
Abstract
The lysosomal degradation pathway of macroautophagy (herein referred to as autophagy) plays a crucial role in cellular physiology by regulating the removal of unwanted cargoes such as protein aggregates and damaged organelles. Over the last five decades, significant progress has been made in understanding the molecular mechanisms that regulate autophagy and its roles in human physiology and diseases. These advances, together with discoveries in human genetics linking autophagy-related gene mutations to specific diseases, provide a better understanding of the mechanisms by which autophagy-dependent pathways can be potentially targeted for treating human diseases. Here, we review mutations that have been identified in genes involved in autophagy and their associations with neurodegenerative diseases.
Similar content being viewed by others


Introduction
The original definition of autophagy (Greek, “self-eating”) is the delivery of cytoplasmic cargoes to the lysosome for degradation. This process is conserved in all eukaryotic organisms, occurs at basal levels in nearly all cell types, and is tightly regulated by diverse intracellular and extracellular cues. Autophagy is mediated by autophagy-related genes required for the efficient formation and maturation of autophagosomes (double-membraned structures) that are targeted for degradation and recycling after fusion with lysosomes.
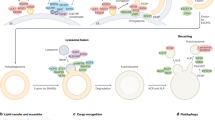
The first morphologically recognizable autophagic structures in mammalian cells are cup-shaped double membraned structures called phagophores. After the edges of the phagophores extend and fuse, they become autophagosomes. At the initial steps of autophagy, the ULK1 kinase complex (comprising of ULK1, FIP200, ATG13, and ATG101) plays a major role by phosphorylating multiple downstream effectors1. Two distinct Beclin 1/class III phosphatidylinositol 3-kinase (PI3KC3) complexes generate phosphatidylinositol 3-phosphate (PI3P) to act in autophagosome nucleation (Complex 1: Beclin 1, VPS34, VPS15, and ATG14), or endolysosomal and autophagolysosomal maturation (Complex 2: Beclin 1, VPS34, VPS15, and UVRAG). WIPI (WD repeat domain phosphoinositide-interacting) proteins together with ATG2, function in the early stages of membrane elongation at the site of PI3P generation. Autophagosome membrane expansion and completion involves the formation of the ATG5-ATG12-ATG16L1 complex and LC3/GABARAP family protein lipidation. The core ATG proteins are necessary, but not sufficient for degradative autophagy (Fig. 1). Autophagosomal degradation cannot proceed without successful fusion to functional lysosomes. Research in the past decade has unmasked several key factors required for autophagolysosomal fusion, such as the GABARAP subfamily, as well as the CORVET–HOPS and the STX17–SNAP29–VAMP8/VAMP7 SNARE complexes2,3,4,5.

The ULK1 complex activates the PI3KC3 complexes, resulting in PI3P synthesis and the nucleation of pre-autophagosome structures that may receive membrane from multiple sources, such as the ER, Golgi, plasma membrane, and recycling endosomes. PI3P is the recruitment signal for WIPI proteins and the ATG12–ATG5–ATG16L1 complex, both are required for autophagosome membrane expansion and completion. Subsequent fusion to lysosomes results in degradation of autophagosomal contents.
Autophagy was initially considered as a bulk degradation process, which lacks substrate specificity. However, we now know that there can be specificity in controlling the choice of substrate that will be degraded by autophagosomes. Numerous studies have reported processes enabling the preferential degradation of aggregation-prone misfolded proteins, such as those involved in the pathogenesis of neurodegenerative diseases, and various organelles, such as damaged mitochondria (Fig. 2)6,7,8,9. Substrates either contain an LC3-interacting region (LIR) motif that directly binds LC3 or they are labeled with ubiquitin, which is recognized by receptor proteins (also called autophagy adaptors). Receptor proteins contain both an ubiquitin-binding domain and a LIR motif, thus serving as a bridge between the substrates and the LC3 conjugated to phagophores (Fig. 2). Some autophagy adaptors/receptors, such as the autophagy-linked FYVE protein (ALFY) and optineurin, have a dual role as they can bind cargoes, but they also regulate the recruitment of the ULK1 and PI3KC3 complexes to initiate autophagosome formation10,11,12,13.

Through the action of autophagy receptors, specific cargoes are recognized and incorporated into autophagosomes for selective degradation180. Autophagy receptors can therefore mediate aggrephagy, mitophagy, pexophagy, ER-phagy, lysophagy, xenophagy, or pexophagy. While this figure aims to provide an overview of the many autophagy receptors involved in different forms of selective autophagy, we have focussed in the text on the most important examples involved in Mendelian neurodegenerative diseases, like P62, ALFY, and OPTN.
Autophagy is required for normal maintenance and function of the mammalian nervous system14,15. Multiple studies employing tissue-specific knockout mouse models of core autophagy genes have revealed that autophagy plays a key role in clearing aggregate-prone proteins associated with neurodegeneration and its impairment results in reduces survival and causes progressive neurodegeneration across broad areas of the central and peripheral nervous system14,15. Here, we review the known genetic mutations of molecules involved in bulk and selective autophagy and their associations with neurodegenerative diseases (summarized in Table 1).
Autophagy related 5 (ATG5)
ATG5 is a core autophagy protein that functions in autophagosome formation and is required for both of the ubiquitin-like conjugation systems involved in LC3 lipidation. ATG5 is first irreversibly conjugated to ATG12 by the E1-like ATG7 and E2-like ATG1016. Then, the ATG5–ATG12 conjugate non-covalently interacts with ATG16L1 to form the ATG5–ATG12–ATG16L1 complex, which functions as an E3-like enzyme for the covalent conjugation of the C-terminal glycine of LC3 to phosphatidylethanolamine (PE), resulting to LC3-II formation17 (Fig. 1).
Whole-exome sequencing of two Turkish siblings with childhood ataxia associated with hypoplasia of the cerebellar vermis identified a homozygous missense mutation (E122D) in ATG518. E122 is located in the vicinity of the ATG12–ATG5 interaction surface and the E122D mutation impaired the conjugation of ATG5 to ATG12. In patient‐derived lymphoblastoid cell lines, the levels of ATG5–ATG12 conjugate is severely reduced, therefore leading to impaired autophagosome formation18. Ataxia is a common clinical sign of impaired coordination of movement and balance during voluntary activity, mainly caused by dysfunction of the complex circuitry connecting the basal ganglia, cerebellum, and cerebral cortex. Dysfunction of the cerebellar vermis causes staggering gait, asynergic movements, dysmetria, nystagmus, intentional tremor, and difficulty in forming speech19.
SNPs and variants causing dysregulation of ATG5 expression levels have also been reported. A homozygous ATG5 variant (rs6568431 AA genotype) resulting in lower ATG5 expression was identified to be associated with childhood cerebral palsy (CP)20. It will be important to validate these findings in subsequent replication studies. CP is a group of permanent motor disorders, often accompanied by disturbances of sensation, perception, cognition, communication and behavior, epilepsy, and secondary musculoskeletal problems. It is caused by nonprogressive disturbances that occurred in the developing fetal or infant brain21. A heterozygous ATG5 variant, located in the promoter region (106774459T > A), which leads to increased expression levels of ATG5, was identified in one Parkinson’s disease (PD) patient22. However, it is unclear how and if this variant contributes to the PD pathogenesis.
Vacuolar protein sorting-associated protein 15 (VPS15)
VPS15 is the regulatory subunit and membrane targeting factor of the PI3KC3 complex, which regulates the production of PI3P23. VPS15 and VPS34 together with beclin 1 form three distinctive PI3K complexes. These core components together with ATG14L form the PI3KC3 complex 1, which is essential for the induction of canonical autophagy. In PI3KC3 complex 2, ATG14L is replaced by UVRAG, and the activity is directed towards regulation of endocytosis and endosome fusion, and in complex 3 Rubicon binds to UVRAG to negatively regulate the activity of this complex24,25.
A VPS15 missense mutation (L1224R) has been identified in a patient that presented with severe cortical and optic nerve atrophy, localized cortical dysplasia, intellectual impairment, spasticity, ataxia, psychomotor delay, muscle wasting, pseudobulbar palsy, a mild hearing deficit, and late-onset epilepsy26. Human fibroblasts carrying the L1224R mutation have an accumulation of autophagy substrates, as well as reduced levels of VPS15, VPS34, and beclin126, indicating that the PI3KC3 complex assembly and therefore PI3P production is impaired.
WD repeat domain phosphoinositide-interacting protein 4 (WIPI4)
WIPI proteins (WIPI1–WIPI4) are the only known PtdIns3P-binding proteins with conserved PI3P effector functions that recruit downstream regulators and facilitate the formation of autophagosomes27. WIPI4 associates with ATG2, a lipid-transfer protein that functions in autophagosome expansion and closure28. The recruitment of ATG2–WIPI4 complex onto phagophores requires ATG9 and the TRAPPIII complex that is required for autophagosome closure29.
Mutations of WIPI4 (encoded by the WDR45 gene), resulting in truncated proteins that are prone to degradation, were found to cause an X-linked dominant subtype of neurodegeneration with iron accumulation (NBIA), known as BPAN (beta-propeller protein-associated neurodegeneration)30,31,32,33,34. Central nervous system (CNS)-specific Wdr45 knockout mice recapitulate some phenotypes of BPAN and show ubiquitin-positive protein aggregates and p62/SQSTM1 accumulation in neurons, particularly in axons, suggesting a blockage of autophagy flux35. Lymphoblastoid cell lines derived from BPAN patients showed impaired autophagy flux and accumulation of LC3-positive autophagosome membranes that were also positive for ATG931. BPAN patients are characterized by neurological movement disorder, progressive degeneration of the nervous system, and strong iron deposition in the substantia nigra (SN) and globus pallidus (GP).
Sequestosome 1 (SQSTM1; also known as p62)
p62/SQSTM1 is a multifunctional scaffold protein, which plays important roles as an adaptor/receptor for selective autophagy, but it also mediates activation of the mammalian target of rapamycin complex 1 (mTORC1) on lysosomes and the Keap1–Nrf2 pathway, a major cellular defense mechanism against oxidative stress36. Among its multiple domains, Phox1 and Bem1p domain (PB1), LIR, and the C-terminal ubiquitin associated domain (UBA) domains are required for selective autophagy37,38,39, whereas the Kelch-like ECH associated protein 1 (Keap1)-interacting region is required for Keap1 binding40.
Several mutations in p62/SQSTM1 have been identified in cases of familial and sporadic amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD), leading to loss of function of p62/SQSTM1 in selective autophagy41,42, or a decreased interaction with Keap143. In the brains of the ALS–FTLD patients, p62/SQSTM1 is detected in ubiquitin intracellular protein aggregates, suggesting that p62/SQSTM1 and ubiquitin contributes to the formation of inclusions44. The familial ALS-associated P392L mutation45, located in the UBA domain of p62/SQSTM1, impairs the binding ability of p62/SQSTM1 to polyubiquitin chains, resulting in loss of basal autophagy46. Moreover, the L341V mutation in the LIR domain found in sporadic ALS47 impairs its binding to LC3, therefore inhibiting p62/SQSTM1-mediated delivery of ubiquitinated substrates to autophagosomes48. Also, the ALS–FTLD-associated p62/SQSTM1 P348L and G351A mutations impair Keap1 binding, resulting in reduced Nrf2 signaling and aberrant expression of oxidative response genes48. ALS and FTLD are highly related neurodegenerative disorders on the same pathological disease spectrum. ALS clinical signs include motor neuron degeneration, resulting in progressive muscle weakness and paralysis, eventually leading to fatal respiratory failure. The term FTLD covers a range of clinical syndromes associated by atrophy of the frontal temporal lobes in the brain, characterized by behavioral and personality changes41
Autophagy-linked FYVE protein (ALFY)
ALFY (encoded by the WDFY3 gene) is a member of the Beige and Chendiak-Higashi (BEACH) domain-containing protein family involved in vesicle transport, membrane fission and fusion events, and autophagy49. ALFY is required for the recruitment of ubiquitinated proteins to cytosolic p62/SQSTM1 bodies and nuclear promyelocytic leukemia nuclear bodies (PML-NBs)10. ALFY, together with p62/SQSTM1 and interactions with LC3 and the ATG5–ATG12 complex, drives the formation of larger aggregates that are targeted for autophagic degradation10,11. In addition to its role as an autophagy receptor, ALFY binds PI3P in the autophagic membrane through its FYVE domain and facilitates the binding of ATG5–ATG12 to ATG1610, therefore acting as an autophagosome nucleation inducer.
WDFY3 is a recently identified disease gene where mutations are seen in autosomal dominant primary microcephaly (MCPH)50. Primary microcephaly is a group of autosomal disorders characterized by a reduction in the number of neurons, resulting in a smaller brain volume coupled with a head circumference of at least three standard deviations below the age- and sex-specific means50,51. Individuals with MCPH usually have intellectual disability and speech delay, with varying degrees of motor delay50,51. The heterozygous mutation R2637W in ALFY, located in the PH domain, is directly related to its ability to remove protein aggregates from the cytosol and flies expressing this mutation have smaller and malformed brains with clusters of cells containing ALFY-positive aggregates52. Lack of ALFY activity leads to aberrant activation of the Wnt signaling pathway, resulting in reduced neuronal differentiation and microcephaly52. Similarly, another mutation located in the PH domain (G2558S) was found to be associated with intellectual disability and microcephaly53. By contrast, a missense mutation in the BEACH domain (R2823W), as well as several de novo pathogenic variants of WDFY3 leading to haploinsufficiency of the gene, result in macrocephaly and cause an autosomal dominant neurodevelopmental disorder with mild-to-moderate neurodevelopmental delay and intellectual disability associated with deficits in motor coordination, as well as autism spectrum disorders53. Interestingly, studies on heterozygous Wdfy3+/lacZ mice showed that Wnt signaling is inhibited53. The proposed mechanism for this discrepancy describes that mutations on the PH domain of ALFY would lead to aberrant upregulation of Wnt signaling and microcephaly, whereas truncating variants and mutations in the BEACH domain will lead to downregulation of Wnt signaling and macrocephaly53.
Parkin and PINK1
The E3 ligase Parkin and the serine–threonine kinase PINK1 (encoded by the PARK2 and PARK6 genes, respectively) play important roles in the maintenance of mitochondrial quality and the degradation of damaged mitochondria through mitophagy54,55. PINK1, located in the inner mitochondrial membrane, is constitutively cleaved in the inner mitochondrial membrane by the presenilin-associated rhomboid-like protein and is degraded by the proteasome56,57,58. However, when mitochondrial membrane potential is disrupted, PINK1 is no longer imported into mitochondria and accumulates on the outer mitochondrial membrane56,57. PINK1 on the outer membrane phosphorylates Parkin as well as ubiquitin located on mitochondria, thereby releasing Parkin’s autoinhibition and inducing its E3 ligase activity, as well as promoting its recruitment to damaged mitochondria59,60,61. The assembly of Parkin-mediated polyubiquitin chains on damaged mitochondria initiates the recruitment of autophagy receptors, such as p62/SQSTM1 and optineurin (OPTN), as well as activation of the TANK-binding kinase1 (TBK1)37,62,63. PINK1, independently of Parkin, also recruits OPTN and NDP52/CALCOCO2 to mitochondria to activate mitophagy, which also induce the recruitment of the components of the autophagosome formation machinery, such as ULK1 and WIPI proteins64.
Parkin and PINK1 mutations are a major cause of early-onset PD65,66,67,68,69,70. Considerations have been raised regarding the roles of Parkin and PINK1 in mitophagy, as studies using primary neurons or neurons derived from induced pluripotent stem cells resulted in several controversies71,72,73. However, studies, using patient derived fibroblasts have revealed aberrant mitochondrial function and morphology74,75,76,77,78, indicative of mitophagy dysfunction. Patients with PARK2 mutations show the typical signs characterizing PD, which are rigidity, bradykinesia, postural instability, and tremor, as well as additional signs such as freezing gait and lower foot dystonia. In contrast to typical idiopathic PD, cognitive dysfunction, hallucination, and REM behavioral disorder are not seen. Patients with PARK6 mutations show clinical symptoms similar to sporadic PD79. However, the accumulation of Lewy body aggregates, a pathognomonic feature of sporadic PD, is not a typical feature of Parkin-associated Parkinsonism80.
Optineurin (OPTN)
OPTN is a highly conserved and ubiquitously expressed cytoplasmic protein that contains several domains including a LIR motif and an ubiquitin-binding region81,82,83. OPTN is an autophagy adaptor/receptor protein that participates particularly in the selective autophagy of protein aggregates/pathological inclusions, damaged mitochondria, and intracellular pathogens12,62,84,85,86,87. OPTN is recruited by PINK1 to damaged mitochondria and subsequently recruits ULK1 in an ubiquitin-binding dependent manner12. OPTN also promotes autophagosomal elongation by mediating the recruitment of the ATG12–ATG5–ATG16L1 complex to phagophores13. OPTN also regulates autophagosomal maturation through its interaction with the GTPase Rab1a88, and autophagosome–lysosome fusion through its interaction with the myosin VI complex89,90.
Mutations and polymorphisms in OPTN have been linked to neurodegenerative diseases, including ALS91,92,93 and glaucoma82,83,94. A number of studies have shown that glaucoma-associated OPTN mutations induce defective autophagy and increased cell death both in vitro as well as in vivo (in mice expressing the glaucoma-linked E50K mutation)95,96,97,98. In ALS, OPTN mutations have pathogenic effects as a consequence of deletions of the exon 4 or 5 or nonsense mutations encoding truncated proteins lacking its C-terminal85,92,99. More than 20 ALS-linked missense OPTN mutations have been reported. However, the role of most mutations on disease progression is still poorly understood. The ALS-linked OPTN mutations Q398X and E478G lead to failure in myosin VI binding and Myb 1 (Tom1) targeting, causing inhibition of autophagosome–lysosome fusion90,100. Another heterozygous missense mutation in OPTN (V295F) induces ALS-like cellular phenotypes, like Golgi fragmentation and increased ER stress101.
TANK-binding kinase 1 (TBK1)
TBK1 is a ubiquitously expressed protein kinase and contains a serine/threonine kinase domain, an ubiquitin-like domain and two coiled-coil domains (CCD1 and CCD2) that mediate homodimerization102. TBK1 has a critical role in autophagy by regulating the phosphorylation of the autophagy receptors OPTN, NDP52/CALCOCO2, and p62/SQSTM162,102. Phosphorylation of OPTN by TBK1 increases its affinity to LC3 and its ubiquitin binding62,84. The ALS-associated E696K TBK1 mutant, which is deficient in OPTN binding, fails to be recruited to damaged mitochondria103, suggesting that mitophagy by OPTN is mediated by TBK1. In addition, TBK1 phosphorylates p62/SQSTM1 on a residue essential for its role in autophagic clearance104. Recent studies have suggested that TBK1 is also critical for autophagy initiation by regulating syntaxin 17 (Stx17) phosphorylation and the formation of FIP200-ATG13 pre-autophagosomal structures105. TBK1 has also been implicated in the maturation of autophagosomes into autolysosomes104, probably through regulation of the cytoplasmic levels of dynein106 that regulates autophagosome transport via microtubules to lysosomes.
Mutations in TBK1 have been shown to cause ALS, FTD, and a combination of both (ALS–FTD). From a number of genetics studies with ALS, ALS–FTD, and FTD patients, more than 40 mutations have been found in the TBK1 gene that cause either a frameshift or a premature stop leading to truncated products102,107. Several ALS-causing TBK1 mutations induce protein truncation resulting in loss of the CCD2 domain in TBK1, decreasing its kinase activity107. Some TBK1 mutations also result in decreased mRNA and protein levels107, which may decrease activation of autophagy receptors, resulting in decreased autophagic substrate clearance, and accumulation of protein aggregates in neurons108.
Vacuolar protein sorting 13D (VPS13D)
VPS13D belongs to the VPS13 family, which in humans comprises of 4 proteins (VPS13A-D) that appear to be critical for the non-vesicular transport of lipids between adjacent organelles109,110. Studies in Drosophila intestinal cells reveal that VPS13D plays an important role in mitochondrial fission and mitophagy through its putative ubiquitin-binding UBA domain111. VPS13D functions downstream of the recruitment of the fission factor Drp1 to control mitochondrial fission, and upstream of ATG8A to control mitophagy111. However, little is known about the specific molecular function of VPS13D.
Several heterozygous mutations in VPS13D have been identified in families with neurological disorders112,113. Patient-derived fibroblasts and muscle biopsies show severe alterations in mitochondrial morphology and energy production112,113, indicating accumulation of damaged mitochondria. The disease associated with rare recessive VPS13D variants is a novel, complex, hyperkinetic neurological disorder of variable age of onset that can be associated with developmental delay and progressive spastic ataxia or paraplegia. Individuals can present with motor delays and gait instability, as well as cognitive impairment. Individuals have also presented with dysarthria, and eye movement abnormalities. The disorder progresses to spastic ataxia or generalized dystonia, which can lead to loss of independent ambulation112,113.
Peroxisomal membrane protein 13 (PEX13)
PEX13 is a transmembrane protein that forms part of the core of the docking/translocation module in peroxisomes and regulates protein import during peroxisome biogenesis114,115. PEX13 loss-of-function impairs the clearance of damaged mitochondria and the degradation of Sindbis virus, without disturbing general autophagy116,117. Although PEX13 seems to be important for mitophagy and virophagy, the mechanism of PEX13 in selective autophagy remains elusive.
Mutations in PEX13, lead to Zellweger syndrome (ZS), associated with impaired neuronal migration, neuronal positioning, and brain development118. Several mutations has been described in PEX13, including missense, nonsense, deletions, and splice site mutations, that lead to complete or partial impairment in peroxisome function118,119,120. As peroxisomes are involved in the biosynthesis of phospholipids, oxidation of fatty acids and detoxification, patients with ZS accumulate long-chain fatty acids, pristanic acid, phyntanic acid, plasmalogens, and C27-bile acid intermediates121,122. Mice lacking PEX13 exhibit many of the clinical features of ZS patients, as they lack morphologically intact peroxisomes and have abnormal mitochondrial distribution and morphologies117,123. Fibroblasts from PEX13-deficient mice or patient fibroblasts with PEX13 mutations show increased levels of mitochondrial superoxide and membrane depolarization, suggesting that PEX13 loss-of-function can cause aberrant mitochondrial dynamics and impaired mitophagy116,117,124,125. Clinically, ZS patients present with severe hypotonia, ocular abnormalities, such as cataracts or glaucoma, and often show seizures, renal cysts, and hepatic dysfunction122,126.
Valosin-containing protein (VCP; also known as p97 or Cdc48)
VCP is a member of the AAA + -ATPase family of chaperone-like proteins and is ubiquitously expressed in all tissues and throughout the brain127. Through its interaction with ubiquitinated proteins, VCP regulates diverse cellular functions, including protein degradation through the proteasome and autophagy128. VCP is essential for autophagosome maturation through a yet undefined mechanism. Cells that lack VCP or that express disease-associated VCP mutants have an accumulation of immature autophagic vesicles and fail to degrade autophagic substrates, whereas patient-derived myoblasts accumulate aberrant autophagosomes and endo-lysosomes129,130,131.
Mutations in VCP cause a rare and complex multisystem degenerative disease that manifests with inclusion body myopathy, Paget’s disease of bone, and frontotemporal dementia, as well as ALS132,133. VCP disease-associated mutations are found throughout the protein sequence, but mostly reside within the N-terminal and D1/2 domains, which are regions important for substrate and cofactor association, as well as VCP ATPase activity, resulting to either imbalanced cofactor binding or aberrant ATPase activity, as well as reduced protein expression134.
Ubiquilin 2 (UBQLN2)
UBQLN2 is a chaperone protein that delivers ubiquitinated cargo for proteosomal degradation via its ubiquitin-associated (UBA) and UBL domains135. However, a role for UBQLN2 in regulating autophagic flux has now also emerged. Initial studies showed that UBQLN2 binds LC3 and cells depleted from UBQLN2 have impaired autophagosome degradation136,137. Recent studies in flies showed that Ubqn/UBQLN is required for Torc1/mTORC1 activity and lysosomal acidification through its interaction with v-ATPase and its loss leads to defective autophagy flux138. Consistent with these data, depletion of UBQLN1, UBQLN2, and UBQLN4 (UBQLNs) in human cells also cause reduced mTORC1 activity and impaired autophagic degradation138.
Mutations of UBQLN2 gene are found in cases of familial ALS and ALS–FTD135 and the majority reside within its proline-rich domain that is important for protein–protein interactions139. Expression of the ALS-associated UBQLN2 P497H mutation in flies results in impaired autophagic degradation, due to a failure in lysosomal acidification138, whereas rat spinal motor neurons expressing the UBQLN2 P497H mutation have decreased LC3 lipidation140. Interestingly, expression of the P497H or P506T ALS-associated UBQLN2 mutations in human neuroblastoma cells leads to a deficiency in protein degradation through the proteasome system139. Therefore, further work is needed to elucidate the involvement of UBQLN2 in autophagy and disease.
Spastizin, spatacsin, and tectonin β-propeller repeat-containing protein 2 (TECPR2)
Spastic gait genes (SPG), identified by genetic studies of hereditary spastic paraplegia (HSP) patients, regulate several pathways, including ER function, intracellular membrane trafficking, mitochondrial regulation, myelination, lipid metabolism, and autophagy141,142.
Spastizin and spatacsin (encoded by the SPG15 and SPG11 genes, respectively) physically interact and have similar subcellular distributions, including endosomes, lysosomes, and ER143,144,145,146,147. Spastizin and spatacsin play critical roles in autophagic lysosome reformation (ALR), a mechanism of lysosomal biogenesis that maintains a pool of lysosomes able to fuse with autophagosomes after prolonged starvation148,149. However, spastizin and spatacsin are differentially involved in autophagosome–endosome fusion. Both proteins interact with the AP-5 complex and the small GTP-binding proteins Rab5A and Rab11, which regulate endosome trafficking and maturation146,150, whereas spastizin interacts with the beclin1–UVRAG complex and regulates autophagosome maturation151,152. Interestingly, although spastizin mutations impair autophagosome–endosome fusion, spatacsin loss of function had no effect, implying that only spastizin acts at the intersection between endocytosis and autophagy150,153.
Tectonin β-propeller repeat-containing protein 2 (TECPR2) has a C-terminal LIR motif which binds to LC3/GABARAP proteins and acts as positive regulator of autophagy154,155,156. Its association with several trafficking components, including COPII coat proteins, is required for the maintenance of functional ER exit site and efficient ER export in an LC3 binding-depending manner156. However, the exact functions of TECPR2 are still unclear.
HSPs are rare inherited disorders characterized by age-dependent neurodegeneration of motor neurons, manifesting with weakness of the legs (paraplegia) and involuntary spasms and muscle stiffness (spasticity). Spastic paraplegia type 11 (SPG11), type 15 (SPG15), and type 49 (SPG49) are classified as complex HSPs, because they involve all four limbs as well as additional features, including abnormalities of the brain, such as a thin corpus callosum157. SPG11 and SPG15 are the two most common types of HSP. The onset of symptoms varies greatly. However, abnormalities usually become noticeable in adolescence. So far, more than 100 SPG11 and SPG15 mutations have been described, being either nonsense or insertions or deletions leading to a frameshift, suggesting a loss-of-function mechanism. Indeed, mice lacking spastizin or spatacsin develop symptoms consistent with spastic paraplegia145,149. The clinical features in SPG11 and SPG15 are often indistinguishable, with distinctive features of early onset Parkinsonism, cognitive impairment, white matter changes, mild cerebellar ataxia, retinal abnormalities, and lens opacities157,158,159. SPG49 is caused by mutated TECPR2, resulting in a premature stop codon and expression of truncated, rapidly degraded TECPR2 protein155. SPG49 often begins in childhood, starting with weak muscle tone, which gradually worsens, causing difficulty in walking. In addition, affected individuals have moderate to severe intellectual disability and distinctive physical features, including short stature and microcephaly. In SPG49 patients, autonomic neurons are affected, causing abnormalities in involuntary body functions such as heart rate, digestion, and breathing. Patients often experience gastroesophageal reflux, which could lead to life-threatening bacterial lung infections.
Ectopic P granules protein 5 (EPG5)
EPG5 is involved in autophagosome maturation and fusion to the lysosome160,161,162. EPG5 interacts with Rab7 on late endosomes, co-localizes with LC3-positive autophagosomes and the assembled SNARE complex STX17–SNAP29–VAMP8/VAMP7163, which is essential for fusion of autophagosomes to lysosomes3. These observations, together with convincing in vitro data, suggest that GTP-bound Rab7 recruits EPG5 to late endosomes, enabling endosome–autophagosome fusion followed by recruitment and assembly of the STX17–SNAP29–VAMP8/VAMP7 SNARE complex163. Consequently, EPG5 functions as a tether to specifically direct fusion of autophagosomes and endosomes and subsequent fusion to the lysosome.
EPG5 mutations cause Vici syndrome164,165,166, a rare, recessive neurodevelopmental disorder with multisystem involvement, involving underdeveloped corpus callosum, cataracts, cardiomyopathy, combined immunodeficiency, developmental delay, hypopigmentation, and failure to thrive165. Most of the mutations identified in patients result in a non-functional EPG5 protein due to truncation, frameshift, premature stop codons or splicing errors. Consistent with a role for EPG5 in autophagosome fusion to the lysosome, studies in patient fibroblasts revealed an accumulation of LC3 and autophagic substrates due to impaired autophagic flux164,165. Endocytotic uptake and degradation was normal in the patient cells, indicating that lysosomal function is maintained and that the EPG5 mutation affects an earlier step in the autophagosome maturation process165. Furthermore, genetic variants of EPG5 have been associated with altered age of onset in Alzheimer’s disease167, consistent with the importance of autophagy in protection against neurodegeneration.
Vacuolar protein sorting-associated protein 11 (VPS11)
VPS11 is part of the CORVET–HOPS complex, which is essential for fusion of endosomes or autophagosomes with lysosomes168. The C846G missense mutation in VPS11 causes aberrant ubiquitination, accelerated turnover of the protein and impaired protein complex stability169. A very rare mutation has been also found (reported in two siblings) that involves a 9 amino acid deletion in VPS11 (ΔL387_G395) leading to reduced protein levels170. The HOPS complex instability (due to loss or reduced levels of VPS11) is associated with dysfunctional autophagy–lysosome trafficking169.
Mutations in VPS11 cause genetic leukoencephalopathies (gLEs), a group of heterogeneous disorders with abnormalities affecting the CNS. Patients affected with gLEs manifest variable neurologic phenotypes, including motor impairment, hypotonia, pyramidal dysfunction, dystonia, and/or dyskinesia, ataxia, seizures, cortical blindness, optic atrophy, and impaired cognitive development171. The neuro-imaging features include severe hypomyelination with clusters of membranous cytoplasmic bodies in dermal unmyelinated nerve axons170, similar to what it has been seen in classic lysosomal storage diseases.
Sorting nexin-14 (SNX14)
SNX14 belongs to the PXA–RGS–PX–PXC subfamily, along with SNX13, SNX19, and SNX25.41 Similar to SNX13, SNX14 contains a putative double transmembrane domain including a short cytoplasmic leader sequence and an RGS domain (regulator of G protein signaling)172, implying they share similar functions. Different hypotheses have been formulated about the cellular localization and function of SNX14. Although SNX14 is co-expressed with endosomal sorting markers173, it is localized in lysosomes, but not in endosomal or Golgi compartments174,175. Moreover, a lipid-binding assay using the PX domain of SNX14 shows binding to PI(3,5)P2176, the predominant phosphoinositide associated with lysosomes. SNX14 also localizes at ER–lipid droplet contact sites and it may play a role in fatty acid homeostasis, which when perturbed contributes to neurological disease175,177. Altogether, these findings support the notion that SNX14 is required for proper lysosomal function and lipid homeostasis.
Mutations in SNX14 cause autosomal recessive spinocerebellar ataxia 20 (SCAR20), a neurodevelopmental disorder characterized by severely delayed psychomotor development with poor or absent speech, absent gait and cerebellar atrophy. At least 18 different point mutations or deletions have been described, with most resulting in either truncation or complete loss of the SNX14 protein175. Fibroblasts from patients carrying a homozygous splice site SNX14 mutation show accumulation of vacuoles with fine nonspecific granular material and electron-dense laminated inclusions178, suggesting that loss of function of SNX14 leads to lipid degeneration and structures similar to multilamellar bodies. In addition, SNX14 mutant fibroblasts show abnormal accumulation of p62/SQSTM1-positive structures, indicating defective autophagosome degradation178. Consistent with a role in lysosomal function, mutant fibroblasts have reduced levels of cathepsin D174,175 and accumulation of cholesterol in lysosomes179.
Concluding remarks
Accumulating evidence indicates that impaired autophagy results in human disease. Here, we have reviewed the genetic variants found in autophagy related proteins and their association to neurodegenerative diseases. These proteins act steps throughout the autophagy itinerary and their loss-of-function results in diverse neurodegenerative conditions, ranging from developmental disorders to motor neuron diseases and dementia-associated conditions. Some of these studies are restricted to individual small families (not large enough to generate significant genetic linkage), or small genetic association studies that do not have genome-wide significance. Also, most of the disease-associated genetic variants have not been characterized in animal models and the only available functional data are from in vitro studies. Hence, additional work is required to provide robust genetic support, although in some cases the functional data are consistent with disease causality. Further studies towards understanding how autophagy can be modulated in normal and disease conditions and how autophagy perturbation leads to such a diverse conditions are fundamental to understand how to treat human diseases. Drug discovery efforts towards the development of pharmacological agents that will restore normal levels of autophagy in human brains are also required. This is a challenging task as we currently have no assay to measure autophagy flux in human brains.
Change history
16 June 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Morishita, H. & Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol.35, 453–475 (2019).
Kriegenburg, F., Ungermann, C. & Reggiori, F. Coordination of autophagosome-lysosome fusion by Atg8 family members. Curr. Biol.28, R512–R518 (2018).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell151, 1256–1269 (2012).
Balderhaar, H. J. & Ungermann, C. CORVET and HOPS tethering complexes—coordinators of endosome and lysosome fusion. J. Cell Sci.126, 1307–1316 (2013).
Yu, L., Chen, Y. & Tooze, S. A. Autophagy pathway: cellular and molecular mechanisms. Autophagy14, 207–215 (2018).
Chu, C. T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis.122, 23–34 (2019).
Evans, C. S. & Holzbaur, E. L. F. Quality control in neurons: mitophagy and other selective autophagy mechanisms. J. Mol. Biol.432, 240–260 (2020).
Fritsch, L. E., Moore, M. E., Sarraf, S. A. & Pickrell, A. M. Ubiquitin and receptor-dependent mitophagy pathways and their implication in neurodegeneration. J. Mol. Biol. (2019) Epub ahead of print.
Conway, O., Akpinar, H. A., Rogov, V. V. & Kirkin, V. Selective autophagy receptors in neuronal health and disease. J. Mol. Biol. (2019) Epub ahead of print.
Filimonenko, M. et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell38, 265–279 (2010).
Schläfli, A. M., Isakson, P., Garattini, E., Simonsen, A. & Tschan, M. P. The autophagy scaffold protein ALFY is critical for the granulocytic differentiation of AML cells. Sci. Rep.7, 12980 (2017).
Lazarou, M. et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature524, 309–314 (2015).
Bansal, M. et al. Optineurin promotes autophagosome formation by recruiting the autophagy-related Atg12-5-16L1 complex to phagophores containing the Wipi2 protein. J. Biol. Chem.293, 132–147 (2018).
Menzies, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron93, 1015–1034 (2017).
Menzies, F. M., Fleming, A. & Rubinsztein, D. C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci.16, 345–357 (2015).
Mizushima, N. et al. A protein conjugation system essential for autophagy. Nature395, 395–398 (1998).
Ichimura, Y. et al. A ubiquitin-like system mediates protein lipidation. Nature408, 488–492 (2000).
Kim, M. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife5, e12245 (2016).
Pavone, P. et al. Ataxia in children: early recognition and clinical evaluation. Ital. J. Pediatr.43, 6 (2017).
Xu, J. et al. A variant of the autophagy-related 5 gene is associated with child cerebral palsy. Front. Cell Neurosci.11, 407 (2017).
Graham, H. K. et al. Cerebral palsy. Nat. Rev. Dis. Prim.2, 15082 (2016).
Chen, D. et al. A novel and functional variant within the ATG5 gene promoter in sporadic Parkinson’s disease. Neurosci. Lett.538, 49–53 (2013).
Backer, J. M. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J.410, 1–17 (2008).
Itakura, E., Kishi, C., Inoue, K. & Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell19, 5360–5372 (2008).
Matsunaga, K. et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol.11, 385–396 (2009).
Gstrein, T. et al. Mutations in Vps15 perturb neuronal migration in mice and are associated with neurodevelopmental disease in humans. Nat. Neurosci.21, 207–217 (2018).
Proikas-Cezanne, T., Takacs, Z., Donnes, P. & Kohlbacher, O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J. Cell Sci.128, 207–217 (2015).
Osawa, T. et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol.26, 281 (2019).
Stanga, D. et al. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to preautophagosomal membranes. Traffic20, 325–345 (2019).
Haack, T. B. et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet.91, 1144–1149 (2012).
Saitsu, H. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet.45, 445–449 (2013).
Zarate, Y. A. et al. Lessons from a pair of siblings with BPAN. Eur. J. Hum. Genet.24, 1080–1083 (2016).
Carvill, G. L. et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia59, E5–E13 (2018).
Akcakaya, N. H. et al. A novel and mosaic WDR45 nonsense variant causes beta-propeller protein-associated neurodegeneration identified through whole exome sequencing and X chromosome heterozygosity analysis. Neuromol. Med.21, 54–59 (2019).
Zhao, Y. G. et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy11, 881–890 (2015).
Katsuragi, Y., Ichimura, Y. & Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J.282, 4672–4678 (2015).
Pankiv, S. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem.282, 24131–24145 (2007).
Moscat, J., Diaz-Meco, M. T. & Wooten, M. W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci.32, 95–100 (2007).
Ciuffa, R. et al. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep.11, 748–758 (2015).
Liu, W. J. et al. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett.21, 29 (2016).
Rea, S. L., Majcher, V., Searle, M. S. & Layfield, R. SQSTM1 mutations-bridging Paget disease of bone and ALS/FTLD. Exp. Cell Res.325, 27–37 (2014).
van der Zee, J. et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol.128, 397–410 (2014).
Goode, A. et al. ALS-FTLD associated mutations of SQSTM1 impact on Keap1-Nrf2 signalling. Mol. Cell Neurosci.76, 52–58 (2016).
Nakano, T., Nakaso, K., Nakashima, K. & Ohama, E. Expression of ubiquitin-binding protein p62 in ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis with dementia: analysis of five autopsy cases with broad clinicopathological spectrum. Acta Neuropathol.107, 359–364 (2004).
Fecto, F. et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol.68, 1440–1446 (2011).
Hiruma, Y. et al. A SQSTM1/p62 mutation linked to Paget’s disease increases the osteoclastogenic potential of the bone microenvironment. Hum. Mol. Genet.17, 3708–3719 (2008).
Chen, Y. et al. SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging35, 726 e727–726 e729 (2014).
Goode, A. et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy12, 1094–1104 (2016).
Cullinane, A. R., Schäffer, A. A. & Huizing, M. The BEACH is hot: a LYST of emerging roles for BEACH‐domain containing proteins in human disease. Traffic14, 749–766 (2013).
Jayaraman, D., Bae, B. I. & Walsh, C. A. The genetics of primary microcephaly. Annu. Rev. Genomics Hum. Genet.19, 177–200 (2018).
Létard, P. et al. Autosomal recessive primary microcephaly due to ASPM mutations: an update. Hum. Mutat.39, 319–332 (2018).
Kadir, R. et al. ALFY-controlled DVL3 autophagy regulates Wnt signaling, determining human brain size. PLoS Genet.12, e1005919 (2016).
Le Duc, D. et al. Pathogenic WDFY3 variants cause neurodevelopmental disorders and opposing effects on brain size. Brain142, 2617–2630 (2019).
Narendra, D., Tanaka, A., Suen, D. F. & Youle, R. J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol.183, 795–803 (2008).
Abou-Sleiman, P. M., Muqit, M. M. & Wood, N. W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci.7, 207–219 (2006).
Jin, S. M. et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol.191, 933–942 (2010).
Meissner, C., Lorenz, H., Weihofen, A., Selkoe, D. J. & Lemberg, M. K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem.117, 856–867 (2011).
Yamano, K. & Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy9, 1758–1769 (2013).
Kondapalli, C. et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol.2, 120080 (2012).
Shiba-Fukushima, K. et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep.2, 1002 (2012).
Koyano, F. et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature510, 162–166 (2014).
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J. & Harper, J. W. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell60, 7–20 (2015).
Richter, B. et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl Acad. Sci. USA113, 4039–4044 (2016).
Vargas, J. N. S. et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell74, 347–362 e346 (2019).
Shimura, H. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet.25, 302–305 (2000).
Imai, Y., Soda, M. & Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem.275, 35661–35664 (2000).
Valente, E. M. et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science304, 1158–1160 (2004).
Matsumine, H. et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet.60, 588–596 (1997).
Kitada, T. et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature392, 605–608 (1998).
Valente, E. M. et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet.68, 895–9002 (2001).
Van Laar, V. S. et al. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet.20, 927–940 (2011).
Rakovic, A. et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem.288, 2223–2237 (2013).
Ashrafi, G., Schlehe, J. S., LaVoie, M. J. & Schwarz, T. L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol.206, 655–670 (2014).
Grunewald, A. et al. Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp. Neurol.219, 266–273 (2009).
Rakovic, A. et al. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum. Mol. Genet.19, 3124–3137 (2010).
Hoepken, H. H. et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis.25, 401–411 (2007).
Auburger, G. et al. Primary skin fibroblasts as a model of Parkinson’s disease. Mol. Neurobiol.46, 20–27 (2012).
Zanellati, M. C. et al. Mitochondrial dysfunction in Parkinson disease: evidence in mutant PARK2 fibroblasts. Front. Genet.6, 78 (2015).
Kumazawa, R. et al. Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch. Neurol.65, 802–808 (2008).
Doherty, K. M. & Hardy, J. Parkin disease and the Lewy body conundrum. Mov. Disord.28, 702–704 (2013).
Li, F. et al. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun.7, 12708 (2016).
Rezaie, T. et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science295, 1077–1079 (2002).
Chalasani, M. L., Swarup, G. & Balasubramanian, D. Optineurin and its mutants: molecules associated with some forms of glaucoma. Ophthalmic Res.42, 176–184 (2009).
Wild, P. et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science333, 228–233 (2011).
Maruyama, H. et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature465, 223–226 (2010).
Hortobagyi, T. et al. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol.121, 519–527 (2011).
Osawa, T. et al. Optineurin in neurodegenerative diseases. Neuropathology31, 569–574 (2011).
Song, G. J., Jeon, H., Seo, M., Jo, M. & Suk, K. Interaction between optineurin and Rab1a regulates autophagosome formation in neuroblastoma cells. J. Neurosci. Res.96, 407–415 (2018).
Tumbarello, D. A. et al. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat. Cell Biol.14, 1024–1035 (2012).
Sundaramoorthy, V. et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum. Mol. Genet.24, 3830–3846 (2015).
Iida, A. et al. Novel deletion mutations of OPTN in amyotrophic lateral sclerosis in Japanese. Neurobiol. Aging33, 1843 e1819–1824 (2012).
Tumer, Z. et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol. Aging33, 208 e201–205 (2012).
van Blitterswijk, M. et al. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging33, 1016 e1011–1016 e1017 (2012).
Minegishi, Y., Nakayama, M., Iejima, D., Kawase, K. & Iwata, T. Significance of optineurin mutations in glaucoma and other diseases. Prog. Retin Eye Res.55, 149–181 (2016).
Sirohi, K. & Swarup, G. Defects in autophagy caused by glaucoma-associated mutations in optineurin. Exp. Eye Res.144, 54–63 (2016).
Ryan, T. A. & Tumbarello, D. A. Optineurin: a coordinator of membrane-associated cargo trafficking and autophagy. Front. Immunol.9, 1024 (2018).
Tseng, H. C. et al. Visual impairment in an optineurin mouse model of primary open-angle glaucoma. Neurobiol. Aging36, 2201–2212 (2015).
Sayyad, Z., Sirohi, K., Radha, V. & Swarup, G. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci. Rep.7, 16855 (2017).
Beeldman, E. et al. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph. Lateral Scler. Frontotemporal Degener.16, 410–411 (2015).
Shen, W. C., Li, H. Y., Chen, G. C., Chern, Y. & Tu, P. H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy11, 685–700 (2015).
Fifita, J. A. et al. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph. Lateral Scler. Frontotemporal Degener.18, 126–133 (2017).
Oakes, J. A., Davies, M. C. & Collins, M. O. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain10, 5 (2017).
Moore, A. S. & Holzbaur, E. L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl Acad. Sci. USA113, E3349–E3358 (2016).
Pilli, M. et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity37, 223–234 (2012).
Kumar, S. et al. Phosphorylation of syntaxin 17 by TBK1 controls autophagy initiation. Dev. Cell49, 130–144 e136 (2019).
Pillai, S. et al. Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat. Commun.6, 10072 (2015).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci.18, 631–636 (2015).
Pottier, C. et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol.130, 77–92 (2015).
Bean, B. D. M. et al. Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J. Cell Biol.217, 3593–3607 (2018).
Kumar, N. et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol.217, 3625–3639 (2018).
Anding, A. L. et al. Vps13D encodes a ubiquitin-binding protein that is required for the regulation of mitochondrial size and clearance. Curr. Biol.28, 287–295 e286 (2018).
Gauthier, J. et al. Recessive mutations in >VPS13D cause childhood onset movement disorders. Ann. Neurol.83, 1089–1095 (2018).
Seong, E. et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol.83, 1075–1088 (2018).
Gould, S. J. et al. Pex13p is an SH3 protein of the peroxisome membrane and a docking factor for the predominantly cytoplasmic PTs1 receptor. J. Cell Biol.135, 85–95 (1996).
Reguenga, C., Oliveira, M. E., Gouveia, A. M., Sa-Miranda, C. & Azevedo, J. E. Characterization of the mammalian peroxisomal import machinery: Pex2p, Pex5p, Pex12p, and Pex14p are subunits of the same protein assembly. J. Biol. Chem.276, 29935–29942 (2001).
Lee, M. Y. et al. Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep.18, 48–60 (2017).
Rahim, R. S., Chen, M., Nourse, C. C., Meedeniya, A. C. & Crane, D. I. Mitochondrial changes and oxidative stress in a mouse model of Zellweger syndrome neuropathogenesis. Neuroscience334, 201–213 (2016).
Shimozawa, N. et al. Nonsense and temperature-sensitive mutations in PEX13 are the cause of complementation group H of peroxisome biogenesis disorders. Hum. Mol. Genet.8, 1077–1083 (1999).
Waterham, H. R. & Ebberink, M. S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta1822, 1430–1441 (2012).
Liu, Y. et al. PEX13 is mutated in complementation group 13 of the peroxisome-biogenesis disorders. Am. J. Hum. Genet.65, 621–634 (1999).
Gould, S. J. & Valle, D. Peroxisome biogenesis disorders: genetics and cell biology. Trends Genet.16, 340–345 (2000).
Wanders, R. J. & Waterham, H. R. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin. Genet.67, 107–133 (2005).
Maxwell, M. et al. Pex13 inactivation in the mouse disrupts peroxisome biogenesis and leads to a Zellweger syndrome phenotype. Mol. Cell. Biol.23, 5947–5957 (2003).
Baumgart, E. et al. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol.159, 1477–1494 (2001).
Krysko, O. et al. Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J. Neurosci. Res.85, 58–77 (2007).
Weller, S., Gould, S. J. & Valle, D. Peroxisome biogenesis disorders. Annu. Rev. Genomics Hum. Genet.4, 165–211 (2003).
Hirabayashi, M. et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ.8, 977–984 (2001).
Stolz, A., Hilt, W., Buchberger, A. & Wolf, D. H. Cdc48: a power machine in protein degradation. Trends Biochem. Sci.36, 515–523 (2011).
Tresse, E. et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy6, 217–227 (2010).
Papadopoulos, C. et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J.36, 135–150 (2017).
Ju, J. S. et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol.187, 875–888 (2009).
Johnson, J. O. et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron68, 857–864 (2010).
Watts, G. D. et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet.36, 377–381 (2004).
Abrahao, A. et al. One family, one gene and three phenotypes: A novel VCP (valosin-containing protein) mutation associated with myopathy with rimmed vacuoles, amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Sci.368, 352–358 (2016).
Zhang, K. Y., Yang, S., Warraich, S. T. & Blair, I. P. Ubiquilin 2: a component of the ubiquitin-proteasome system with an emerging role in neurodegeneration. Int. J. Biochem. Cell Biol.50, 123–126 (2014).
N’Diaye, E. N. et al. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep.10, 173–179 (2009).
Rothenberg, C. et al. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet.19, 3219–3232 (2010).
Senturk, M. et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol.21, 384–396 (2019).
Deng, H. X. et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature477, 211–215 (2011).
Chen, T., Huang, B., Shi, X., Gao, L. & Huang, C. Mutant UBQLN2(P497H) in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol. Commun.6, 122 (2018).
Noreau, A., Dion, P. A. & Rouleau, G. A. Molecular aspects of hereditary spastic paraplegia. Exp. Cell Res.325, 18–26 (2014).
Blackstone, C. Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol.51, 139–146 (2018).
Hanein, S. et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am. J. Hum. Genet.82, 992–1002 (2008).
Murmu, R. P. et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol. Cell Neurosci.47, 191–202 (2011).
Khundadze, M. et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet.9, e1003988 (2013).
Hirst, J. et al. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell24, 2558–2569 (2013).
Slabicki, M. et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol.8, e1000408 (2010).
Chang, J., Lee, S. & Blackstone, C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Invest.124, 5249–5262 (2014).
Varga, R. E. et al. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet.11, e1005454 (2015).
Vantaggiato, C. et al. ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis. Autophagy15, 34–57 (2019).
Vantaggiato, C. et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain136, 3119–3139 (2013).
Sagona, A. P. et al. A tumor-associated mutation of FYVE-CENT prevents its interaction with Beclin 1 and interferes with cytokinesis. PLoS ONE6, e17086 (2011).
Renvoise, B. et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol.1, 379–389 (2014).
Behrends, C., Sowa, M. E., Gygi, S. P. & Harper, J. W. Network organization of the human autophagy system. Nature466, 68–76 (2010).
Oz-Levi, D. et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet.91, 1065–1072 (2012).
Stadel, D. et al. TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol. Cell60, 89–104 (2015).
Goizet, C. et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology73, 1111–1119 (2009).
Finsterer, J. et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci.318, 1–18 (2012).
Schule, R. et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J. Neurol. Neurosurg. Psychiatry80, 1402–1404 (2009).
Zhao, H. et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol.200, 731–741 (2013).
Zhao, Y. G., Zhao, H., Sun, H. & Zhang, H. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy9, 1258–1262 (2013).
Tian, Y. et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell141, 1042–1055 (2010).
Wang, Z. et al. The Vici syndrome protein EPG5 Is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell63, 781–795 (2016).
Cullup, T. et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet.45, 83–87 (2013).
Hori, I. et al. Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement. Sci. Rep.7, 3552 (2017).
Byrne, S. et al. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain139, 765–781 (2016).
Wang, K. S., Liu, X., Xie, C., Liu, Y. & Xu, C. Non-parametric survival analysis of EPG5 gene with age at onset of Alzheimer’s disease. J. Mol. Neurosci.60, 436–444 (2016).
Wartosch, L., Gunesdogan, U., Graham, S. C. & Luzio, J. P. Recruitment of VPS33A to HOPS by VPS16 is required for lysosome fusion with endosomes and autophagosomes. Traffic16, 727–742 (2015).
Zhang, J. et al. A founder mutation in VPS11 causes an autosomal recessive leukoencephalopathy linked to autophagic defects. PLoS Genet.12, e1005848 (2016).
Hortnagel, K. et al. The second report of a new hypomyelinating disease due to a defect in the VPS11 gene discloses a massive lysosomal involvement. J. Inherit. Metab. Dis.39, 849–857 (2016).
Parikh, S. et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol. Genet. Metab.114, 501–515 (2015).
Worby, C. A. & Dixon, J. E. Sorting out the cellular functions of sorting nexins. Nat. Rev. Mol. Cell Biol.3, 919–931 (2002).
Miller, J. A. et al. Transcriptional landscape of the prenatal human brain. Nature508, 199–206 (2014).
Akizu, N. et al. Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat. Genet.47, 528–534 (2015).
Bryant, D. et al. SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum. Mol. Genet27, 1927–1940 (2018).
Teasdale, R. D. & Collins, B. M. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease. Biochem. J.441, 39–59 (2012).
Datta, S., Liu, Y., Hariri, H., Bowerman, J. & Henne, W. M. Cerebellar ataxia disease-associated Snx14 promotes lipid droplet growth at ER-droplet contacts. J. Cell Biol.218, 1335–1351 (2019).
Thomas, A. C. et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am. J. Hum. Genet.95, 611–621 (2014).
Fraldi, A. et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J.29, 3607–3620 (2010).
Kirkin, V. & Rogov, V. V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell76, 268–285 (2019).
Klein, C. & Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med.2, a008888 (2012).
Minegishi, Y. et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet.22, 3559–3567 (2013).
Acknowledgements
We are grateful for funding from the UK Dementia Research Institute (funded by the MRC, Alzheimer’s Research UK and the Alzheimer’s Society), National Institute for Health Research Cambridge Biomedical Research Centre, The Roger de Spoelberch Foundation, and Alzheimer’s Research UK, to D.C.R. L.W. receives support from an EMBO long-term fellowship (ALTF 135-2016). M.F. receives funding from the Takeda Science Foundation. Y.Z. is supported by China Scholarship Council. S.M.H. receives support by the Swedish Natural Research Council (reference 2016-06605) and from an EMBO long-term fellowship (ALTF 1024-2016). J.V. is supported from a Marie Curie fellowship within the EU’s Horizon 2020 research and innovation program (grant agreement No. 842919). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D.C.R. is CSO of Aladdin Healthcare Technologies.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stamatakou, E., Wróbel, L., Hill, S.M. et al. Mendelian neurodegenerative disease genes involved in autophagy. Cell Discov 6, 24 (2020). https://doi.org/10.1038/s41421-020-0158-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-020-0158-y
This article is cited by
-
Ubr1-induced selective endophagy/autophagy protects against the endosomal and Ca2+-induced proteostasis disease stress
Cellular and Molecular Life Sciences (2022)
-
Fibronectin type III domain-containing protein 5 promotes autophagy via the AMPK/mTOR signaling pathway in hepatocellular carcinoma cells, contributing to nab-paclitaxel chemoresistance
Medical Oncology (2022)
-
Autophagy regulation by acetylation—implications for neurodegenerative diseases
Experimental & Molecular Medicine (2021)
-
Simvastatin accelerated motoneurons death in SOD1G93A mice through inhibiting Rab7-mediated maturation of late autophagic vacuoles
Cell Death & Disease (2021)
-
AMPK: a key regulator of energy stress and calcium-induced autophagy
Journal of Molecular Medicine (2021)
