
Abstract
A large number of studies in recent years indicate that osteocytes are the orchestrators of bone remodeling by regulating both osteoblast and osteoclast activities. Oxidative stress-induced osteocyte apoptosis plays critical roles in the pathological processes of postmenopausal osteoporosis. Resveratrol is a natural polyphenolic compound that ameliorates postmenopausal osteoporosis. However, whether resveratrol regulates osteocyte apoptosis via autophagy remains largely unknown. The effects of resveratrol on regulating osteocyte apoptosis and autophagy were analyzed both in vivo and in vitro. In vitro, cultured MLO-Y4 cells were exposed to H2O2 with or without resveratrol. In vivo, an ovariectomy-induced osteoporosis model was constructed in rats with or without daily intraperitoneal injection of 10 mg/kg body weight resveratrol. It was found that resveratrol attenuated H2O2-induced apoptosis through activating autophagy in cultured MLO-Y4 cells, which was mediated by the dissociation of Beclin-1/Bcl-2 complex in AMPK/JNK1-dependent pathway, ultimately regulating osteocytes function. Furthermore, it was shown that resveratrol treatment reduced osteocytes oxidative stress, inhibited osteocytes apoptosis and promoted autophagy in ovariectomized rats. Our study suggests that resveratrol protects against oxidative stress by restoring osteocytes autophagy and alleviating apoptosis via AMPK/JNK1 activation, therefore dissociating Bcl-2 from Beclin-1.
Similar content being viewed by others

Introduction
Postmenopausal osteoporosis is characterized by mechanical unloading and low estrogen levels due to a decrease in bone density and micro-architectural deterioration of bone tissues [1, 2]. Bone-forming osteoblasts and bone-resorbing osteoclasts, accompanied with osteocytes, are involved in the process of bone remodeling, which can be embedded within the mineralized matrix of the bones [3]. Numerous studies now show that osteocytes, as the most abundant cells in bone, can serve as the orchestrators of bone remolding by modulating the functions of osteoclasts and osteoblasts [4]. The viable osteocytes are capable to repair bone damage, maintain bone density, and ensure an efficient remodeling process [5, 6]. Thus, improving the viability of osteocytes may serve as a potential strategy for treating postmenopausal osteoporosis.
Growing evidence suggests that oxidative stress is increased with estrogen deficiency and aging, thus leading to the development of postmenopausal osteoporosis [7, 8]. ROS induces the apoptotic levels of osteocytes and osteoblasts, which suppresses osteogenesis or bone mineralization and promotes osteoclastogenesis [9]. Oxidative stress can induce apoptosis in osteocytes, which leads to an imbalance between bone formation by osteoblasts and bone resorption by osteoclasts and ultimately impairs bone remodeling and promotes bone loss [10]. Besides, the apoptotic osteocytes are involved in the recruitment of osteoclasts to the bone sites, thereby improving bone resorption [11]. Autophagy is a crucial evolutionarily conserved intracellular process that affects cell growth, differentiation, development, and homeostasis. It can degrade and recycle damaged or long-lived cellular organelles and proteins, as well as maintain osteocyte homeostasis [12]. Numerous adverse factors, such as hypoxia and ROS, can affect osteocyte survival via regulation of autophagy [13]. Previous evidence has demonstrated that multiple signaling pathways such as ROS/AMPK and ROS/JNK/c-Jun are involved in oxidative stress-induced autophagy and apoptosis in bone remodeling [14]. Additionally, the interaction between autophagic protein Beclin1 and the anti-apoptotic protein Bcl-2 represents an important link between autophagy and apoptosis [15]. MLO-Y4 cells after fluid shearing force exhibit AMPK signaling activation Using RNA-seq [16]. AMPK regulated RANKL and sclerostin expression through the mevalonate pathway in osteocytes [17]. However, the regulatory effect of autophagy mediated by AMPK/JNK1 signaling pathway on osteocyte apoptosis is less known.
Resveratrol is a naturally occurring antioxidant compound derived from grapes, peanuts, berries and white hellebore [18]. Previous data from our lab and from other investigators have suggested that resveratrol activates AMPK under different conditions [19,20,21,22,23]. Ayumu Takeno et al. demonstrated in vitro that AMPK activation protects against oxidative stress-induced apoptosis of osteocytes by regulating the expressions of NADPH oxidase 1 (Nox1) and Nox2 [24]. However, whether resveratrol modulates survival and function of osteocytes to inhibit postmenopausal osteoporosis is unknown and needs to be clearly elucidated. The present research aimed to assess whether resveratrol has a protective effect on postmenopausal osteoporosis via inducing osteocytes autophagy and inhibiting apoptosis. The target molecules involved in mediating these effects, such as AMPK and JNK1, were also assessed. We found resveratrol restored autophagy and inhibited apoptosis by disrupting the interaction between Beclin1 and Bcl-2 via AMPK/JNK1- mediated signaling pathways, thus contributing to the improvement of postmenopausal osteoporosis.
Results
Effects of resveratrol on osteocyte viability and oxidative stress
To assess whether resveratrol can affect the viability of osteocytes, MLO-Y4 cells were exposed to different doses (0, 30, 60, 120 μM) of H2O2 and pretreated with resveratrol, osteocyte viability was detected by using the CCK-8 assay. As shown in Fig. 1A, resveratrol had no significant effect on MLO-Y4 cell survival, but H2O2 reduced the viability of MLO-Y4 cells in a concentration- and time-dependent fashion, compared to the control group (Fig. 1B). When 25, 50, 100 μM resveratrol were exposed to MLO-Y4 cells for 24 hr prior to 120 μM H2O2 intervention for 14 d, viable MLO-Y4 cells significantly elevated compared to the untreated cells (Fig. 1C). To investigate whether the protective function of resveratrol is related to its antioxidant activity, the levels of MDA and ROS in MLO-Y4 cells were detected. Our results demonstrated that H2O2 increase oxidant activity and induced oxidative stress to MLO-Y4 cells. After pretreatment with resveratrol at various doses, the levels of MDA and ROS were dose-dependently inhibited (Fig. 1D, E). These findings indicated that resveratrol, in part, protected MLO-Y4 cells from H2O2-induced oxidative damage.

A Cell viability of MLO-Y4 cells following treatment with different concentrations of resveratrol from 7 d to 14 d. B Cell viability of MLO-Y4 cells in different concentrations of H2O2 from 7 d to 14 d. C Cell viability of MLO-Y4 cells administered with resveratrol for 24 h before 120 μM H2O2 administration for 14 d. D, E Effects of different concentrations of resveratrol on MDA and intracellular ROS content in MLO-Y4 cells supernatants after treatment with 120 μM H2O2. n = 6, #P < 0.05 vs control, ##P < 0.01 vs control, *P < 0.05 vs H2O2, **P < 0.01 vs H2O2.
Effects of resveratrol on osteocyte apoptosis and autophagy
In order to evaluate the effect of resveratrol on H2O2-triggered MLO-Y4 cells apoptosis, FITC-Annexin V, PI staining and flow cytometric analysis were carried out. As demonstrated in Fig. 2A, B, the proportion of apoptotic MLO-Y4 cells treated with H2O2 120 μM was 63.7 ± 8.0 %, which was remarkably higher than that of the untreated cells (P < 0.01). After pretreated with various concentrations of resveratrol (25-100 μM), the proportion of apoptotic MLO-Y4 cells were severely decreased. And the apoptotic cells were prominently decreased by 100 μM resveratrol compared with that of 25 μM resveratrol. Bcl-2/Bax ratio indicates the direction of apoptotic cells. Next, western blot analysis was used to evaluate the expression of Bcl-2 and Bax. Compared with the normal group, the ratio of Bcl-2/Bax in the H2O2 120 μM group was significantly decreased. After resveratrol pretreatment, a ratio of Bcl-2/Bax elevated gradually. These results suggested that resveratrol could inhibit apoptosis in MLO-Y4 cells induced by H2O2 in a concentration-dependent fashion (Fig. 2C, D).

Cells were exposed to H2O2 (120 μM) and treated with resveratrol (Res, 25, 50, 100 μM) for 14 d. A Representative diagram of annexin V/PI staining. B The apoptotic cells were calculated as the ratio of annexin V + /PI- cells to total cells. C, D The protein expression of Bcl-2, Bax and Caspase-3 in cell lysates were detected by western blotting. E, F MLO-Y4 cells were stained for LC3 (red arrows) and with the nuclear dye DAPI (blue) following the different treatments for 14 d. G, H The protein expression of LC3-II/I and Beclin1 in cell lysates were detected by western blotting. n = 6, #P < 0.05 vs control, ##P < 0.01 vs control, *P < 0.05 vs H2O2, **P < 0.01 vs H2O2.
To explore whether resveratrol activates autophagy in MLO-Y4 cells, the intracellular level of the autophagy marker LC3 was detected by immunofluorescence. Our results showed that MLO-Y4 cells in the H2O2 120 μM group had diffuse, weak cytoplasmic staining with red fluorescent protein (LC3), however MLO-Y4 cells pretreated with resveratrol showed increased fluorescence intensity, indicating that LC3-II is distributed within resveratrol-treated cells (Fig. 2E, F). To further confirm the autophagic role of resveratrol, the expressions of LC3-II, Beclin-1 were detected in MLO-Y4 cells of all groups by western blotting. Similarly, the H2O2-treated MLO-Y4 cells for 48 h expressed lower levels of LC3-II/LC3-I, Beclin-1 protein than that of the control group. Resveratrol treatment dose-dependently enhanced the expressions of LC3- II, Beclin-1 in MLO-Y4 cells (P < 0.01) (Fig. 2G, H).
Resveratrol inhibits H2O2-trigerred apoptosis via activation of autophagy
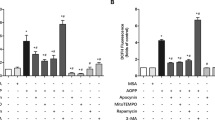
To examine the interactions between apoptosis and autophagy in resveratrol’ effect on MLO-Y4 cells, MLO-Y4 cells was exposed to the autophagy inhibitor 3-methylademine that mainly plays an inhibitory role in the formation of autophagosomes. Notably, resveratrol reversed H2O2-triggered decreases in P62-deposition and LC3- II production, which could offseted by adding 3-methylademine (Fig. 3A). Simultaneously, resveratrol reduced the levels of PARP and cleaved caspase-3 (the apoptotic markers) after exposure H2O2 to MLO-Y4 cells, which could be neutralized by 3-methylademine (Fig. 3B). This suggests that inhibition of autophagy can accelerate H2O2-triggered MLO-Y4 cell death, and the inhibition apoptosis of resveratrol is markedly associated with the activation of autophagy.

MLO-Y4 cells were pretreated with 3-methyladenine (3-MA, 10 μM) for 30 min before exposure to H2O2 (120 μM) and resveratrol (Res, 100 μM) for 14 d. A The protein expression of LC3-II/I and P62 in cell lysates were detected by western blotting. B Apoptosis markers including cleaved caspase-3 (C-caspase-3) and cleaved PARP (C-PARP) in cell lysates were detected by western blotting. n = 3; #P < 0.05 vs. control, *P < 0.05 vs. H2O2, &P < 0.05 vs. H2O2/Res.
Resveratrol activated AMPK/JNK1 to dissociate Bcl-2 and Beclin-1 in MLO-Y4 cells
It has been reported that resveratrol can activate AMPK and JNK1 in a variety of cells [19,20,21,22,23]. Similarly, our results indicated that resveratrol dose-dependently elevated AMPK/JNK1 phosphorylation in MLO-Y4 cells (Fig. 4A). We first confirmed that AMPK and JNK1 were induced by resveratrol in H2O2-exposed MLO-Y4 cells. Moreover, the results of co-immunoprecipitation assays demonstrated that resveratrol remarkably enhanced the activation of AMPK/JNK1 after H2O2 intervention (Fig. 4B). Next, we determined if resveratrol caused JNK1 phosphorylation in AMPK-dependent manner. Compound C (CC), an AMPK inhibitor, can block the activation of AMPK. Consequently, reduced JNK1 phosphorylation was detected in CC-treated MLO-Y4 cells after exposure to H2O2 (Fig. 4C). These findings suggested both kinases could act coordinatively to regulate apoptosis and autophagy in resveratrol-treated MLO-Y4 cells.

MLO-Y4 cells were pretreated with compound C (CC, 20 μM, AMPK inhibitor), or SP600125 (SP, 10 μM, JNK1 inhibitor) for 30 min before exposure to H2O2 (120 μM). MLO-Y4 Cells were treated with resveratrol for 14 d. A Resveratrol (Res, 25, 50, 100 μM) activated AMPK and JNK1 in MLO-Y4 cells exposed to H2O2. The levels of phosphorylated AMPKɑ (Thr172) and phosphorylation of JNK1 (Thr183/Tyr185) in cells lysates were measured by western blotting. B Resveratrol (Res, 100 μM) enhanced the interaction of AMPKɑ with JNK1 in MLO-Y4 cells exposed to H2O2. The association of AMPKɑ and JNK1 was assessed by immunoprecipitation (IP). C MLO-Y4 cells were pretreated with compound C (CC, 20 μM) for 30 min before exposure to 120 μM H2O2 and treated with resveratrol (Res, 100 μM) for 14 d, JNK1 and phosphorylation of JNK1 (Thr172) protein levels were measured by western blotting. D, E Beclin-1 or Bcl-2 was immunoprecipitated (IP) from cell lysates, and Bcl-2 or Beclin1 in the immunoprecipitate was detected by western blotting. F MLO-Y4 cells were pretreated with SP600125 (SP, 10 μM, JNK1 inhibitor) for 30 min before exposure to H2O2 (120 μM) and treated with resveratrol (Res, 100 μM) for 14 d, The association of Beclin-1 and Bcl-2 was assessed by immunoprecipitation (IP). G, H LC3-II/I protein levels and apoptosis markers (C-Casp3 and C-PARP) in cells lysates were measured by western blotting. n = 3; #P < 0.05 vs. control, *P < 0.05 vs. H2O2, &P < 0.05 vs. H2O2/Res.
Bcl-2 and Beclin-1 is a switch between apoptosis and autophagy [25, 26]. Research shows that activation of AMPK leads to phosphorylation of threonine 388 site of autophagy gene Beclin-1 [27], and the activated JNK1 may result in Bcl-2 phosphorylation [28, 29], which leads to dissociation of autophagy protein Beclin-1 and the members of the anti-apoptotic Bcl-2 family [30]. Immunoprecipitation assays showed that H2O2 strengthened the relationship between Bcl-2 and Beclin-1, and resveratrol could disrupt this interaction (Fig. 4D, E). Additionally, we used JNK1 inhibitors to further explore whether the action of resveratrol is related to the activation of JNK1. Our data suggested that the disruption of Beclin-1/Bcl-2 complex by resveratrol could be reversed by treatment with the JNK1 inhibitor SP600125 (Fig. 4F). Importantly, resveratrol-enhanced autophagy and inhibited apoptosis were attenuated by SP600125 treatment (Fig. 4G, H). Therefore, the effect of resveratrol to inhibit apoptosis and promote autophagy is mediated by dissociation of Beclin-1/Bcl-2 complex in AMPK/JNK1-dependent pathway.
Protective effect of resveratrol on osteoporosis in ovariectomized rats
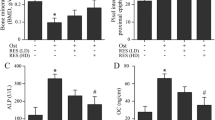
The HE staining showed that trabeculae significantly decreased and thinned in OVX group compared with the sham group, while resveratrol effectively reversed the alterations by a high number of trabecular bone and a decrease in trabecular bone separation (Fig. 5A). Consistent with the results of HE staining, micro-CT data showed that ovariectomy reduced caused a significant trabecular bone loss and deteriorated bone microarchitecture, which was indicated through the decreases of indicators such as BMD, BS/BV, BV/TV, Conn.D, SMI Tb.N, Tb.Sp and Tb.Th, compared to the normal rats. However, the administration of ovariectomized rats with 10 mg/kg resveratrol significantly reversed these bone indicators and enhanced the microstructural features of trabecular bones (Fig. 5B-J). In addition, serum levels of BALP and osteocalcin, bone formation markers, and Tracp 5b and β-CTX, bone resorption markers, were detected for assessing the turnover of the bones. BALP and osteocalcin levels were lower, but β-CTX and TRACP-5b levels were obviously higher in the OVX rats than those in the Sham rats (P < 0.01). The increased levels of BALP and osteocalcin and decreased levels of Tracp 5b and β-CTX were found in resveratrol-treated rats compared with those in OVX rats (P < 0.01) (Fig. 5K-N).

A The hematoxylin-eosin staining in distal femur among groups. B The 2D images of sagittal and transverse section. C–J The trabecula parameters of cancellous bone in the proximal tibial metaphysis static parameters: BMD bone mineral density, BV/TV bone volume/tissue volume, Tb.N trabecular number, Tb.Th trabecular thickness, Tb.Sp trabecular separation, BS/BV Bone surface/bone volume, SMI structure model index, and Conn.D connectivity Density. K–N serum bone alkaline phosphatase (BALP), osteoprotegerin (OPG), tartrate resistant acid phosphatase (TRACP-5b), and β-cross-linked c-telopeptide of type I collagen (β-CTX), n = 5, ##P < 0.01 vs. Sham group; *P < 0.05 vs. OVX group and **P < 0.01 vs. OVX group.
Effects of resveratrol on osteocytes redox balancein in ovariectomized rats
To determine whether postmenopausal prevention of resveratrol is related to a low oxidative stress of osteocytes, the activity and expression of antioxidant enzymes and ROS were examined in cortical bone of proximal tibias. Our results showed that ROS levels were obviously elevated, whereas superoxide dismutase (SOD) activity, catalase (CAT), total antioxidant capacity (tAOC), SOD1/SOD2 expression levels were markedly decreased in OVX group compared to sham group, but resveratrol treatment restored the changes of the above indicators (Fig. 6A-D). These results revealed that estrogen deficiency induced oxidative stress in bone tissue, and resveratrol administration reduced the oxidative stress of osteocytes in OVX rats.

A–C total antioxidant capacity (T-AOC) (A), catalase (CAT) (B), and superoxide dismutase (SOD) (C) activity in proximal tibias. (D) The protein expression levels of SOD1 and SOD2 were detected by western blot. Data were the means ± SD (n = 6 for each group). ##P < 0.01 vs. Sham group; **P < 0.01 vs. OVX group.
Resveratrol administration inhibited apoptosis and induced autophagy in the osteocytes of ovariectomized rats
HE staining results showed that the amount of osteocytes was remarkably decreased (P < 0.05) in OVX rats compared with sham rats, and the number of osteocytes increased significantly after resveratrol treatment (Fig. 7A). The ultrastructural analysis revealed osteocytes exhibited scarce cytoplasm with few organelles, irregular shape of nucleus and nearly all nuclei filled with condensed chromatin in the OVX rats. Resveratrol treatment obviously improved the pathological morphology of osteocytes (Fig. 7B). Tunel staining results showed that an obvious increase in Tunel-positive osteocytes in OVX rats compared with sham rats, whereas resveratrol could attenuate Tunel-positive osteocytes in the proximal tibia (Fig. 7C). Meanwhile, immunohistochemical staining results demonstrated that the numbers of caspase-3-positive and P62-positive osteocytes were increased and the numbers of LC3 + osteocytes were decreased in the proximal tibia of OVX rats, and resveratrol administration prominently reduced the number of caspase-3-positive and P62-positive osteocytes, and enhanced the number of LC3 + osteocytes in tibia shafts (Fig. 7D, E). This indicates that resveratrol has protective effects on osteocytes in ovariectomized rats by inducing autophagy and inhibiting apoptosis.

A Several osteocytes inside the lacunae are observed by hematoxylin-eosin staining. B Ultrastructural architecture of osteocytes in the proximal tibia. C The percentage of apoptotic osteocytes was assessed by Tunel staining in the proximal tibia. D Osteocyte-enriched bone sections were analyzed by immunohistochemistry for Cleaved caspase-3, LC3, P62. E Histomorphometric quantifications of the osteocytes number, ##P < 0.01 vs. Sham group; **P < 0.01 vs. OVX group.
Discussion
Osteoporosis is characterized by bone fragility and increased vulnerability to fractures due to a decrease in bone density and micro-architectural deterioration of bone tissues. Primary pharmacological strategies targeting osteoblasts and osteoclasts for the treatment of osteoporosis, including hormone replacement therapy (HRT), and alendronate therapies may not be recommended for long-term usage due to adverse side-effects [31]. Therefore, agents that simultaneously stimulate osteocytes activity and functions are needed. Numerous clinical trials and animal studies have shown that resveratrol can improve bone quality, regulate bone remodeling and delay osteoporosis [32], however, the exact mechanism involved has not been fully elucidated. In this work, the efficacy and mechanism of resveratrol on osteocytes were investigated using oxidative stress cell model and a rat model of osteoporosis. In this study, we found that resveratrol can suppress postmenopausal osteoporosis by regulating the switch between autophagy and apoptosis mechanism through activating AMPK and JNK1 pathway, and thereby protecting osteocytes from oxidative stress.
Osteocytes, as the most abundant cells in mammalian bone and regulate bone metabolism in coordination with osteoblasts and osteoclasts, play critical roles in maintaining bone homeostasis by acting as orchestrators and mechanosensors during the bone remodeling processes [33, 34]. Because osteocytes are long-lived cells residing within the bone matrix, they may be more susceptible to hypoxic conditions, ROS accumulation and nutrient deprivation [35].
Estrogen deficiency can increase oxidative damage and reduce antioxidant enzyme activity, which are the proximal culprits of osteoporosis [8, 36]. In this experiment, we established a hydrogen peroxide (H2O2) - mediated oxidative stress model of osteocytic cell lines in vitro. Our present study showed H2O2 stimulation induced excessive aggregation of reactive oxygen species in MLO-Y4 cells. Owing to the antioxidant effects, resveratrol ameliorated oxidative stress by reducing the levels of ROS. These excess free radicals may damage the cytomembrane and mitochondria, thus resulting in cell apoptosis [37]. Moreover, we also found that H2O2 induced apoptosis of MLO-Y4 cells, which was consistent with previous reports [38], and resveratrol suppressed the pathways of mitochondrial apoptosis by upregulating Bcl-2 expression and downregulating caspase-3 and Bax expression. Autophagy is not only important for the removal of damaged proteins and organelles, but also involved in the recycling of cellular components for maintaining energy balance [39]. Since osteocytes have long life-span and live under low-oxygen and -nutrient conditions, autophagy plays a key role in the survival of osteocytes [40]. Suppression of autophagy leads to the accumulation of damaged mitochondria which produce elevated levels of reactive oxygen species [41,42,43]. Furthermore, autophagy can protect against cell apoptosis [44]. Previous studies reported that autophagy was suppressed in response to chronic oxidative stress [45]. Our results demonstrated that prolonged exposure to oxidative stress could induce obvious autophagy inhibition in MLO-Y4 cells, and resveratrol can induce a beneficial autophagic process to protect the MLO-Y4 cells against H2O2-induced damage. Meanwhile, our results demonstrated that resveratrol could prevent MLO-Y4 cells from H2O2-triggered apoptosis by enhancing autophagic activities.
AMPK is a sensor of cellular energy status that plays a crucial role in the maintenance of energy homeostasis [46]. Moreover, activation of AMPK is involved in determining multiple cellular processes, including protein synthesis, cell growth, autophagy and apoptosis [47,48,49,50,51,52]. Previous studies have revealed that activation of AMPK stimulated JNK1–Bcl-2 signaling and disrupted the Beclin-1/Bcl-2 complex [53]. Bcl-2 / Beclin-1 complex plays a “switch” role in the development of autophagy and apoptosis. Beclin-1 may interact with Bcl-2 [54], and such interaction through BH3 domain suppresses Beclin-1-dependent autophagy by sequestering it away from class 3 PI3K [27, 28]. Our present study revealed that resveratrol activated AMPK/JNK1 pathways to dissociate Beclin-1/Bcl-2 complex. The free Beclin-1 binds to class 3 PI3K to generate a kinase complex and promote autophagy, while the free Bcl-2 can bind to pro-apoptotic molecules to suppress apoptosis [55]. Therefore, the current study suggested that resveratrol activated AMPK/JNK1, and thereby dissociating the Bcl-2/Beclin-1 complex, which may be an important mechanism of restoring autophagy and preventing apoptosis in response to oxidative stress.
The ovariectomized rat is a widely employed animal model for studying estrogen-deficiency in postmenopausal patients [56]. To understand the mechanisms underlying the in vivo effect of resveratrol on osteocyte of postmenopausal osteoporosis, estrogen-deficient rats were subjected to ovariectomy to establish an OVX model. First, the effect of supplementation of resveratrol on bone turnover parameters in OVX rats was assessed. Histological and micro-CT analyses revealed that resveratrol could positively affect trabecular bone microarchitecture, bone mineral density. Similarly, biochemical results confirmed that resveratrol decreased the serum levels of TRACP-5b and β-CTX, and a marked increase in the serum BALP and OPG in rats, thus indicating a dual therapeutic effect on stimulating bone formation and suppressing bone resorption following treatment with resveratrol. Consistent with the results of in vitro experiments, resveratrol administration for 12 weeks reversed, in part, OVX-induced reduction of antioxidative biomarkers in osteocyte. In addition, in accordance with previous study [57], the decreased number of osteocytes and the increased levels of TUNEL-positive osteocytes and cleaved caspases-3, but the reduced expression of beclin-1 and a significant increase of p62 were found in the proximal tibia of OVX groups rats, and resveratrol treatment effectively inhibit apoptosis and enhance autophagy of osteocyte in OVX rats. However, contrary to our findings, estrogen deficiency and replacement therapy could induce and suppress autophagic processes in the osteocytes of ovariectomized rats [58]. The reason for the contradictory conclusion is that autophagy modulation has differential effects on various cells, even in the same cell lineage over time [13]. The latter experiment was done 45 days after ovariectomy, while our experiment time was 12 weeks after modeling. These divergent results may also be attributed to the fact that osteocyte can undergo different autophagic processes depending on estrogen levels.
In summary, our findings demonstrate that resveratrol prevents ovariectomy osteoporosis by inhibiting osteocytes apoptosis through promoting autophagy in rats. This effect is attributed by AMPK/JNK1-mediated disassociation of Beclin-1/Bcl-2 complexes (Fig. 8). Although the accurate switch between apoptotic and autophagic machinery in osteocytes needs to be further investigated in the future, and other signal pathways may also be involved in the protective effect of resveratrol on osteocyte autophagy activation and apoptosis inhibition, our results provide new evidence for the protective roles of resveratrol in postmenopausal osteoporosis.

Resveratrol protects osteocytes by regulating the switch between autophagy and apoptotic machinery under estrogen deficiency conditions, which is attributed by AMPK/JNK-mediated dissociation of Beclin-1-Bcl-2. ROS reactive oxygen species, AMPK Adenosine 5‘-monophosphate (AMP)-activated protein kinase, JNK1 c-Jun N-terminal kinase1.
Materials and methods
Cell culture and treatment
MLO-Y4 cells were purchased from iCell Bioscience Inc (Shanghai, China; Product No.: iCell-m037), grown in 0.15 mg/ml rat tail collagen type I-coated culture plates. α-MEM containing 2.5% bovine calf serum and 2.5% fetal bovine serum was added, followed by incubation at 37 °C in a 5% CO2 incubator [59]. In this experiment, the chronic stress state was used to simulate the physiological state of human body after estrogen deficiency. For chronic oxidative stress studies, MLO-Y4 cells were exposed to 0, 30, 60, or 120 μM H2O2 with or without resveratrol 25, 50, 100 μM (Sangon Biological Engineering Technology Company, Shanghai, China) as indicated daily for 14 d. The cells were pre-treated with 10 μM SP600125, 20 μM compound C (CC) and 10 μM 3-methyladenine 30 min before exposure to H2O2.
Cell viability and oxidative stress assay
Osteocyte viability was evaluated by CCK8 assays. The cells (1 × 103 cells/ml) were grown in 96-well plates, and the cell proliferation activity was measured at 24, 48, and 72 hr after inoculation using CCK8 (Dojindo, Kumamoto, Japan) with the accordance of manufacturer’s instruction. The absorbance values at OD450 were recorded using a microplate spectrophotometer (Thermo Labsystems, Vantaa, Finland). Oxidative stress was assessed by detecting malondialdehyde (MDA) and ROS levels according to the manufacturer′ instructions. ROS levels were assessed with DCHF-DA probes.
FITC-annexin V and PI staining
Osteocyte apoptosis was detected with FITC-Annexin V and PI staining by following the kit’s protocols (Clontech, CA). After growing in a 35-mm plate at 37 °C overnight, MLO-Y4 cells were trypsinized and incubated with PI solution (5 μl) and FITC-Annexin (5 μl) in the dark for 15 min. MLO-Y4 cells apoptosis rates were analyzed by FACS.
Immunofluorescence confocal microscopy
The expression of LC3 in MLO-Y4 cells was determined via immunofluorescence. MLO-Y4 cells were grown in a 24-well plate on slides. After treatment, the cells were fixed in paraformaldehyde (4%), and permeabilized with Triton X-100 (0.1%). The cells were incubated with anti-LC3 primary antibody (Code n. sc-135836, Santa Cruz Biotechnology, USA); and then probed with fluorophore-conjugated secondary antibodies and DAPI. The confocal imaging was analyzed using an FV1000 Olympus laser scanning confocal microscope (Olympus, Japan).
Immunoprecipitation and immunoblotting
MLO-Y4 cells were homogenized in lysis buffer, and protein concentration was quantified using the Bradford assay. After separation through 15% SDS-PAGE and transferred onto PVDF membranes, cell lysate was subjected to immunoprecipitation with specific antibodies for 1 h at RT, followed by immunoblotting according to the kit’s protocols (Roche, Basel, Switzerland). The primary antibodies used for immunostaining were: rabbit anti-JNK1 (1:1000, Abcam, ab199380), rabbit anti-p-JNK1 ab47337, rabbit anti-AMPK (1:1000, Abcam, ab32382), rabbit anti-p-AMPK (1:1000, Abcam, ab133448), rabbit anti-Beclin 1(1:1000, Abcam, ab207612), rabbit anti-Bcl-2 (1:1000, Abcam, ab182858), rabbit anti-LC3 (1:1000, Abcam, ab128025), rabbit anti-Cleaved Caspase-3 (1:1,000, Abcam, ab32042), rabbit anti-Cleaved PARP (1:1000, Abcam, ab32064), rabbit anti-SOD1 (1:1000, Abcam, ab179843), rabbit anti-SOD2 (1:1000, Abcam, ab74231), rabbit anti-Actin (1:3000, Abcam, ab8227). The secondary antibodies used for immunostaining were: mouse HRP (1:2000, Abcam, ab6728), rabbit HRP (1:2000, Abcam, ab6721).
Animals and treatments
Female SD rats (6 months old) in this experiment were supplied by the Laboratory Animal Center of Zhengzhou University. The rats were raised in a 12-h day and night cycle with free water intake at 23 °C, and then andomized into 3 groups (n = 8 per group): ovariectomized model group (OVX), sham surgery with intact ovaries group (Sham), and resveratrol group (Res). The sample size was determined by considering statistical considerations and ethical concerns. All animals that participate in the experiment were included for analysis, unless animal death. The rats in OVX and Res groups underwent bilateral ovariectomy (OVX) under anesthesia with an intraperitoneal injection of 0.3% sodium pentobarbita per 0.1–0.2 ml/10 g of body weight. An additional 8 rats underwent sham surgery. Two weeks after ovariectomy, rats in OVX and Res groups were received respectively resveratrol solution at 10 mg/kg body weight by daily intraperitoneal injection or saline. After 12 weeks of treatment, the rats were sacrificed, blood, proximal tibias, distal femurs, lumbar vertebrae were collected. The study procedures were approved by the Experimental Animal Ethics Committee at orthopedics hospital of Henan province (Animal Protocol Approval No.: 201803010), and conducted in accordance with ethical requirements and ARRIVE guidelines.
Serum markers of bone formation and resorption
Blood specimens were withdrawn from the abdominal aorta after 8 weeks of treatment. Separation of the serum was accomplished by centrifugation (3000 rpm, 15 min, 4 °C). The levels of serum TRACP-5b, β-CTX, OPG, and BALP were measured with ELISA kits by following the kit’s protocols.
Measurements of SOD, CAT, and tAOC
The proximal tibial bone tissue (100 mg) was homogenized with the aid of dry ice. As cortical bone has a more concentration of osteocytes, it would be more suitable to collect homogenates only from cortical bone compartment. After centrifugation (1000 g, 4 °C, 10 min), the levels of SOD, CAT, and tAOC in supernatant were determined with commercial kits (Beyotime, Jiangsu, China) by following the kit’s protocols. The protein levels of SOD1 and SOD2 were assessed by immunoblotting.
Micro CT analysis
Micro CT (μCT 80, Scanco Medical, Zurich, Switzerland) was used to evaluate the microstructure of trabeculae at the epiphysis of femoral shaft. Bone trabeculae are abundant in the distal femur. Each selected sample was scanned from the distal to the proximal at a resolution of 16 μm. The region of interest (ROI) of trabecular bone started from 0.15 mm proximal to distal growth plate, and extended proximally for 0.4 mm. The parameters in the ROI, such as bone volume over total volume (BV/TV), bone mineral density (BMD), trabecular bone number (Tb.N), trabecular bone thickness (Tb.Th), and trabecular bone separation (Tb.Sp), were automatically calculated using the micro-CT system’s software, and the analysis threshold was 245.
Histological analysis
The proximal tibias were fixed in paraformaldehyde (4%) for 48 hr, decalcified in 15% EDTA at room temperature for 2 weeks, and paraffin-embedded. Then, 4 μm slices were coronally sectioned, followed by hematoxylin-eosin (HE) staining. The stained proximal tibias were examined using a light microscope (Nikon Eclipse 80i, Tokyo, Japan).
TEM assay
The proximal tibial tissues were fixed in glutaraldehyde (2%) for 24 hr, followed by demineralization in EDTA (5%) for 14 days. After cutting into 1mm3, the specimens were postfixed in 1% OsO4 and embedded with epoxy resin. Next, the specimens were sectioned at 80-nm sections, followed by uranyl acetate and lead citrate staining. Lastly, the ultrathin slides were subjected to TEM analysis (Phillips CM-80, Netherlands).
Tunel assay
Tunel assay was conducted with ApopTag Plus Peroxidase in situ apoptosis detection kit by following the kit’s protocol (KeyGEN Biotech, Nanjing, China). Briefly, the sections of proximal tibias were dewaxed and hydrated by routine methods. After incubation with Tunel reaction mixture at 37˚C for 1 h, the sections were stained with DAB substrate at room temperature for 10 min. Apoptotic cells were observed using a fluorescence microscope (CX41RF; Olympus Corporation, Tokyo, Japan).
Immunohistochemical staining
The paraffin-embedded femur tissues were sectioned at 3-μm thickness. After incubation with cleaved caspase-3 antibody (Cell Signalling Technology, Boston, MA, 1:100), LC3 antibody (Abcam, Cambridge, MA, 1: 200) and P62 antibody (Santa Cruz Biotechnology, CA, 1: 100) at 4 °C overnight, then incubated in HRP-labeled goat anti-rabbit secondary antibody (Beyotime Institute of Biotechnology, Inc., Jiangsu, China) for 30 min at 37 °C. After another 30-min incubation with Vectastain ABC-AP kit (Vector, CA), the specimens were rinsed and exposed to 3,3-diamino-benzidine (DAB) substrate for 3–10 min.
Statistical analysis
Data were obtained from at least three separate experiments performed in triplicate. All values are shown as means ± standard deviations (SD). Statistical analysis was undertaken only for studies where each group size was at least n = 5. The difference in means between two groups was compared by two-tailed Student’s t test. One-way analysis of variance (ANOVA) and two-way ANOVA was performed for multiple comparisons. The variance was similar between the groups that were being statistically compared. All statistical tests were conducted with GraphPad Prism software, and P < 0.05 was deemed statistically significant.
Blind statement
The investigator was blinded to the group allocation during the experiment and/or when assessing the outcome.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Farr JN, Rowsey JL, Eckhardt BA, Thicke BS, Fraser DG, Tchkonia T, et al. Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: evidence in young adult mice and older humans. J Bone Miner Res. 2019;34:1407–18.
Watson SL, Weeks BK, Weis LJ, Harding AT, Horan SA, Beck BR. High-intensity resistance and impact training improves bone mineral density and physical function in postmenopausal women with osteopenia and osteoporosis: The LIFTMOR randomized controlled trial. J Bone Miner Res. 2018;33:211–20.
Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285:25103–8.
Plotkin LI, Bellido T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat Rev Endocrinol. 2016;12:593–605.
Qin L, Liu W, Cao H, Xiao G. Molecular mechanosensors in osteocytes. Bone Res. 2020;8:23.
Tresguerres FGF, Torres J, López-Quiles J, Hernández G, Vega JA, Tresguerres IF. The osteocyte: a multifunctional cell within the bone. Ann Anat. 2020;227:151422.
Domazetovic V, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Oxidative stress in bone remodeling: role of antioxidants. Clin Cases Miner Bone Metab. 2017;14:209–16.
Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38.
Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem. 2011;112:2412–23.
Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50:1115–22.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Yin X, Zhou C, Li J, Liu R, Shi B, Yuan Q, et al. Autophagy in bone homeostasis and the onset of osteoporosis. Bone Res. 2019;7:28.
Zhu C, Shen S, Zhang S, Huang M, Zhang L, Chen X. Autophagy in bone remodeling: a regulator of oxidative stress. Front Endocrinol. 2022;13:898634.
Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–51.
Govey PM, Kawasawa YI, Donahue HJ. Mapping the osteocytic cell response to fluid flow using RNA-Seq. J Biomech. 2015;48:4327–32.
Yokomoto-Umakoshi M, Kanazawa I, Takeno A, Tanaka K, Notsu M, Sugimoto T. Activation of AMP-activated protein kinase decreases receptor activator of NF-κB ligand expression and increases sclerostin expression by inhibiting the mevalonate pathway in osteocytic MLO-Y4 cells. Biochem Biophys Res Commun. 2016;469:791–6.
Lorenz P, Roychowdhury S, Engelmann M, Wolf G, Horn TF. Oxyresveratrol and resveratrol are potent antioxidants and free radical scavengers: effect on nitrosative and oxidative stress derived from microglial cells. Nitric Oxide. 2003;9:64–76.
Qin N, Wei L, Li W, Yang W, Cai L, Qian Z, et al. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J Pharmcol Sci. 2017;134:166–74.
Pineda-Ramírez N, Alquisiras-Burgos I, Ortiz-Plata A, Ruiz-Tachiquín ME, Espinoza-Rojo M, Aguilera P. Resveratrol activates neuronal autophagy through AMPK in the ischemic brain. Mol Neurobiol. 2020;57:1055–69.
Xu K, Liu XF, Ke ZQ, Yao Q, Guo S, Liu C. Resveratrol modulates apoptosis and autophagy induced by high glucose and palmitate in cardiac cells. Cell Physiol Biochem. 2018;46:2031–40.
Zhao H, Chen S, Gao K, Zhou Z, Wang C, Shen Z, et al. Resveratrol protects against spinal cord injury by activating autophagy and inhibiting apoptosis mediated by the SIRT1/AMPK signaling pathway. Neuroscience. 2017;348:241–51.
Borriello A, Bencivenga D, Caldarelli I, Tramontano A, Borgia A, Zappia V, et al. Resveratrol: from basic studies to bedside. Cancer Treat Res. 2014;159:167–84.
Takeno A, Kanazawa I, Tanaka K, Notsu M, Yokomoto M, Yamaguchi T, et al. Activation of AMP-activated protein kinase protects against homocysteine-induced apoptosis of osteocytic MLO-Y4 cells by regulating the expressions of NADPH oxidase 1 (Nox1) and Nox2. Bone 2015;77:135–41.
Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–6.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39.
Zhang D, Wang W, Sun X, Xu D, Wang C, Zhang Q, et al. AMPK regulates autophagy by phosphorylating BECN1 at Threonine 388. Autophagy. 2016;12:1447–59.
Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–78.
Blagosklonny MV. Unwinding the loop of Bcl-2 phosphorylation. Leukemia. 2001;15:869–74.
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88.
Chen LR, Ko NY, Chen KH. Medical treatment for osteoporosis: from molecular to clinical opinions. Int J Mol Sci. 2019;20:2213.
Raut N, Wicks SM, Lawal TO, Mahady GB. Epigenetic regulation of bone remodeling by natural compounds. Pharmacol Res. 2019;147:104350.
Schaffler MB, Cheung WY, Majeska R, Kennedy O. Osteocytes: master orchestrators of bone. Calcif Tissue Int. 2014;94:5–24.
Florencio-Silva R, Sasso GR, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int. 2015;2015:421746.
Prideaux M, Findlay DM, Atkins GJ. Osteocytes: the master cells in bone remodeling. Curr Opin Pharmacol. 2016;28:24–30.
Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. 2010;21:369–74.
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–62.
Kikuyama A, Fukuda K, Mori S, Okada M, Yamaguchi H, Hamanishi C. Hydrogen peroxide induces apoptosis of osteocytes: involvement of calcium ion and caspase activity. Calcif Tissue Int. 2002;71:243–8.
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21.
Zahm AM, Bohensky J, Adams CS, Shapiro IM, Srinivas V. Bone cell autophagy is regulated by environmental factors. Cells Tissues Organs. 2011;194:274–8.
Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA. 2009;106:2770–5.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30.
Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38.
Thorburn A. apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2008;13:1–9.
Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10:1989–2005.
Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–83.
Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–9.
Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–9.
Shibata R, Ouchi N, Kihara S, Sato K, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis in response to tissue ischemia through stimulation of amp-activated protein kinase signaling. J Biol Chem. 2004;279:28670–4.
Hickson-Bick DL, Buja LM, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol. 2000;32:511–9.
Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy. 2007;3:238–40.
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22.
He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2–Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–81.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6.
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39.
Jee WSS, Yao W. Overview: animal models of osteopenia and osteoporosis. J Musculoskelet Neuronal Interact. 2001;1:193–207.
Florencio-Silva R, Sasso GRS, Sasso-Cerri E, Simões MJ, Cerri PS. Effects of estrogen status in osteocyte autophagy and its relation to osteocyte viability in alveolar process of ovariectomized rats. Biomed Pharmacother. 2018;98:406–15.
Yang Y, Zheng X, Li B, Jiang S, Jiang L. Increased activity of osteocyte autophagy in ovariectomized rats and its correlation with oxidative stress status and bone loss. Biochem Biophys Res Commun. 2014;451:86–92.
Kato Y, Windle JJ, Koop BA, Mundy GR, Bonewald LF. Establishment of an osteocyte-like cell line, MLO-Y4. J Bone Miner Res. 1997;12:2014–23.
Acknowledgements
This study was supported by the Natural Science Foundation of China (No. 82174413) and Health Commission of Henan Provincial, China (HNSWJW-2021016). The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services provided.
Author information
Authors and Affiliations
Contributions
LW and SC performed the cultured cells and animal experiments, Annexin V FITC and PI staining, immunofluorescence staining, immunoprecipitation, western blotting, collected data, analyzed the data, and produced all figures. CY, HZ, and JL performed the immunohistochemical staining, Tunel assay and participated in the animal surgery. NQ acquired funding, designed the experiments, oversaw the collection of the results and data interpretation and drafted the reports. All authors have seen and approved the final version of this paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, L., Chai, S., Yue, C. et al. Resveratrol protects osteocytes against oxidative stress in ovariectomized rats through AMPK/JNK1-dependent pathway leading to promotion of autophagy and inhibition of apoptosis. Cell Death Discov. 9, 16 (2023). https://doi.org/10.1038/s41420-023-01331-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01331-2
This article is cited by
-
The effects of resveratrol in animal models of primary osteoporosis: a systematic review and meta-analysis
Journal of Orthopaedic Surgery and Research (2024)
