels of rnf146 in MRL/lpr MSCs. e Western blot analysis showed that both MSCs and apoptotic bodies contained RNF146. f Immunofluorescent staining showed that systemically infused EGFP-RNF146-labeled apoptotic bodies were engulfed by MRL/lpr MSCs at 24 h post-infusion. g Immunofluorescent staining showed that EGFP-RNF146-labeled apoptotic bodies were engulfed by culture-expanded MRL/lpr MSCs after 24 h co-culture. h Western blot analysis showed that β-catenin siRNA treatment efficiently knocked down the expression level of β-catenin and blocked apoptotic body-induced upregulation of Wnt/β-catenin in MRL/lpr MSCs. i β-catenin siRNA treatment blocked apoptotic body-induced increase in mineralized nodule formation, as assessed by alizarin red staining, and upregulation of Runx2 and ALP, as assessed by Western blot. j Western blot analysis showed that treatment with Wnt pathway inhibitor XAV939 blocked apoptotic body-induced upregulation of Wnt/β-catenin and RNF146 and downregulation of Axin 1 in MRL/lpr MSCs. k XAV939 treatment blocked apoptotic body-induced increase in mineralized nodule formation, as assessed by alizarin red staining, and upregulation of Runx2 and ALP, as assessed by Western blot. All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. **P < 0.01, N.S., no significance. Scale bar, 10 μm (f), (g)
To verify that RNF146 and Wnt/β-catenin signaling activation contribute to apoptotic body-induced rescue of impaired MRL/lpr MSCs, we used a siRNA approach to examine whether knocking down β-catenin expression could abolish apoptotic body-induced rescue of MRL/lpr MSCs. Western blot analysis showed that siRNA treatment knocked down β-catenin and active-β-catenin expression efficiently (Fig. 3h). Apoptotic body treatment could upregulate RNF146 and downregulate Axin1 levels, but failed to significantly upregulate active-β-catenin expression in the β-catenin knockdown conditions, and had no effect on osteogenic differentiation in MRL/lpr MSCs, as indicated by calcium nodule formation and expression of osteogenic markers ALP and Runx2 when cultured under the osteogenic inductive conditions (Fig. 3h, i). To further confirm these findings, we used XAV939, an Axin stabilizing agent capable of blocking enzymes tankyrase1/2 to abolish RNF146’s function, to block the Wnt/β-catenin pathway in MRL/lpr and Casp3−/− MSCs. Western blot analysis showed that XAV939 treatment upregulated the expression level of Axin1 and downregulated the expression levels of RNF146 and activated-β-catenin (Fig. 3j; Supplementary information, Figure S4h). Moreover, XAV939 treatment blocked apoptotic body-induced upregulation of osteogenic differentiation in MRL/lpr and Casp3−/− MSCs, as indicated by calcium nodule formation and expression of Runx2 and ALP when cultured under the osteogenic inductive conditions (Fig. 3k; Supplementary information, Figure S4i). These experimental data suggest that apoptotic bodies may directly provide RNF146 to MSCs for Wnt/β-catenin activation.
To further confirm the role of RNF146 in apoptotic body-induced rescue of impaired MRL/lpr and Casp3−/− MSCs, we examined whether blockage of RNF146 could abolish the apoptotic body-induced therapeutic effect. After using siRNA to knock down RNF146 expression in MSCs (Supplementary information, Figure S5a), allogenic apoptotic bodies newly generated by STS treatment contained significantly reduced levels of RNF146 (Supplementary information, Figure S5b). After 4 weeks of systemic infusion, these RNF146-deficient apoptotic bodies failed to upregulate RNF146 or active-β-catenin expression, and had no effect on the expression level of Axin1 in MRL/lpr and Casp3−/− MSCs (Fig. 4a; Supplementary information, Figure S5c). As expected, RNF146 knockdown apoptotic bodies failed to rescue impaired MRL/lpr and Casp3−/− MSCs in terms of their proliferation and population doubling rates (Fig. 4b; Supplementary information, Figure S5d); their capacity for osteogenic differentiation, as assessed by calcium nodule formation and expression of Runx2 and ALP when cultured under the osteogenic inductive conditions (Fig. 4c; Supplementary information, Figure S5e); new bone formation when implanted into immunocompromised mice (Fig. 4d; Supplementary information, Figure S5f); and capacity for adipogenic differentiation, as assessed by Oil red O staining and expression of PPARγ and LPL (Fig. 4e; Supplementary information, Figure S5g). Furthermore, we used in vitro cultured MRL/lpr and Casp3−/− MSCs to verify that RNF146 knockdown apoptotic bodies failed to rescue the proliferation, population doubling, osteogenic and adipogenic differentiation of impaired MSCs when compared with the control groups (Fig. 4f–h, Supplementary information, Figure S5h–j). These data indicate that RNF146 plays a critical role in apoptotic body-induced rescue of MRL/lpr and Casp3−/− MSCs.

RNF146 played an essential role in apoptotic body-mediated rescue of impaired MRL/lpr MSCs. a Western blot analysis showed that systemic infusion of apoptotic bodies derived from RNF146 knockdown MSCs failed to upregulate active-β-catenin expression and downregulate Axin1 expression in MRL/lpr MSCs when compared to the control group at 4 weeks post-infusion. β-Actin was used as a protein loading control. b BrdU labeling and continuous passage assay showed that apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced proliferation and population doubling rates in MRL/lpr MSCs when compared to the control group. c Apoptotic bodies derived from RNF146 knockdown MSCs also failed to rescue reduced capacities to form mineralized nodules in MRL/lpr MSCs at 4 weeks post-infusion when cultured under the osteogenic inductive conditions, as assessed by alizarin red staining (n = 5), and expression of osteogenic markers ALP and Runx2, as assessed by Western blot, when compared with the control group. d Apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced capacities to generate new bone in MRL/lpr MSCs at 4 weeks post-infusion when implanted into immunocompromised mice subcutaneously using HA/TCP as a carrier (n = 5). H&E staining showed newly formed bone (B) and HA/TCP (HA) carrier. e Apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced capacities to form adipocytes under the adipogenic inductive conditions, as assessed by Oil red O staining (n = 5), and expression of adipogenic markers PPARγ and LPL, as assessed by Western blot, at 4 weeks post-infusion. f BrdU labeling and continuous passage assay confirmed that apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced proliferation and population doubling rates of culture-expanded MRL/lpr MSCs. g Apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced capacities to form mineralized nodules under the osteogenic inductive conditions, as assessed by alizarin red staining (n = 5), and expression of osteogenic markers Runx2 and ALP, as assessed by Western blot, in culture-expanded MRL/lpr MSCs. h Apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced capacities to differentiate into adipocytes under the adipogenic inductive conditions, as assessed by Oil red O staining (n = 5), and expression of adipogenic markers PPARγ and LPL, assessed by Western blot, in culture-expanded MRL/lpr MSCs. All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. ***P < 0.001; **P < 0.01; *P < 0.05. Scale bar, 50 μm (d). KD, knockdown
Apoptotic body infusion ameliorated osteopenia phenotype and rescued impaired MSCs in OVX mice
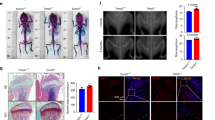
Since apoptotic body infusion was able to ameliorate the osteopenia phenotype in apoptosis-deficient mice, we asked whether apoptotic body treatment could rescue bone loss and impaired MSCs in a mouse model in which osteoporosis is not related to apoptosis. Therefore, we used ovariectomized (OVX) mice to verify the role of apoptotic body infusion in a clinically related osteoporosis animal model.50,51 After 4 weeks of apoptotic body infusion, microCT analysis showed that the reduced bone mineralized density (BMD) and bone volume/tissue volume (BV/TV) seen in the distal femur of OVX mice were restored (Fig. 5a). Histological analysis confirmed that apoptotic body infusion rescued trabecular bone volume in the distal femur of OVX mice (Fig. 5b). However, apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced BMD, BV/TV, and trabecular bone volume in OVX mice (Fig. 5a, b). The expression of bone formation marker genes Bglap (bone gamma carboxyglutamate protein, osteocalcin) and Alp (alkaline phosphatase) from the whole-bone tissue decreased in OVX group compared with the sham group (Fig. 5c).52 After 4 weeks of apoptotic body infusion, the expression levels of Bglap and Alp were elevated (Fig. 5c). However, apoptotic bodies derived from RNF146 knockdown MSCs failed to affect the expression levels of Bglap and Alp (Fig. 5c), suggesting RNF146 is required for apoptotic body-induced elevation of bone formation. Moreover, we showed that the treatment with apoptotic bodies from wild-type MSCs, but not from RNF146 knockdown MSCs, rescued the reduced new bone formation rate in OVX mice, as assessed by bone formation rate per bone surface (BFR/BS) values using double calcein labeling (Fig. 5d).

Apoptotic body treatment ameliorated osteoporotic phenotype and rescued impaired MSC function in OVX mice. a Compared with the sham group, the femurs of OVX mice showed significantly reduced bone mineral density (BMD) and bone volume/total volume (BV/TV), as assessed by microCT (n = 5). After 4 weeks of apoptotic body treatment, the reduced BMD and BV/TV in the femurs of OVX mice were rescued (n = 5), but apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced BMD and BV/TV (n = 5). b Histogram showed that OVX resulted in reduced trabecular bone area in distal femurs of OVX mice, when compared with the sham group (n = 5). After 4 weeks of apoptotic body treatment, the reduced trabecular bone area in distal femurs of OVX mice was rescued, but apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced trabecular bone area (n = 5). c Real-time PCR analysis showed that the expression levels of Bglap and Alp mRNA levels decreased in the femurs bone marrow of OVX mouse compared with the sham group (n = 5). After 4 weeks of apoptotic body treatment, the reduced expression levels of Bglap and Alp mRNA in OVX mice were elevated in wild-type MSC-derived, but not RNF146 knockdown MSC-derived, apoptotic body group (n = 5). d Calcein double labeling of the metaphyseal trabecular bone in the distal femora showed that bone formation rate per bone surface (BFR/BS) values decreased in OVX group compared with the sham group (n = 5). This reduced new bone formation rate in OVX group was rescued 4 weeks after treatment with apoptotic bodies derived from wild-type MSCs, but not from RNF146 knockdown MSCs (n = 5). e Western blot analysis showed that the level of active-β-catenin was decreased and the level of Axin1 was slightly increased in OVX MSCs, when compared with sham MSCs. After 4 weeks of apoptotic body treatment, the levels of RNF146 and active-β-catenin were increased and level of Axin1 was decreased, but apoptotic bodies from RNF146 knockdown MSCs failed to upregulate active-β-catenin expression and downregulate Axin1 expression in OVX MSCs. β-Actin was used as a protein loading control. f BrdU labeling and continuous passage assay showed that OVX MSCs (n = 5) had increased proliferation and reduced population doubling rates when compared to the sham group. After 4 weeks of apoptotic body treatment, proliferation and population doubling rates were rescued in OVX MSCs (n = 5), but apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue increased proliferation and reduced population doubling rates in OVX MSCs (n = 5). g Compared to Sham MSCs, OVX MSCs showed reduced capacities to form mineralized nodules when cultured under the osteogenic inductive conditions, assessed by alizarin red staining (n = 5), and reduced expression of osteogenic markers Runx2 and ALP, assessed by Western blot. After 4 weeks of apoptotic body treatment, reduced mineralized nodule formation and expression of Runx2 and ALP were rescued in OVX MSCs (n = 5), but apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue the reduced mineralized nodule formation and expression of Runx2 and ALP. h OVX MSCs showed reduced capacities to form new bone when implanted into immunocompromised mice subcutaneously using HA/TCP as a carrier. After 4 weeks of apoptotic body treatment, reduced bone formation capacity was rescued in OVX MSCs (n = 5), but apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue reduced capacities to generate new bone in OVX MSCs (n = 5). H&E staining showed new bone (B) and HA/TCP (HA) carrier. All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. ***P < 0.001; **P < 0.01; *P < 0.05. Scale bar, 200 μm (a), 25 μm (d), 50 μm (h). KD, knockdown
These data indicate that apoptotic body treatment is able to improve bone formation in OVX mice. To examine whether apoptotic body infusion is able to rescue impaired MSCs in OVX mice, we showed that apoptotic body infusion elevated the expression levels of RNF146 and active-β-catenin, whereas it reduced the expression levels of Axin1 in OVX MSCs (Fig. 5e). Moreover, we showed that apoptotic body infusion reduced the proliferation rate and increased the population doubling rate in OVX MSCs, as assessed by BrdU labeling assay and continuous passage assay, respectively (Fig. 5f). Reduced osteogenic differentiation in OVX MSCs was also rescued by apoptotic body infusion as indicated by increased mineralized nodule formation, expression of Runx2 and ALP, and in vivo bone formation when implanted into immunocompromised mice (Fig. 5g, h). As expected, apoptotic bodies derived from RNF146 knockdown MSCs failed to activate Wnt/β-catenin pathway and rescue the impaired MSCs in OVX mice (Fig. 5e–h).
Since bone mass is maintained by the balance between osteoblast-mediated bone formation and osteoclast-mediated bone resorption, we examined osteoclast activity in apoptotic body-treated OVX mice and found that apoptotic body infusion was able to reduce the number of tartrate-resistant acid phosphatase (TRAP)-positive osteoclasts in the distal femur of OVX mice (Supplementary information, Figure S6a). We next determined whether apoptotic body treatment directly inhibits osteoclastogenesis. We isolated osteoclast precursors from bone marrow and induced them to differentiate into osteoclasts with adding macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor κB ligand (RANKL). We used an in vitro co-culture system to show that apoptotic body treatment failed to inhibit osteoclastogenesis as assayed by TRAP staining (Supplementary information, Figure S6b). Since MSCs can induce osteoclast apoptosis via the FasL pathway53 and activation of the β-catenin pathway upregulates FasL expression,54 we hypothesized that apoptotic body treatment improved MSCs’ capacity to induce osteoclast apoptosis. Our experimental data showed that apoptotic body treatment indeed improved MSC-mediated reduction of osteoclast numbers (Supplementary information, Figure S6c). However, apoptotic bodies derived from RNF146 knockdown MSCs failed to exert the same function (Supplementary information, Figure S6c). Moreover, we showed that the treatment of apoptotic bodies from wild-type MSCs, but not from RNF146 knockdown MSCs, significantly increased the expression level of FasL, but not the levels of OPG or RNAKL, as assessed by Western blot analysis (Supplementary information, Figure S6d). Next, we showed that knockdown of FasL expression abolished apoptotic body-induced elevation of MSC’s capacities to reduce the number of osteoclasts (Supplementary information, Figure S6e and f).
In addition, apoptotic body infusion reduced the serum levels of type I collagen cross-linked telopeptide (CTX), TRAP5b and RANKL, whereas it elevated the serum level of OPG in OVX mice (Supplementary information, Figure S6g–j). However, apoptotic bodies derived from RNF146 knockdown MSCs failed to rescue the levels of CTX-1, TRAP5b, RANKL and OPG in OVX mice (Supplementary information, Figure S6g–j). These experimental data suggest that apoptotic body treatment can directly improve bone formation and indirectly inhibit osteoclast activity.
MRL/lpr and Casp3 −/− MSCs engulfed miR-328-3p from apoptotic bodies to upregulate Wnt/β-catenin pathway
Since it was reported that apoptotic bodies could horizontally transfer microRNAs (miRNAs),12 we next asked whether miRNAs, which are noncoding ~22 nucleotide RNAs capable of silencing gene expression at post-transcriptional level, also contribute to upregulation of Wnt/β-catenin pathway via downregulation of Axin1 in apoptotic body-induced rescue of MRL/lpr and Casp3−/− MSCs. We used online software, DIANA and Targetscan, to computationally search miRNA targets on Axin1 and predicted 16 miRNAs that may target Axin1 gene expression (Supplementary information, Table S1). Subsequently, real-time PCR revealed that miR-328-3p, miR-22-3p, miR-124-3p and miR-148a-3p showed elevated expression in apoptotic body-treated MRL/lpr MSCs (Fig. 6a). We next used microRNA mimics to show that miR-328-3p, miR-22-3p, and miR-148a-3p, but not miR-124-3p, could downregulate Axin1 expression in MRL/lpr MSCs at 24 h post-treatment (Fig. 6b). Then we knocked down these miRNAs in normal MSCs (Fig. 6c) and used STS treatment to generate apoptotic bodies. Western blot analysis showed that apoptotic bodies from miR-328-3p, but not from miR-22-3p or miR-148a-3p, knockdown MSCs exhibited abolished capacity to downregulate Axin1 and upregulate active-β-catenin (Fig. 6d). These data suggest that miR-22-3p, miR-328-3p, and miR-148a-3p mimics could downregulate Axin1 expression, but only miR-328-3p knockdown showed a significant effect on the apoptotic body-mediated Axin1/β-catenin cascade. Thus, we selected miR-328-3p as a functional miRNA for the following study. We next transfected MSCs with Cy3-labeled miR-328-3p mimics to generate Cy3-miR328-3p-positive apoptotic bodies. Immunofluorescent staining showed that cultured MSCs could engulf apoptotic bodies containing Cy3-miR328-3p (Fig. 6e). When apoptotic bodies from miR-328-3p knockdown MSCs were systemically infused into MRL/lpr mice, their rescue effects were partially abolished when compared to control apoptotic bodies in terms of regulating proliferation and population doubling rates (Fig. 6f); capacities for osteogenic differentiation, as indicated by calcium nodule formation and expression of Runx2 and ALP when cultured under the osteogenic inductive conditions (Fig. 6g); and capacities for adipogenic differentiation, as indicated by Oil red O staining and expression of PPARγ and LPL (Fig. 6h). We further used immunostaining to verify whether MSCs are able to engulf apoptotic body-derived EGFP-RNF146 and Cy3-miR-328-3p both in vitro and in vivo (Fig. 6i). When treated with apoptotic bodies that contain EGFP-labelled RNF146 and Cy3-labelled miR-328-3p, majority of MSCs engulfed EGFP-RNF146 (91.2%), Cy3-miR-328-3p (84.5%), or both of EGFP-RNF146 and Cy3-miR-328-3p (79.3%) (Fig. 6i). After systemic infusion of apoptotic bodies containing EGFP-RNF146 and Cy3-miR-328-3p, we found that 27.8% of CD105-positive MSCs in bone marrow colocalized with EGFP-RNF146 signal, 24.7% of them colocalized with Cy3-miR-328-3p signal, and 21.8% of them showed colocalization with both EGFP-RNF146 and Cy3-miR-328-3p signals (Fig. 6i). These data suggest that apoptotic bodies can be engulfed by MSCs in vivo and in vitro. Collectively, these findings imply that MSCs from apoptosis-deficient mice can reuse multiple cellular factors, including RNF146 and miR-328-3p, from apoptotic bodies to generate “chimeric” cells to rescue impaired MSCs.

miR-328-3p contributed to apoptotic body-mediated rescue of impaired MRL/lpr MSCs. a qPCR analysis showed the levels of miR-328-3p, miR-22-3p, miR-124-3p and miR-148a-3p were increased after apoptotic body treatment in MRL/lpr MSCs. b Western blot showed that mimics of miR-328-3p, miR-22-3p, and miR-148a-3p, but not miR-124-3p, reduced the level of Axin1 in apoptotic body-treated MRL/lpr MSCs. c qPCR showed that siRNA knockdown efficiently reduced the levels of miR-328-3p, miR-22-3p and miR-148a-3p in MSCs. d Western blot showed that apoptotic bodies from miR-328-3p-treated MSCs showed reduced capacity to downregulate Axin1 as well as to upregulate active-β-catenin when compared to miR-22-3p and miR-148a-3p groups. e Immunostaining showed that Cy3-miR-328-3p-labeled apoptotic bodies (Red) could be engulfed by MRL/lpr MSCs. f BrdU labeling and continuous passage assay showed that miR-328-3p knockdown partially abolished apoptotic body-induced rescue of the decreased proliferation and population doubling rates in MRL/lpr MSCs, but not as effectively as in the RNF146 knockdown group. g miR-328-3p knockdown partially abolished apoptotic body-induced rescue of mineralized nodule formation, as assessed by alizarin red staining, and expression of Runx2 and ALP, as assessed by Western blot, in MRL/lpr MSCs, but not as effectively as in the RNF146 knockdown group. h miR-328-3p knockdown partially abolished apoptotic body-induced rescue of adipogenic differentiation, as assessed by Oil red O staining, and expression of adipogenic markers, PPARγ and LPL, assessed by Western blot, in MRL/lpr MSCs, but not as effectively as in the RNF146 knockdown group. i Immunostaining showed that MRL/lpr MSCs engulfed both EGFP-RNF146-labeled apoptotic bodies and Cy3-miR-328-3p-labeled apoptotic bodies in an in vitro co-culture system. After intravenous infusion of PKH67-labeled apoptotic bodies for 24 h, immunofluorescent staining showed that both EGFP-RNF146- and Cy3-miR-328-3p-labeled apoptotic bodies co-localized with CD105-positive cells in the femurs of MRL/lpr mice. All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. **P < 0.01; *P < 0.05; N.S., no significance. Scale bar, 20 μm (e), (i). KD, knockdown
Apoptotic bodies participate in circulation to regulate distant MSCs
Since our data revealed that systemic infusion of apoptotic bodies is capable of rescuing impaired MSCs in apoptosis-deficient mice, we hypothesized that apoptotic bodies may participate in circulation to regulate distant MSCs. We used a parabiosis mouse model, which has been used to examine circulatory factors transferring from one animal to another (Fig. 7a; Supplementary information, Figure S7a),55,56 to show that connecting GFP mice with MRL/lpr or Casp3−/− mice resulted in an increased amount of apoptotic bodies in the bone marrow of MRL/lpr and Casp3−/− mice at 4 weeks post-surgical connection (Fig. 7b; Supplementary information, Figure S7b). After parabiosis with GFP mice, GFP and Annexin V double-positive apoptotic bodies were detected in the bone marrow of MRL/lpr and Casp3−/− mice, as assessed by flow cytometric analysis (Fig. 7c; Supplementary information, Figure S7c). Immunofluorescent staining also showed that MRL/lpr and Casp3−/− MSCs were able to engulf GFP-positive apoptotic bodies (Fig. 7d; Supplementary information, Figure S7d), suggesting that apoptosis-deficient MSCs possess the capacity to reuse cellular factors from circulating apoptotic bodies. Moreover, immunofluorescent staining showed that most of the GFP signals detected in the bone marrow of MRL/lpr and casp3−/− mice were colocalized with apoptotic maker C1q (Supplementary information, Figure S7e), indicating that most of the GFP-positive particles were apoptotic bodies but not cells. When MRL/lpr and Casp3−/− mice were paired with wild-type mice in parabiosis for 4 weeks, MRL/lpr and Casp3−/− MSCs showed elevated levels of RNF146 and active-β-catenin, along with reduced expression of Axin1 (Fig. 7e; Supplementary information, Figure S7f). Moreover, impaired MSCs in MRL/lpr and Casp3−/− mice were rescued, as indicated by elevated proliferation and population doubling rates (Fig. 7f; Supplementary information, Figure S7g); improved capacity for osteogenic differentiation, as assessed by calcium nodule formation, expression of Runx2 and ALP when cultured under the osteogenic inductive conditions, and in vivo bone formation when implanted into immunocompromised mice (Fig. 7g, h; Supplementary information, Figure S7h and i); and elevated capacity for adipogenic differentiation, as assessed by Oil red O staining and expression of PPARγ and LPL (Fig. 7i; Supplementary information, Figure S7j). Taken together, these data indicate that apoptotic bodies participate in the circulation system to regulate distant MSCs.

Circulating apoptotic bodies rescued impaired MSCs in MRL/lpr mice. a Scheme illustrating parabiosis. MRL/lpr-MRL/lpr, MRL/lpr-GFP, and MRL/lpr-WT parabiosis models were used. b Compared to the control MRL/lpr-MRL/lpr parabiosis model, the amount of apoptotic bodies was significantly increased in MRL/lpr mice in the MRL/lpr-GFP parabiosis model. Flow cytometric analysis confirmed that the number of apoptotic bodies was increased in the bone marrow of MRL/lpr mice in the MRL/lpr-GFP parabiosis model. c Flow cytometric analysis showed that GFP and Annexin V double-positive apoptotic bodies were detected in the bone marrow of MRL/lpr mice in the MRL/lpr-GFP parabiosis model. The total number of Annexin V-positive apoptotic bodies was increased in MRL/lpr mice in the MRL/lpr-MRL/lpr parabiosis model. d Immunofluorescent staining showed that CD105-positive cells engulfed GFP apoptotic bodies in the bone marrow of MRL/lpr mice in the MRL/lpr-GFP parabiosis model. e Western blot showed that the levels of RNF146 and active-β-catenin were increased and the levels of Axin1 were decreased in MRL/lpr MSCs in the MRL/lpr-WT parabiosis model at 4 weeks post-parabiotic surgery. f BrdU labeling and continuous passage assay showed that MRL/lpr MSCs from the MRL/lpr-WT parabiosis model had increased proliferation and population doubling rates compared to MRL/lpr MSCs from the MRL/lpr-MRL/lpr parabiosis model. g MRL/lpr MSCs from the MRL/lpr-WT parabiosis model showed increased capacities to form mineralized nodules under the osteogenic inductive conditions, assessed by alizarin red staining (n = 5), and increased expression levels of ALP and Runx2, assessed by Western blot, when compared to MRL/lpr MSCs from the MRL/lpr-MRL/lpr parabiosis model. h MRL/lpr MSCs from the MRL/lpr-WT parabiosis model showed increased capacities to form new bone when implanted into immunocompromised mice compared to MRL/lpr MSCs from the MRL/lpr-MRL/lpr parabiosis model. i MRL/lpr MSCs from the MRL/lpr-WT parabiosis model showed increased capacities to differentiate into adipocytes under the adipogenic inductive conditions, as assessed by Oil red O staining (n = 5), and increased expression levels of PPAR-γ and LPL, as assessed by Western blot, when compared to MRL/lpr MSCs from the MRL/lpr-MRL/lpr parabiosis model. All results are representative of data generated in three independent experiments. Error bars represent the S.D. from the mean values. ***P < 0.001; **P < 0.01; *P < 0.05. Scale bar, 10 μm (d), 50 μm (h)
Discussion
Apoptosis is a tightly regulated cell death process. It plays a crucial role in the maintenance of tissue homeostasis. Apoptotic cells may stimulate progenitor cell proliferation to improve tissue regeneration and replace damaged cells.57,58 In this study, we showed that deficient apoptosis in MRL/lpr and Casp3−/− mice impaired self-renewal and osteo-/adipo-genic differentiation of MSCs. Systemic infusion of exogenous apoptotic bodies was able to rescue impaired MSCs via reusing apoptotic body-derived RNF146 and miR-328-3p to activate the Wnt/β-catenin pathway (Supplementary information, Figure S8).
Extracellular vesicles (EVs), including exosomes, microvesicles and apoptotic bodies, play important roles in cell-cell communication and signaling regulation.59,60,61 Apoptotic bodies are specifically generated during cell apoptosis capable of encapsulating cellular factors generated during apoptosis. Apoptotic bodies can be recognized and engulfed by macrophages, fibroblasts, and specific phagocytes (Sertoli cells) for clearance purposes.62,63 In addition, engulfment of apoptotic cells may prime macrophages to generate molecular memory.19 Our data indicate that bone marrow MSCs are able to engulf apoptotic bodies to maintain their stem cell properties. PtdSer recognition receptors play an important role in apoptotic body recognition and engulfment.44 It is well-known that MSCs express a variety of integrins.64 Here we found MSCs express integrin αvβ3 and αvβ5, but only integrin αvβ3 contributes to apoptotic body engulfment in MSCs.
Our previous study showed that MSC transplantation is able to regulate Notch signaling in MRL/lpr MSCs to rescue a Fas/miR-29-controlled epigenetic cascade.41 Here we found apoptosis-deficient MSCs were able to simultaneously engulf RNF146 and miR-328-3p from exogenous apoptotic bodies to block Axin1, resulting in activation of the Wnt/β-catenin pathway and rescue of impaired MSCs in MRL/lpr and Casp3−/− mice. These rescued MSCs became chimeric cells containing donor cellular factors, including apoptotic body-derived RNF146 and miRNAs. Although we can’t exclude potential effect of Fas reuse in apoptotic body-mediated rescue of MRL/lpr MSCs, our data show that apoptotic bodies derived from RNF146 or miR-328-3p knockdown MSCs fail to rescue MRL/lpr and Casp3−/− MSCs. These data suggest that RNF146 and miR-328-3p play a critical role in apoptotic body-mediated rescue of impaired MSCs. The microenvironment surrounding MSCs, including inflammatory cytokines and immune cells, may affect MSC-mediated tissue regeneration and immunomodulation function.24,27,65,66,67 MRL/lpr mice represent a SLE disease model with a severe osteopenia phenotype due to deficient apoptosis and immune system disorders. Casp3−/− mice also show immune system disorders68 and Caspase 3 deletion can induce necrosis,69 which may trigger an inflammatory response. MSC transplantation is able to ameliorate the immune and osteopenia phenotypes in MRL/lpr mice and patients by increasing regulatory T cells and reducing Th17 cells.25 In this study, we find that MRL/lpr and Casp3−/− mice showed reduced apoptotic body formation in bone marrow. Apoptotic body treatment was capable to rescuing osteopenia phenotype and impaired MSCs in MRL/lpr and Casp3−/− mice, supporting the notion that impaired MSCs in apoptosis-deficient mice can be rescued by apoptotic body treatment. We also find that CD11b-positive immune cells engulf apoptotic bodies, implying that the interplay between apoptotic bodies and immune cells may regulate immune response. Although we are not able to exclude the possibility that apoptotic body-regulated immune response may contribute to the rescue of impaired MSCs in MRL/lpr, Casp3−/−, and OVX mice, our in vitro experimental data, in which all potential influences of the surrounding microenvironment are excluded, show that apoptotic bodies are capable of directly regulating WNT/β-catenin pathway to maintain MSC homeostasis. OVX mice are widely used as a postmenopausal osteoporotic model.48 We showed that apoptotic body treatment is able to ameliorate the osteoporotic phenotype in this model, suggesting the potential use of apoptotic bodies to treat osteoporosis. It appears that apoptotic body treatment directly improves the function of osteogenic cells to enhance bone formation and indirectly inhibits osteoclast activity by upregulating FasL expression in MSCs to cause osteoclast apoptosis.
Wnt/β-catenin signaling pathway plays a crucial role in regulating MSC self-renewal and differentiation.70,71,72,73,74 Exogenous application of Wnt3a increased self-renewal and decreased apoptosis of MSCs.71,72 Both the canonical and non-canonical Wnt signaling pathways control early differentiation of MSCs. Canonical Wnt/β‐catenin signaling promotes osteogenesis of MSCs by directly stimulating Runx2 gene expression.70,75,76 However, canonical Wnt signaling may also suppress osteogenic differentiation of MSCs, and non-canonical Wnt5a has been showed to promote osteogenic differentiation.71 The inconsistent effects of canonical Wnt signaling on MSC osteogenesis may due to the levels of Wnt activity, different sources of the cells, and differing experimental conditions. Previous studies showed that apoptotic bodies could horizontally transfer microRNAs and mRNAs.12,77 We found that apoptotic bodies transferred E3 ligase RNF146 and miR-328-3p to target Axin1 in MSCs and rescue impaired stem cell properties through activation of the Wnt/β-catenin pathway. RNF146, a RING-domain E3 ubiquitin ligase, can activate Wnt/β-catenin signaling by mediating tankyrase-dependent degradation of Axin. Axin1 and Axin2 were identified as negative regulators of the Wnt/β-catenin pathway. We found that apoptotic body treatment significantly downregulated Axin1, but not Axin2, in apoptosis-deficient MSCs. This may be due to the fact that Axin1 expression is ubiquitous in most tissues whereas Axin2 expression is more restricted to specific cell types.78
In addition to transferring RNF146, apoptotic bodies transferred miR-328-3p to downregulate Axin1 expression in MRL/lpr MSCs. Knockdown of miR-328-3p, but not other Axin-targeted microRNAs including miR-22-3p and miR-148a-3p, partially abolished apoptotic body treatment-induced rescue of impaired MSC functions. These data suggest that recipient cells may selectively engulf cellular factors, such as miRNAs, from apoptotic bodies to improve a specific signaling pathway for the purpose of rescuing stem cell function.79 Different microRNAs converging on the same target has been reported previously80,81; however, we found reused proteins and microRNAs from apoptotic bodies are able to work together to regulate a specific pathway, implying that recipient MSCs may participate in the reuse process. The advantage of cell therapy is that infused cells exert therapeutic effects in multiple ways, which may depend on the host microenvironments.65,82 These properties may explain why MSC transplantation has shown therapeutic effects in a variety of disease conditions. It will be interesting to assess whether apoptotic bodies may also play multiple regulatory roles when used in a therapeutic approach. In addition, our experimental data imply that apoptotic body component reuse may be a common biological process in physiological conditions for the maintenance of tissue homeostasis.
To elucidate the biological role of apoptotic bodies in normal physiological conditions, we used a parabiosis model, in which GFP and MRL/lpr mice were surgically joined to share a circulation system after the formation of microvasculature between the two organisms.56,83,84 In heterochronic parabiosis, a young mouse shares circulation with an old one. Some key molecules in circulation have been shown to regulate rejuvenation of the old mouse.85 Circulating extracellular vesicles including apoptotic bodies were recognized as major mediators in intercellular communication. Although the shared circulating blood cells in parabiosis mice may home to the bone marrow, circulating apoptotic bodies are engulfed by bone marrow cells. In this study, we paired an apoptosis-deficient mouse with a wild-type one to show that apoptotic bodies participate in circulation to regulate distant MSCs. This suggests that the reuse of apoptotic body components may be a common in vivo biological event.
Materials and methods
Mice
Female C3MRL-Faslpr/J (MRL/lpr), C3H/HeJ, C57BL/6-Tg (CAG-EGFP)1Osb/J and a pair of Casp3tm1Flv (Casp3+/−) mice were purchased from the Jackson Laboratory. Female immunocompromised mice (Beige XIDIII nude/nude) were purchased from Envigo. Casp3+/− mice were intercrossed to generate the homozygous Casp3−/− mice. All animal experiments were performed under institutionally approved protocols for the use of animal research (University of Pennsylvania IACUC# 805478 and University of Southern California IACUC #11953).
In vivo apoptosis rate assay
100 μL Annexin V and 30 µL 10× Annexin V binding buffer (Southern Biotech) were diluted in 170 µL distilled water for tail vein injection. At 2 h post-infusion, mice were sacrificed and the femurs were fixed in 4% PFA and then decalcified with 5% ethylenediaminetetraacetic acid (EDTA), followed by optimal cutting temperature compound (OCT, Sakura Finetek, Torrance, CA, USA) embedding. Frozen sections were prepared and slides were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). Apoptosis rate was measured as the percentage of Annexin V-positive signals among the DAPI-positive cells.
Reagents and chemicals
Staurosporine (ALX-380-014) was purchased from Enzo Life Sciences (Farmingdale, NY, USA). XAV-939 (S1180) (1 μM for working solution) was purchased from Selleck Chemicals (Houston, TX, USA). Hoechst 33342 (14533) was purchased from Sigma (St. Louis, MO, USA). Flow Cytometry Size Calibration Kit (F-13838), Lipofectamine RNAiMAX (13778) and Lipofectamine LTX with Plus (15338) were purchased from ThermoFisher Scientific (Waltham, MA, USA). Label IT miRNA Labeling Cy-3 Kit was purchased from Mirus Bio (Madison, WI, USA). Liquid Counting Beads (4.5 μm size) for flow cytometric analysis (335925) was purchased from BD Bioscience (San Jose, CA, USA). PKH26(MINI67) and PKH67(MINI67) cell linker were purchased from Sigma (St. Louis, MO, USA). M-CSF and RANKL were purchased from PeproTech (Rocky Hill, NJ, USA).
Antibodies
Anti-mouse Runx2 (8486), anti-mouse Smurf2 (12024S) and anti-mouse Axin1 (2087S) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-rat Alexa Fluor 647 (A21247) and anti-rat Alexa Flour 568 (A11077) antibodies were purchased from Invitrogen (Carlsbad, CA, USA). Anti-mouse ALP (sc-28904), anti-mouse PPAR-γ (sc-7273), anti-mouse APC (sc-896), anti-mouse TSP1 (sc-95886), anti-mouse αvβ3(sc-6627), anti-mouse αvβ5 (sc-13588), anti-mouse MerTK (sc-365499) anti-mouse FasL (C-178), anti-mouse OPG (H-249), anti-mouse RANKL (FL-317) and anti-mouse frizzled (sc-9169) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-mouse MFGE8 (D199-3) was purchased from MBL (Woburn, MA, USA). Anti-mouse LPL (SAB2700761) and anti-β-actin antibody (A1978) were purchased from Sigma (St. Louis, MO, USA). Anti-mouse CD73 (550738), anti-mouse CD105 (550546) and anti-mouse CD44 (553131) antibodies were purchased from BD Biosciences (San Jose, CA, USA). Annexin V-FITC (10039-02) and Annexin V-APC (10040-11) were purchased from Southern Biotech (Birmingham, AL, USA). CD62P-PE (12-0626) was purchased from eBioscience (San Diego, CA, USA). Anti-mouse USP34 (A300-824A-T) antibody was purchased from Bethyl Laboratories. Anti-mouse RNF146 (ab201212) and Anti-mouse Axin2 (ab32197) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-mouse active-β-catenin (05665) and anti-mouse β-catenin (06734) antibodies were purchased from EMD Millipore (Billerica, MA, USA). Anti-mouse C1q (CL7501F) antibody was purchased from CEDARLANE (Burlington, NC, USA).
Apoptotic body identification and calculation by flow cytometric analysis
Apoptotic bodies were isolated using a sequential centrifugation followed by a sequential filtering. Briefly, after 300× g centrifugation for 10 min to remove cell debris, the supernatant was subsequently filtered with 5 and 1 μm filters to collect the supernatant containing particles between 1 and 5 μm in diameter. Next, the supernatant was centrifuged at 2000× g for 20 min to pellet the apoptotic body-sized extracellular vesicles (Supplementary information, Figure S2a). Apoptotic body-sized extracellular vesicles derived from bone marrow and STS-treated MSCs were isolated after subsequent centrifugation, and then stained with 1 μg of CD62P-PE antibodies on ice for 30 min. Apoptotic body-sized extracellular vesicles were pelleted and stained with 1 μg of Annexin V-APC in binding buffer. Apoptotic body-sized extracellular vesicles were pelleted to remove the supernatant for the next step analysis. Apoptotic body-sized extracellular vesicles were re-suspended in 500 μL PBS. 10 μL apoptotic body-sized extracellular vesicles, 50 µL counting beads, 10 μL 1μm-sized and 10 µL 10 μm-sized calibration beads were added to 500 μL PBS. 1~5 μm apoptotic body-sized extracellular vesicles were gated by calibration beads and counting beads. Apoptotic bodies were defined as Annexin V-positive and CD62P-negative events. The number of counting beads was calculated according to the manufacturer’s instructions. The number of apoptotic bodies was calculated by the number of counting beads multiplied by the ratio of apoptotic body events to counting bead events in flow cytometry plots.
Isolation of mouse bone marrow mesenchymal stem cells (MSCs)
A single suspension of bone marrow-derived all nucleated cells (ANCs) from mouse femurs and tibias was seeded at a density of 1.5 × 107 cells per 10 cm culture dish (Corning, NY, USA) at 37 °C in 5% CO2. Non-adherent cells were removed after 48 h and attached cells were maintained for 16 days in alpha minimum essential medium (α-MEM, Invitrogen) supplemented with 20% fetal bovine serum (FBS, Equitech-Bio, Kerrville, TX, USA), 2 mM l-glutamine, 55 μM 2-mercaptoethanol, 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen). Colony-forming attached cells were passed once for further experimental use.
Cell proliferation assay
Proliferation of each mesenchymal stem cell population was performed by bromodeoxyuridine (BrdU) incorporation assay. Briefly, MSCs (1.0 × 104 cells/well) were seeded on 2-well chamber slides (Nunc) and cultured for 2–3 days. The cultures were incubated with BrdU solution (1:100) (Invitrogen) for 20 h and stained with a BrdU Staining Kit (Invitrogen) according to the manufacturer’s instructions. BrdU-positive and total cell numbers were counted in ten image areas per subject. The number of BrdU-positive cells was indicated as a percentage of the total number of cells. The BrdU assay was repeated in 4 or 5 independent samples for each experimental group.
Population doubling (PD) assay
Single colony cluster cells (P0 cells) were trypsinized and seeded at 0.5 × 106 on 60 mm dish (Corning) at the first passage. When they reached confluence, cells were harvested and seeded at the same density. The PD number was calculated at every passage according to the equation: PD = log2 (number of harvested cells/number of seeded cells). The PD numbers were determined by cumulative addition of total numbers generated from each passage until cells ceased dividing. The PD assay was repeated with five independent isolated cells for each experimental group.
Osteogenic differentiation assay
BMMSCs were loaded at 1 × 106 cells per well into a 6-well plate. When cells reached 100% confluence and stopped proliferating, we started to induce osteogenic differentiation to eliminate potential influence caused by the altered proliferation rates of the MRL/lpr and Casp3−/− MSCs. MSCs were cultured in osteogenic medium containing 2 mM β-glycerophosphate (Sigma), 100 μM L-ascorbicacid 2-phosphate (Sigma) and 10 nM dexamethasone (Sigma). Ten days after osteogenic induction, total protein was extracted from cultured MSCs and the expression of Runx2 and ALP was assayed by Western blot analysis. After four weeks of osteogenic induction, the cultures were stained with 1% alizarin red-S (Sigma). Alizarin red-positive area was analyzed using Image-J software (NIH) and shown as a percentage of the total area.
Western blot analysis
Total protein was extracted using M-PER mammalian protein extraction reagent (Thermo, Rockford, IL, USA). Protein was applied and separated on 4%–12% NuPAGE BT gel or 3%–8% NuPAGE TA gel (Invitrogen) and transferred to ImmobilonTM-P membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% non-fat dry milk and 0.1% tween-20 for 1 h, followed by incubation with the primary antibodies (1:200-1,000 dilution) at 4 °C overnight. Horseradish peroxidase-conjugated IgG (Santa Cruz Biosciences; 1:10,000) was used to treat the membranes for 1 h, after which the membranes were enhanced with a SuperSignal West Pico Chemiluminescent Substrate (Thermo). The bands were detected on films (Bioland, Paramount, CA, USA). β-actin antibody was used to quantify the amount of loaded protein.
MSC-mediated bone formation in vivo
Approximately 4.0 × 106 MSCs were mixed with hydroxyapatite/tricalcium phosphate (HA/TCP) ceramic particles (40 mg, Zimmer, Warsaw, IN, USA) as a carrier and subcutaneously implanted into the dorsal surface of 8- to 10-week-old immunocompromised mice. At 8 weeks post-implantation, the implants were harvested and fixed with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), decalcified with 5% EDTA in PBS, then embedded in paraffin. The 6-μm-thick sections were stained with hematoxylin and eosin (H&E). Images of the implants were analyzed using Image J software (NIH). Five fields were selected and the newly formed mineralized tissue area in each field was calculated as a percentage of the total tissue area.
Adipogenic differentiation
BMMSCs were loaded at 1 × 106 cells per well into a 6-well plate. When cells reached 100% confluence and stopped proliferating, we started to induce adipogenic differentiation to eliminate potential influence caused by the altered proliferation rates of the MRL/lpr and Casp3−/− MSCs. MSCs were cultured under adipogenic inductive conditions, in growth medium containing 500 nM isobutylmethylxanthine (Sigma-Aldrich), 60 μM indomethacin (Sigma-Aldrich), 500 nM hydrocortisone (Sigma-Aldrich), 10 μg/mL insulin (Sigma-Aldrich), and 100 nM l-ascorbic acid phosphate. At 7 days post-induction, the adipocytes were stained with Oil red O (Sigma-Aldrich), and positive cells were quantified under microscopy and shown as a number out of the total number of cells.
MicroCT and analysis
After being harvested and fixed in 4% paraformaldehyde (PFA), femurs were imaged and analyzed using a high-resolution Scanco μCT35 scanner (Scanco Medical AG, Bruttisellen, Switzerland). The specimens were scanned using a voxel size of 20 μm at 70 kVp and 200 μA. Scanned data was reconstructed using Scanco software. Datasets were loaded into Amira 5.3.1 software (Visage Imaging, Berlin, Germany) for visualization and analysis. Bone mineral density (BMD) and bone volume/total volume (BV/TV) for each specimen were also calculated by using Amira software.
Systemic infusion of apoptotic bodies
After using STS to induce culture-expanded MSCs to undergo apoptosis for 15 h, apoptotic bodies were isolated and purified. For in vivo apoptotic body infusion, 4 × 106 apoptotic bodies were suspended in 200 μL PBS and infused into MRL/lpr, Casp3−/−, or OVX mice intravenously through the tail vein once a week. Four weeks after the treatment, mice were sacrificed for further analysis. For in vivo tracking of apoptotic bodies, PKH-67-labeled apoptotic bodies were infused into MRL/lpr or Casp3−/− mice intravenously through the tail vein.
Immunofluorescent staining
To detect the in vivo engulfment of apoptotic bodies, PKH67-labeled apoptotic bodies were infused into MRL/lpr or Casp3−/− mice intravenously through the tail vein. At 24 h post-infusion, the femurs were fixed in 4% PFA and then decalcified with 5% EDTA, followed by embedding in OCT compound (Sakura Finetek, Torrance, CA, USA). Frozen sections were prepared and slides were stained with CD105, CD73 or CD44 antibodies, followed by secondary antibody staining. For detection of in vitro engulfment of apoptotic bodies, the MSCs were cultured on 4-well chamber slides (Nunc, Rochester, NY, USA) (2 × 103/well) and treated with 4 × 106 PKH26 pre-labeled apoptotic bodies per 1 × 106 MSCs. The cells were then fixed with 4% paraformaldehyde. ActinGreen 488 Ready Probes Reagent (Life Technologies) was used for cytoskeleton staining. Finally, slides were mounted with Vectashield mounting medium containing DAPI.
Real-time polymerase chain reaction (PCR)
Total RNA was isolated from the cultures using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. For real-time PCR of mRNA, the cDNA was synthesized using SuperScript III (Life Technologies). The real-time PCR was performed using SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and gene-specific primers. The primers included Rnf146: forward, 5′GCTGAAGCCAATGGAGATG-3′ and reverse, 5′-TCAGAGAAGGTGCAGTATTGG-3′; Gapdh: forward, 5′-CACCATGGAGAAGGCCGGGG-3′ and reverse, 5′-GACGGACACATTGGGGGTAG-3′; Bglap: forward, 5′-CTGACCTCACAGATGCCAAG-3′ and reverse, 5′-GTAGCGCCGGAGTCTGTTC-3′; Alp: forward, 5′-GGACAGGACACACACACACA-3′ and reverse, 5′-CAAACAGGAGAGCCACTTCA-3′. For real-time PCR of miRNA, the cDNA was synthesized using SuperScript III (Life Technologies). For real-time PCR of mature miRNA, cDNA was synthesized using the miScript II RT Kit (Qiagen). Real-time PCR was performed using the miScript SYBR Green PCR Kit (Qiagen). RUN6 was used as an endogenous control for MSCs. Real-time PCR was detected on a CFX96™ Real-Time PCR System (Bio-Rad).
RT2 profiler PCR arrays
The RT2 Profiler PCR Array (PAMM-405Z) was purchased from Qiagen (Germantown, MD, USA) to examine the expression patterns of 84 genes involved in mouse stem cell identification, proliferation and differentiation. The manufacturer’s instructions were strictly followed. Gene expression levels were analyzed using the web-based software RT2 Profiler PCR Array Data Analysis. The P-values were calculated based on a Student’s t-test of the replicate 2−ΔΔCT values for each gene in the untreated group and the treated group. All data were normalized to an average of five housekeeping genes, Gusb, Hprt, Hsp90ab1, Gapdh, and Actb.
Plasmids
EGFP-RNF146 fusion protein expression plasmids (BC050795) (Origene, Rockville, MD, USA) were used. Empty plasmids with the same backbone were used as a control. The cells were transfected with plasmids using Lipofectamine LTX with Plus reagent (Life Technologies) according to the manufacturer’s instruction.
siRNA knockdown
siRNAs for RNF146, β-catenin, αvβ3, αvβ5 and FasL were used to treat MSCs according to the manufacturer’s instructions (Santa Cruz Biotechnology). Non-targeting control siRNAs (Santa Cruz Biotechnology) were used as negative controls.
Calcein labeling assay
For double calcein labeling histomorphometric analysis, calcein (Sigma, 15 mg/Kg body weight) prepared in 2% sodium bicarbonate solution was intraperitoneally injected at 10 days and 3 days before sacrifice. Bone dynamic histomorphometric analyses for BFR/BS were performed according to the standardized nomenclature for bone histomorphometry under a fluorescence microscope (Olympus IX71, Japan).
TRAP staining
De-paraffinized sections were re-fixed with a mixture of 50% ethanol and 50% acetone for 10 min, and then incubated in freshly made TRAP solution with 1.6% naphthol AS-BI phosphate in N,N-dimethylformamide, 0.14% fast red-violet LB diazonium salt, 0.097% tartaric acid, and 0.04% MgCl2 in 0.2 M sodium acetate buffer at pH 5.0 for 10 min at 37 °C under a shield. Samples were counterstained with toluidine blue. All reagents for TRAP staining were purchased from Sigma-Aldrich.
Osteoclast formation
Bone marrow cells were flushed out of the tibiae and femora from 8-week-old mice, and 0.5 × 106 bone marrow cells were suspended in αMEM containing 15% heat-inactivated FBS, l-glutamine, penicillin and streptomycin and 20 ng/mL M-CSF in a 24-well plate for 48 h. The adherent cells were then collected and cultured with 20 ng/mL M-CSF and 50 ng/mL RANKL for additional 4 days.
Enzyme-linked Immunosorbent (ELISA) Assay
Serum markers of bone turnover, including collagen X link-1 (CTX-1), tartrate-resistant acid phosphatase 5b (TRAP 5b), receptor activator of nuclear factor kappa-B ligand (RANKL), and osteoprotegerin (OPG), were measured with ELISA kits purchased from R&D Systems (Minneapolis, MN, USA) and IDS (Scottsdale, AZ, USA), according to the manufacturers’ instructions.
MicroRNA mimic and inhibitor transfection. miR-22-3p, miR-328-3p, miR-124-3p, miR-148a-3p mimics, inhibitors and negative controls (Genecopia) were transfected into MSCs according to the manufacturer’s instructions.
Parabiosis model
The mice to be joined in parabiosis were anaesthetized and shaved along the opposite lateral flanks. The excess hair was wiped off with an alcohol prep pad. After further disinfection with Betadine solution and 70% alcohol, identical incisions were made on the corresponding lateral aspects from the olecranon to the knee joint of each mouse. The olecranon and knee joints were each attached by a single 4-0 silk suture and tie, and the dorsal and ventral skins were sewed together with continuous 5-0 Vicryl suture. The mice were then kept on heating pads and continuously monitored until full recovery. Buprenorphine was used for analgesic treatment by subcutaneous injection every 8–12 h for 48 h after the operation.
Statistics
Comparisons between two groups were analyzed using independent unpaired two-tailed Student’s t-tests, and comparisons between more than two groups were analyzed using one-way ANOVA with the Bonferroni adjustment. P values less than 0.05 were considered statistically significant.
References
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011).
Bergmann, A. & Steller, H. Apoptosis, stem cells, and tissue regeneration. Sci. Signal 3, re8 (2010).
Davidson, F. F. & Steller, H. Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature 391, 587–591 (1998).
Hochreiter-Hufford, A. E. et al. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497, 263–267 (2013).
Pellettieri, J. & Sanchez Alvarado, A. Cell turnover and adult tissue homeostasis: from humans to planarians. Annu. Rev. Genet. 41, 83–105 (2007).
Arandjelovic, S. & Ravichandran, K. S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 16, 907–917 (2015).
Radi, E., Formichi, P., Battisti, C. & Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 42(Suppl 3), S125–S152 (2014).
Nagata, S. Apoptosis and autoimmune diseases. Ann. N. Y. Acad. Sci. 1209, 10–16 (2010).
Cheng, J. et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 263, 1759–1762 (1994).
Bergsmedh, A. et al. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl Acad. Sci. USA 98, 6407–6411 (2001).
Zernecke, A. et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2, ra81 (2009).
Nawaz, M. et al. Extracellular vesicles: evolving factors in stem cell biology. Stem Cells Int. 2016, 1073140 (2016).
Kinchen, J. M. et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat. Cell Biol. 10, 556–566 (2008).
Hochreiter-Hufford, A. & Ravichandran, K. S. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 5, a008748 (2013).
Juncadella, I. J. et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493, 547–551 (2013).
Elliott, M. R. & Ravichandran, K. S. The dynamics of apoptotic cell clearance. Dev. Cell 38, 147–160 (2016).
Elliott, M. R. et al. Unexpected requirement for ELMO1 in clearance of apoptotic germ cells in vivo. Nature 467, 333–337 (2010).
Weavers, H., Evans, I. R., Martin, P. & Wood, W. Corpse engulfment generates a molecular memory that primes the macrophage inflammatory response. Cell 165, 1658–1671 (2016).
Friedenstein, A. J., Chailakhyan, R. K., Latsinik, N. V., Panasyuk, A. F. & Keiliss-Borok, I. V. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues: cloning in vitro and retransplantation in vivo. Transplantation 17, 331–340 (1974).
Prockop, D. J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 276, 71–74 (1997).
Shi, S. et al. Bone formation by human postnatal bone marrow stromal stem cells is enhanced by telomerase expression. Nat. Biotechnol. 20, 587–591 (2002).
Shi, S. & Gronthos, S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J. Bone Mineral. Res. 18, 696–704 (2003).
Liu, Y. et al. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat. Med. 17, 1594–1601 (2011).
Sun, L. et al. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells 27, 1421–1432 (2009).
Le Blanc, K. et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 363, 1439–1441 (2004).
Ren, G. et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell stem Cell 2, 141–150 (2008).
Uccelli, A., Pistoia, V. & Moretta, L. Mesenchymal stem cells: a new strategy for immunosuppression? Trends Immunol. 28, 219–226 (2007).
Fulda, S. & Debatin, K. M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811 (2006).
Drappa, J., Brot, N. & Elkon, K. B. The Fas protein is expressed at high levels on CD4+CD8+ thymocytes and activated mature lymphocytes in normal mice but not in the lupus-prone strain, MRL lpr/lpr. Proc. Natl Acad. Sci. USA 90, 10340–10344 (1993).
Chu, J. L., Drappa, J., Parnassa, A. & Elkon, K. B. The defect in Fas mRNA expression in MRL/lpr mice is associated with insertion of the retrotransposon, ETn. J. Exp. Med. 178, 723–730 (1993).
Adachi, M., Watanabe-Fukunaga, R. & Nagata, S. Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc. Natl Acad. Sci. USA 90, 1756–1760 (1993).
Poon, I. K., Lucas, C. D., Rossi, A. G. & Ravichandran, K. S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180 (2014).
Buzas, E. I., Gyorgy, B., Nagy, G., Falus, A. & Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 10, 356–364 (2014).
E. L. A., S., Mager, I., Breakefield, X. O. & Wood, M. J. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 12, 347–357 (2013).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Crescitelli, R. et al. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2, 20677 (2013).
Hristov, M., Erl, W., Linder, S. & Weber, P. C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 104, 2761–2766 (2004).
Jayachandran, M., Miller, V. M., Heit, J. A. & Owen, W. G. Methodology for isolation, identification and characterization of microvesicles in peripheral blood. J. Immunol. Methods 375, 207–214 (2012).
Miura, M. et al. A crucial role of caspase-3 in osteogenic differentiation of bone marrow stromal stem cells. J. Clin. Investig. 114, 1704–1713 (2004).
Liu, S. et al. MSC transplantation improves osteopenia via epigenetic regulation of notch signaling in lupus. Cell Metab. 22, 606–618 (2015).
Belmokhtar, C. A., Hillion, J. & Segal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20, 3354–3362 (2001).
Zhang, X. D., Gillespie, S. K. & Hersey, P. Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol. Cancer Ther. 3, 187–197 (2004).
Penberthy, K. K. & Ravichandran, K. S. Apoptotic cell recognition receptors and scavenger receptors. Immunol. Rev. 269, 44–59 (2016).
Huang, S. M. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 (2009).
DaRosa, P. A. et al. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 517, 223–226 (2015).
Zhang, Y. et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 13, 623–629 (2011).
Kim, S. & Jho, E. H. The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J. Biol. Chem. 285, 36420–36426 (2010).
Lui, T. T. et al. The ubiquitin-specific protease USP34 regulates axin stability and Wnt/beta-catenin signaling. Mol. Cell Biol. 31, 2053–2065 (2011).
Xie, H. et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 20, 1270–1278 (2014).
Weitzmann, M. N. & Pacifici, R. Estrogen deficiency and bone loss: an inflammatory tale. J. Clin. Invest. 116, 1186–1194 (2006).
Wang, X. et al. miR-214 targets ATF4 to inhibit bone formation. Nat. Med. 19, 93–100 (2013).
Wang, L. et al. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 22, 1654–1664 (2015).
Chen, C. et al. Telomerase governs immunomodulatory properties of mesenchymal stem cells by regulating FAS ligand expression. EMBO Mol. Med. 6, 322–334 (2014).
Katsimpardi, L. et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634 (2014).
Weissman, I. L. Stem cells are units of natural selection for tissue formation, for germline development, and in cancer development. Proc. Natl Acad. Sci. USA 112, 8922–8928 (2015).
Zhan, S. S. et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 43, 435–443 (2006).
Li, F. et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal 3, ra13 (2010).
IRobbins, P. D. & Morelli, A. E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 14, 195–208 (2014).
Lotvall, J. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 3, 26913 (2014).
Akers, J. C., Gonda, D., Kim, R., Carter, B. S. & Chen, C. C. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 113, 1–11 (2013).
Julian, L. & Olson, M. F. Apoptotic membrane dynamics in health and disease. Cell Health Cytoskelet. 7, 133–142 (2015).
Mesa, K. R. et al. Niche-induced cell death and epithelial phagocytosis regulate hair follicle stem cell pool. Nature 522, 94–97 (2015).
Semon, J. A. et al. Integrin expression and integrin-mediated adhesion in vitro of human multipotent stromal cells (MSCs) to endothelial cells from various blood vessels. Cell Tissue Res. 341, 147–158 (2010).
Bernardo, M. E. & Fibbe, W. E. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell 13, 392–402 (2013).
Shi, Y. et al. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 33, 136–143 (2012).
Uccelli, A., Moretta, L. & Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 8, 726–736 (2008).
Woo, M. et al. Caspase-3 regulates cell cycle in B cells: a consequence of substrate specificity. Nat. Immunol. 4, 1016–1022 (2003).
Grootaert, M. O. et al. Caspase-3 deletion promotes necrosis in atherosclerotic plaques of ApoE knockout mice. Oxid. Med. Cell Longev. 2016, 3087469 (2016).
Ling, L., Nurcombe, V. & Cool, S. M. Wnt signaling controls the fate of mesenchymal stem cells. Gene 433, 1–7 (2009).
Boland, G. M., Perkins, G., Hall, D. J. & Tuan, R. S. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell Biochem. 93, 1210–1230 (2004).
Cho, H. H. et al. Endogenous Wnt signaling promotes proliferation and suppresses osteogenic differentiation in human adipose derived stromal cells. Tissue Eng. 12, 111–121 (2006).
Baksh, D., Boland, G. M. & Tuan, R. S. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J. Cell. Biochem. 101, 1109–1124 (2007).
Baksh, D. & Tuan, R. S. Canonical and non-canonical Wnts differentially affect the development potential of primary isolate of human bone marrow mesenchymal stem cells. J. Cell Physiol. 212, 817–826 (2007).
Gaur, T. et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 280, 33132–33140 (2005).
Bennett, C. N. et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl Acad. Sci. USA 102, 3324–3329 (2005).
Creemers, E. E., Tijsen, A. J. & Pinto, Y. M. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 110, 483–495 (2012).
Mazzoni, S. M. & Fearon, E. R. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 355, 1–8 (2014).
Zangari, J. et al. Rapid decay of engulfed extracellular miRNA by XRN1 exonuclease promotes transient epithelial-mesenchymal transition. Nucleic Acids Res. 45, 4131–4141 (2017).
Gennarino, V. A. et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res. 22, 1163–1172 (2012).
Chhabra, R., Adlakha, Y. K., Hariharan, M., Scaria, V. & Saini, N. Upregulation of miR-23a-27a-24-2 cluster induces caspase-dependent and -independent apoptosis in human embryonic kidney cells. PLoS ONE 4, e5848 (2009).
Akiyama, K. et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell 10, 544–555 (2012).
Steinert, E. M. et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749 (2015).
Kaiser, J. Aging ‘Rejuvenation factor’ in blood turns back the clock in old mice. Science 344, 570–571 (2014).
Bitto, A. & Kaeberlein, M. Rejuvenation: it’s in our blood. Cell Metab. 20, 2–4 (2014).
Acknowledgements
This work was supported by grants from University of Southern California (AMI funding to S.S), from International Science & Technology Cooperation Program of China (2015DFB30040 to S.S. and Y. Z), an Schoenleber Pilot Research Grant (to S.S) from University of Pennsylvania School of Dental Medicine, and National Institute of Dental and Craniofacial Research, National Institutes of Health, Department of Health and Human Services (R01DE017449 to S.S. and K99E025915 to C.C).
Author information
Authors and Affiliations
Contributions
D.L. designed and performed experiments, analyzed data and wrote the manuscript. C.C., X.K., S.L., Y.L., W.Y., R.Y., T.Y and R.W. performed experiments and analyzed data. S.S., and Y.Z. designed experiments, analyzed and interpreted data, wrote the manuscript and supervised the laboratory studies.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Liu, D., Kou, X., Chen, C. et al. Circulating apoptotic bodies maintain mesenchymal stem cell homeostasis and ameliorate osteopenia via transferring multiple cellular factors. Cell Res 28, 918–933 (2018). https://doi.org/10.1038/s41422-018-0070-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-018-0070-2
Keywords
This article is cited by
-
Apoptotic extracellular vesicles derived from hypoxia-preconditioned mesenchymal stem cells within a modified gelatine hydrogel promote osteochondral regeneration by enhancing stem cell activity and regulating immunity
Journal of Nanobiotechnology (2024)
-
The dual role of mesenchymal stem cells in apoptosis regulation
Cell Death & Disease (2024)
-
Tailored apoptotic vesicles promote bone regeneration by releasing the osteoinductive brake
International Journal of Oral Science (2024)
-
Enhancing osteoporosis treatment with engineered mesenchymal stem cell-derived extracellular vesicles: mechanisms and advances
Cell Death & Disease (2024)
-
Umbilical cord mesenchymal stem cell-derived apoptotic extracellular vesicles ameliorate cutaneous wound healing in type 2 diabetic mice via macrophage pyroptosis inhibition
Stem Cell Research & Therapy (2023)
