
Abstract
Cellular transformation is a major event that helps cells to evade apoptosis, genomic instability checkpoints, and immune surveillance to initiate tumorigenesis and to promote progression by cancer stem cell expansion. However, the key molecular players that govern cellular transformation and ways to target cellular transformation for therapy are poorly understood to date. Here we draw key evidences from the literature on K-Ras-driven cellular transformation in the context of apoptosis to shed light on the key players that are required for cellular transformation and explain how aiming p53 could be useful to target cellular transformation. The defects in key apoptosis regulators such as p53, Bax, and Bak lead to apoptosis evasion, cellular transformation, and genomic instability to further lead to stemness, tumorigenesis, and metastasis via c-Myc-dependent transcription. Therefore enabling key apoptotic checkpoints in combination with K-Ras inhibitors will be a promising therapeutic target in cancer therapy.
Similar content being viewed by others
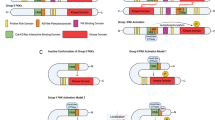

Introduction
Cellular transformation is an important process in tumorigenesis1,2,3. The ability to induce cellular transformation was initially considered as the characteristic feature of oncogenic viruses and then was narrowed down to the individual but multiple oncogenes4,5,6,7,8,9. Cellular transformation which drives evasion of apoptosis, phagocytosis, and accumulation of genomic instability can result in tumorigenesis3,10,11. A huge body of evidence indicates that transcription-driven epithelial to mesenchymal transition in combination with cell death evasion is the main cause for metastasis12, a pivotal event that leads to mortality. Thus, there is a pressing need to understand the pivotal players of cellular transformation and the ways to combat cellular transformation for therapeutic purpose.
Cellular transformation is a process in which the cancer cells (or normal stem cells) acquire the habit of aggregation, cell fusion, and growth into spheroids13,14,15,16. Mere aggregation of cells is not sufficient to complete cellular transformation and this process needs cell fusion through membrane lipids such as cholesterol14. Cellular transformation15 is also mentioned in literature with different names such as spheroid formation17, clonogenic growth18, focus formation19,20, bullet formation21, and so on. Recent studies envisaged that the blebbishield-mediated transformation3,11,12,14,22,23,24,25,26 drives cellular transformation after induction of apoptosis (Fig. 1). If the cells undergo transformation for the first time from normal cells which is accompanied by alterations from normal cell behavior, it is referred to as neoplastic transformation27 and it leads to tumorigenesis depending on the extent of genomic instability it has accumulated11. However, if a cancer stem cell (that is already transformed but not in a transformed/spheroid state) undergoes more rounds of cellular transformation, then it is referred to as malignant transformation3,11,14,22,26 because these additional rounds of transformation often results in further increase in genomic instability and metastasis3. It has been repeatedly shown that cells capable of transformation are able to form tumors in xenograft models and that the cellular transformation activity distinguishes cancer stem cells from bulk cancer cells3,11,14,22,26. Recent studies have materialized the fact that blebbishield emergency program, which includes apoptosis induction and spheroid formation were inevitable steps of K-Ras-driven cellular transformation3,11,14,22,24,26. However, the studies on blebbishield-mediated transformation envisaged that spheroid growth is not a permanent feature of cellular transformation and the spheroids can eventually give rise to polarized monolayer of cancer cells using the exit phase of blebbishield emergency program3,14,26 (Fig. 1). At this point, the cells may have lost the transformed state to various degrees but may retain stemness depending on the stemness regulatory transcription factors present. In blebbishield emergency program, the colonies that exited from spheroid-transformed state express increased c-Myc, a stemness transcription factor14. Furthermore, under certain circumstances that unleash the apoptotic/cell death process, these cells regain the ability to form spheroids28 depending upon death ligands and death receptors involved24. In the case of blebbishield emergency program, c-Myc has been shown to undergo a transient downregulation during the apoptotic phase14,24. In this context, lethal death receptor signaling is suppressed by K-Ras signaling to favor cellular transformation and metastasis29.

Schematic showing the major steps involved in K-Ras-driven cellular transformation by blebbishield emergency program.
Here we draw evidences from the literature on cellular transformation to shed light on the key players that are required for K-Ras-driven cellular transformation that is coupled to apoptosis. Based on the available literature we also discuss that the defects in key apoptosis regulators such as p53 and mitochondrial apoptosis are key to apoptosis evasion, cellular transformation, and genomic instability and discuss the therapeutic vulnerability points that can be exploited for future drug discovery. Of note, many agents discussed in this review are in fact used to understand the therapeutic vulnerable points of K-Ras driven cellular transformation through blebbishield emergency program and therefore are not to be used for clinical purposes without clinical trials.
Molecular targets of cellular transformation in cancer
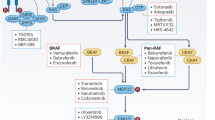
K-Ras driven cellular transformation through blebbishield emergency program has a well-documented apoptotic phase, transformation phase, and an exit phase3,14,22,23,24,25,26,30,31,32,33,34 (Fig. 1). Multiple agents that promote or inhibit these phases were identified to date (Fig. 2). Of which, agents that promote or inhibit the transformation phase are very important as this step of blebbishield emergency program can undermine the therapeutic elimination of cancer cells through apoptosis. Vascular endothelial growth factor (VEGF) signaling is a major driver of transformation phase14,22,26 despite epidermal growth factor (EGF) is being widely implicated35,36. The ability of EGF to induce VEGF secretion37 indicates that EGF might work through VEGF signaling to induce cellular transformation in addition to its role in DNA repair11. Likewise, many receptor tyrosine kinases influence cellular transformation suggesting there should be a central node where these signaling must converge. K-Ras acts as that central node in many instances under the stimulation of multiple RTKs11 (Fig. 2). VEGF mediates cellular transformation under the influence of mutation-activated K-Ras38 and targeting K-Ras activation inhibits VEGFR2 expression and abrogates transformation after apoptosis by blebbishield emergency program24,34. Thus, various RTKs and VEGF might act redundantly. K-Ras, VEGF-A, VEGFR2, p70S6K, FoxM1, Raf-1, ERK-1/2, and p90S6K are directly or indirectly linked to VEGF-mediated cellular transformation. An alternative to the notion that multiple RTKs might converge on K-Ras, different RTKs may play a dominant role depending on the cancer types of tissue/cell type (luminal, basal, epithelial, mesenchymal, etc.39) or even use other Ras family members such as N-Ras40 and H-Ras41. Ras especially K-Ras in collaboration with p47phox and PKC-ζ (and possibly other isoforms of PKC) generates reactive oxygen species (ROS) resulting in sustained activation of PKCs11,25,42 (Fig. 2). PKCs in turn activate p70S6K to promote IRES translation of critical targets that regulate stemness (c-Myc/N-Myc), survival (XIAP, cIAP-1,-2,) to prevent apoptosis11,23,24,26 (Figs. 1 and 2). Notably, K-Ras11 and JNK are implicated in both cellular transformation43 and cell death44 reiterating the point that, cellular transformation is tightly linked to life and death decisions of the cancer cell (Figs. 1 and 2).

Schematic showing the positive (black font) and negative regulators (red font) of major steps involved in K-Ras-driven cellular transformation by blebbishield emergency program. LiCl lithium chloride, AZ-58 Smac mimetic, NEAA non-essential amino acids, MOMP mitochondrial outer membrane permeabilization, PMA phorbol 12-myristate 13-acetate.
Role of blebbishield emergency program in cellular transformation
The next important phase that could be therapeutically exploited is the apoptotic phase. For centuries, apoptosis was considered as the last chapter of cells. It was not known until recently that cancer stem cells can survive after the commitment of morphological and biochemical apoptosis. Apoptosis is an essential intermediate step in K-Ras-driven cellular transformation11,24. Therefore, the apoptotic execution is overwhelmed by survival signals at multiple points to initiate the resurrection process to facilitate cellular transformation (Fig. 2). The main events that are overwhelmed during blebbishield-mediated transformation of cancer stem cells are (1) protection of mitochondria from outer membrane permeabilization (MOMP)23,24 which is primarily done by ROS detoxification systems, (2) protecting or translating the IRES anti-apoptotic target molecules23,24,45,46 including XIAP, c-IAP1, c-IAP2, and so on, (3) overriding secondary necrosis, [a process that follows apoptosis as a result of glycolytic and tricarboxylic acid (TCA) cycle shutdown leading to the paucity of ATP]24,47,48, (4) establishing VEGF autocrine loop through ROS and internal ribosome entry site (IRES) translational elements11,24,26, (5) overriding chromosomal instability checkpoints3 (mainly by suppressing p53-dependent checkpoints), and (6) overriding immunological and phagocytosis checkpoints3 (by promoting galectin-3). K-Ras plays a central role in regulating all these six main events3,11,24, albeit it has been shown in different contexts in addition to blebbishield emergency program.
Cellular transformation is achieved by inactivating Bax and p53-dependent apoptosis49. Selective suppression of p53 happens during the transformation step of blebbishield emergency program3. Cleavage of Bax by proteases into p18-Bax damages the mitochondria by MOMP and cells with MOMP were unable to transform by blebbishield emergency program23,24 (Fig. 2). Notably, Bax-p18 is a more potent mitochondrial outer membrane potential inducer than full-length Bax50. Interestingly, Bax and Bak deficiencies are linked to cellular transformation, demonstrating the importance of Bax and Bak in preventing cellular transformation after apoptosis49,51. The tumor suppressor p53, a well-known inducer of apoptosis is known to suppress transformation52,53 as well as known to transform cells54 depending on Bax status49. Hence Bax-p18 plays an important role downstream of p53 in preventing transformation after the commencement of apoptosis. This is because Bax is a p53 target gene55.
Secondary necrosis a process that occurs in apoptotic cells, spills the intracellular contents outside of the apoptotic cell and culminates in the abrogation of cellular transformation (Fig. 3)24. Secondary necrosis is mostly observed in vitro but it also happens in vivo56. Under in vivo conditions the apoptotic cells are cleared by phagocytosis before reaching the secondary necrotic stage. However, when massive number of cells undergo apoptosis that outnumbers phagocytes, or when apoptosis happens in phagocyte restricted areas of tissues, secondary necrosis can be detected56. Secondary necrosis is a clear indication of the glycolytic shutdown, or to be precise, the necrotic state is triggered by the paucity of intracellular ATP in apoptotic cells48. In blebbishield emergency program, apoptotic cells generate ATP continuously through oligomerization of K-Ras, BAD, p27, Bax, and Bak at mitochondria to boost glycolysis, which overrides secondary necrosis (Fig. 3)24. Oligomerization of Bax is implicated in MOMP induction and cytochrome-C release57, however, identification of Bax oligomers in addition to Bak, BAD, p27, and K-Ras oligomers in non-apoptotic cells convincingly links the oligomers to glycolytic function than to MOMP24. On the other hand, generation of p18-Bax and or p18-Bak is associated with secondary necrosis and abrogation of transformation from apoptotic cells24.

Schematic showing the positive and negative regulation of secondary necrosis during blebbishield emergency program in transforming cancer stem cells. Note that oligomers of K-Ras, pBAD (S-112), p27, Bax, and Bak are present in live cells and are promoted during apoptotic phase, but p18 forms of bax and bak are present only in MOMP and secondary necrosis accompanied by Smac and cytochrome-C release.
In addition to overriding secondary necrosis, apoptotic cancer stem cells also use IRES translation to neutralize pro-apoptotic signals24. Expression of p70S6K is one of the key targets which helps IRES translation by phosphorylating ribosomal S6 proteins24. Apoptotic cells are known to continue IRES translation24,58. Many of the IRES translational targets are strong anti-apoptotic molecules such as c-IAP1/2 (protects cells from extrinsic apoptosis24,45,46), XIAP (protects cells from caspase-3 mediated damage45,46,59,60), c-Myc (multiple survival and apoptotic functions), N-Myc (replenish ribosomal components by transcription61, protect mitochondria62, co-operates with Survivin during malignant transformation63 and drives blebbishield-mediated transformation after the induction of apoptosis24), BCL2 (protects mitochondria57), BCLXL (protects mitochondria57,64), and so on.
Caspase-3 plays a dual role in cellular transformation. It is required for generating blebbishields by inducing apoptosis, however, inhibiting caspase-3 inhibits transformation through loss of N-Myc expression, suggesting that caspase-3 is required for IRES translation of N-Myc24. Notably, the degree of caspase-3 activation is important because full activation results in complete cleavage of PARP24 which can impair DNA repair mechanisms that are essential to reduce DNA-double strand breaks in the genome below the threshold of apoptosis induction. In this context, FasL in combination with Smac mimetic compound AZ-58 that result in partial caspase-3 activation and incomplete PARP cleavage results in cellular transformation after the commencement of apoptosis compared to the combinations of AZ-58 with TNF-α or TRAIL that has full caspase-3 activation and complete PARP cleavage24. Furthermore, Smac and cytochrome-C release from mitochondria can also influence caspase-3 and caspase-9 activation to determine the survival of apoptotic cancer stem cells24. In addition to N-Myc, VEGF-A is also an IRES translational target crucial for cellular transformation26, VEGF autocrine loop is necessary to drive transformation from blebbishields26. Reactive oxygen species (ROS) is known to induce VEGF expression65,66 through induction of base excision repair-mediated VEGF transcription67. Then VEGF-A protein expression is regulated by K-Ras/p47phox/PKC-ζ/p70S6K/IRES translation axis11. Phorbol 12-myristate 13-acetate (PMA) activates PKC-α and PKC-ζ through ROS to enhance VEGF mRNA stability68 and stimulates VEGF-A secretion to promote the exit phase of blebbishield-mediated transformation26. ROS is mainly produced through the p47phox component of NADPH oxidase, which is modulated by the interaction of PKC-ζ with p47phox, and K-Ras25. Inhibiting ROS or inhibiting the expression of PKC-ζ and/or p47phox abrogates blebbishield emergency program25 to reiterate the fact that ROS generation is crucial to establish the VEGF autocrine loop. Although excess ROS can stimulate p53 and induce proper apoptosis, the ROS have to be neutralized to promote cell survival through K-Ras stimulated antioxidant system such as PKC-ζ/PKC-ε/Nrf-2/HO-1 axis11,69,70,71. Hence ROS could play a double role to shift the balance either toward survival or death depending on the status of K-Ras/PKCs/Nrf-2/HO-1 axis activation11 and depolarization of mitochondrial membrane potential23,72.
Accumulation of genomic instability (structural and numeric alterations in chromosomes) is a hallmark of transformed cells (Fig. 4). The degree of genomic instability is a potential indicator of the number of rounds the transformed cells evaded p53-directed genomic checkpoints. Overriding genomic checkpoints are primarily achieved by inactivating p53 by mutations or by suppressing p53 expression at critical stages of cell cycle or during apoptosis (Fig. 4). In the case of blebbishield emergency program, as the cells undergo more rounds of survival after apoptosis, p53 is suppressed and the chromosome number and nuclear size increases reflecting massive ploidy level numeric chromosomal instability (Fig. 4)3. This is primarily achieved by the fusion of apoptotic cells where the merged DNA from multiple apoptotic cells are pooled to a nucleoid state, which then reorganize into individual nuclei and subsequently into individual cells during the exit phase of blebbishield emergency program (Figs. 2 and 4)3.

Schematic showing the mechanisms of apoptotic and genomic checkpoint evasion by cancer stem cells through blebbishield emergency program. Note the self-fusion among blebbishields or blebbishield-immune cell fusion results in ploidy level chromosomal instability in cancer stem cells undergoing blebbishield emergency program. The dark shades in polyploid regions denote increased nuclear size and DNA content. The migratory progenies from blebbishield-immune cell hybrids are known to have high IGFBP5.
When the apoptotic cells are capable of fusion, it fuses with immune cells rather than get phagocytosed by it (Fig. 4). This ability of apoptotic cancer stem cells is demonstrated both in vitro (by co-culturing immune cells with apoptotic cells) and in vivo (by introducing apoptotic cells into phagocytosis competent mice)3. Cancer stem cell immune cells hybrids were demonstrated both in vitro and in vivo (in hepatosplenomegaly). Notoriously, the hybrids acquired vigorous migratory behavior with high IGFBP5 expression (Fig. 4)3. Therefore, blebbishield emergency program orchestrates multiple aspects of tumorigenesis, immune evasion, and metastasis by directing cellular transformation.
Targeting cellular transformation for cancer therapy
While blebbishield emergency program acts as the backbone of cellular transformation after apoptosis, many of the pivotal points of cellular transformation can be exploited as cancer therapeutic targets. Sp1 is a crucial transcription factor that regulates VEGF, VEGFR2 expression to regulate the K-Ras/ROS-driven VEGF autocrine feedback loop, and drives cellular transformation by blebbishield emergency program. In this context, impeding Sp1 node abrogates transformation34. It will be interesting to see if VEGF-trap73 designed to target angiogenesis could complement cytotoxic chemotherapeutics as combination therapy. Furthermore, FoxM1 inhibition targets cellular transformation by inhibiting VEGF expression74,75. At the protein level, heparin blocks VEGF-A to VEGFR2 binding thereby interferes with cellular transformation by abrogating blebbishield emergency program14 (Fig. 2). K-Ras inhibition also inhibits VEGFR activation76 and transformation77. In this context, K-Ras G12C mutant targeted inhibitors (AMG 510 and MRTX 849)78,79 or K-Ras G12D inhibitors (KS-58)80 could augment chemotherapy-induced apoptosis, in particular, it might inhibit survival after induction of apoptosis. K-Ras selectively suppresses p53 expression at protein level during transformation phase of blebbishield emergency program compared to apoptotic cells that are not able to undergo transformation3. This could probably happen through MDM2, an ubiquitin ligase that degrades p5381. Conversely, K-Ras inhibition enables p53 and downregulates MDM282. In this context, agents such as K-Ras inhibitors or quercetin could be useful as these agents can target the K-Ras-directed suppression of p5383. Similarly, inhibition of the K-Ras-associated cascade ERK-1/2, JNK84,85, Raf-1, MEK-1/284, AP185,86,87 also impede or abrogate cellular transformation. In this context, MEK-1/2, ERK-1/2 inhibition with AZD6244 is demonstrated to augment cisplatin efficacy in K-Ras G12D mice background88. Ribosomal S6 kinases (p70S6K, p90S6K, and p52S6K) transduce survival signal downstream to K-Ras/PKC axis to drive IRES translation of vital survival molecules such as c-IAPs (c-IAP1/2, XIAP, c-Myc/N-Myc/Nrf-2, and so on). In this context, BI-D1870 (S6K inhibitor) has been shown to abrogate the transformation phase of blebbishield emergency program in combination with TNF-α. Furthermore, rapamycin, and CF3DODA-Me inhibit cellular transformation by inhibiting or degrading mTOR and p70S6K, respectively34,89. Apart from these agents, multiple drugs are known to target transformation phase of blebbishield emergency program (Fig. 2). However, many of these agents are not tested in combination with standard frontline therapeutics in the context of cancer therapy or not approved for human use. Agents like esomeprazole are already in clinic for other medical conditions and hence have fewer hurdles to be tested as combination agents.
Endocytosis plays a major role in blebbishield formation, transformation, and sorting membranes during the transformed sphere stage26. However, the precise targets that direct endocytosis during these processes have to be identified before aiming therapeutic targeting of endocytosis. Notably, K-Ras is a known driver of membrane reorganization and in turn, membrane reorganization activates K-Ras. N-ethylmalemide interferes with membrane reorganization (Fig. 2) but the use of N-ethylmalemide in the clinic is not feasible due to its high toxicity and non-selectivity. Therefore N-ethylmalemide is restricted to laboratory research alone.
The core apoptosis inducers such as p53, and Bax are potential targets to block cellular transformation. VHL enables p53 to promote apoptosis90, however, in the context of blebbishield emergency program, the p19-VHL and p30-VHL isoforms play oncogenic and tumor suppressor roles, respectively33. Enabling p53 holds the key to target mitochondria damage and inhibition of cellular transformation52. It is very important to know the mutation status of p53 because it affects the activation of caspase-391, a pivotal trigger of blebbishield emergency program24,26. Defects in p53 could also deregulate miRNA-mediated regulation of tumorigenesis and metastasis because defective p53 is linked to chromosome 19 miRNA cluster (C19MC) in hepatocellular carcinoma92. Interestingly, p53 mutations cooperate with C19MC miRNA-520G to reverse interferon-γ signaling through CAAT enhancer-binding protein-β (CEBPB) in hepatocellular carcinoma93. Notably, miR-520G is accumulated ~3.75-fold more under transformed spheroid state than in monolayer growth conditions93. C19MC is also expressed in triple-negative breast cancer94, a known sub-type for therapy resistance.
Conclusion
In conclusion, K-Ras-driven cellular transformation after apoptosis can be targeted by blocking vital signaling events (K-Ras, VEGF/VEGFR2, ERK-1/2, JNK, AP1, ROS, PKCs, p70S6K, IRES translation, Nrf-2/anti-apoptotic factor translation, and ROS neutralization) and by enabling mitochondrial apoptosis regulators such as p53 and Bax-p18. Importantly, K-Ras inhibition has the capability to enable TP53 in cancers. Notably, the ability of cancer cells to generate p18-Bax is an essential aspect to abrogate transformation. Thus developing agents that target cellular transformation after apoptosis especially that are directed against K-Ras in combination with chemotherapeutics may help to combat aggressive therapy-resistant cancers in the future.
References
Kumar, M. S., Lu, J., Mercer, K. L., Golub, T. R. & Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 39, 673–677 (2007).
Ray, D. et al. Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 67, 6605–6611 (2007).
Jinesh, G. G. & Kamat, A. M. The blebbishield emergency program overrides chromosomal instability and phagocytosis checkpoints in cancer stem cells. Cancer Res. 77, 6144–6156 (2017).
Radke, K., Gilmore, T. & Martin, G. S. Transformation by Rous sarcoma virus: a cellular substrate for transformation-specific protein phosphorylation contains phosphotyrosine. Cell 21, 821–828 (1980).
Ressler, S., Connor, L. M. & Marriott, S. J. Cellular transformation by human T-cell leukemia virus type I. FEMS Microbiol. Lett. 140, 99–109 (1996).
Chen, X., Kamranvar, S. A. & Masucci, M. G. Oxidative stress enables Epstein-Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 35, 3807–3816 (2016).
Hashimoto, Y., Kawachi, E., Shudo, K., Sekiya, T. & Sugimura, T. Transforming activity of human c-Ha-ras-1 proto-oncogene generated by the binding of 2-amino-6-methyl-dipyrido[1,2-a: 3’,2’-d]imidazole and 4-nitroquinoline N-oxide: direct evidence of cellular transformation by chemically modified DNA. Jpn. J. Cancer Res. 78, 211–215 (1987).
Rodrigues, G. A., Park, M. & Schlessinger, J. Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO J. 16, 2634–2645 (1997).
Tchernitsa, O. I. et al. Transcriptional basis of KRAS oncogene-mediated cellular transformation in ovarian epithelial cells. Oncogene 23, 4536–4555 (2004).
Moody, C. A. & Laimins, L. A. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10, 550–560 (2010).
Jinesh, G. G., Sambandam, V., Vijayaraghavan, S., Balaji, K. & Mukherjee, S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 37, 839–846 (2018).
Jinesh, G. G. & Brohl, A. S. The genetic script of metastasis. Biol. Rev. Camb. Philos. Soc. 95, 244–266 (2020).
Jiang, L. et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016).
Jinesh, G. G. et al. Blebbishields, the emergency program for cancer stem cells: sphere formation and tumorigenesis after apoptosis. Cell Death Differ. 20, 382–395 (2013).
Gottardi, C. J., Wong, E. & Gumbiner, B. M. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J. Cell Biol. 153, 1049–1060 (2001).
Tran, M. N., Goodwin Jinesh, G., McConkey, D. J. & Kamat, A. M. Bladder cancer stem cells. Curr. Stem Cell Res. Ther. 5, 387–395 (2010).
Sato, M. et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 7, 33297–33305 (2016).
Nomura, Y., Tashiro, H. & Hisamatsu, K. In vitro clonogenic growth and metastatic potential of human operable breast cancer. Cancer Res. 49, 5288–5293 (1989).
Alvarez, A., Barisone, G. A. & Diaz, E. Focus formation: a cell-based assay to determine the oncogenic potential of a gene. J. Vis. Exp. 94, 51742 (2014).
Connan, G., Rassoulzadegan, M. & Cuzin, F. Focus formation in rat fibroblasts exposed to a tumour promoter after transfer of polyoma plt and myc oncogenes. Nature 314, 277–279 (1985).
Lee, E. J. et al. Spherical bullet formation via E-cadherin promotes therapeutic potency of mesenchymal stem cells derived from human umbilical cord blood for myocardial infarction. Mol. Ther. 20, 1424–1433 (2012).
Jinesh, G. G. & Kamat, A. M. Blebbishield emergency program: an apoptotic route to cellular transformation. Cell Death Differ. 23, 757–758 (2016).
Jinesh, G. G., Laing, N. M. & Kamat, A. M. Smac mimetic with TNF-alpha targets Pim-1 isoforms and reactive oxygen species production to abrogate transformation from blebbishields. Biochemical J. 473, 99–107 (2016).
Jinesh, G. G. et al. Mitochondrial oligomers boost glycolysis in cancer stem cells to facilitate blebbishield-mediated transformation after apoptosis. Cell Death Discov. 2, 16003 (2016).
Jinesh, G. G., Taoka, R., Zhang, Q., Gorantla, S. & Kamat, A. M. Novel PKC-zeta to p47 phox interaction is necessary for transformation from blebbishields. Sci. Rep. 6, 23965 (2016).
Jinesh, G. G. & Kamat, A. M. Endocytosis and serpentine filopodia drive blebbishield-mediated resurrection of apoptotic cancer stem cells. Cell Death Discov. 2, 15069 (2016).
Raptis, L. et al. Cellular ras gene activity is required for full neoplastic transformation by the large tumor antigen of SV40. Cell Growth Differ. 8, 891–901 (1997).
Jinesh, G. G., Ganiraju, M., Chinedu, M., Keith, A. B. & Kamat, A. M. Surface PD-L1, E-cadherin, CD24, and VEGFR2 as markers of migrating epithelial cancer stem cells associated with rapid tumorigenesis. Sci. Rep. 7, 9602 (2017).
Hoogwater, F. J. et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 138, 2357–2367 (2010).
Jinesh, G. G., Mokkapati, S., Zhu, K. & Morales, E. E. Pim kinase isoforms: devils defending cancer cells from therapeutic and immune attacks. Apoptosis 21, 1203–1213 (2016).
Jinesh, G. G. & Kamat, A. M. Blebbishields and mitotic cells exhibit robust macropinocytosis. Biofactors 43, 181–186 (2016).
Goodwin Jinesh, G., Willis, D. L. & Kamat, A. M. Bladder cancer stem cells: biological and therapeutic perspectives. Curr. Stem Cell Res. Ther. 9, 89–101 (2014).
Jinesh, G. G. & Kamat, A. M. RalBP1 and p19-VHL play an oncogenic role, and p30-VHL plays a tumor suppressor role during the blebbishield emergency program. Cell Death Discov. 3, 17023 (2017).
Taoka, R., Jinesh, G. G., Xue, W., Safe, S. & Kamat, A. M. CF3DODA-Me induces apoptosis, degrades Sp1, and blocks the transformation phase of the blebbishield emergency program. Apoptosis 22, 719–729 (2017).
Yamamoto, K. et al. Potentiation of epidermal growth factor-mediated oncogenic transformation by sialidase NEU3 leading to Src activation. PLoS ONE 10, e0120578 (2015).
Schnidar, H. et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 69, 1284–1292 (2009).
Saryeddine, L., Zibara, K., Kassem, N., Badran, B. & El-Zein, N. EGF-induced VEGF exerts a PI3K-dependent positive feedback on ERK and AKT through VEGFR2 in hematological in vitro models. PLoS ONE 11, e0165876 (2016).
Okada, F. et al. Impact of oncogenes in tumor angiogenesis: mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc. Natl Acad. Sci. USA 95, 3609–3614 (1998).
Kim, R. K. et al. Activation of KRAS promotes the mesenchymal features of basal-type breast cancer. Exp. Mol. Med. 47, e137 (2015).
Nazarian, R. et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010).
Hah, J. H. et al. HRAS mutations and resistance to the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in head and neck squamous cell carcinoma cells. Head. Neck 36, 1547–1554 (2014).
Jinesh, G. G. Exposing the deadly dark side of apoptotic cancer stem cells. Oncoscience 4, 124–125 (2017).
Liu, Y., Zhang, X., Wang, J., Yang, J. & Tan, W. F. JNK is required for maintaining the tumor-initiating cell-like properties of acquired chemoresistant human cancer cells. Acta Pharm. Sin. 36, 1099–1106 (2015).
Dhanasekaran, D. N. & Reddy, E. P. JNK signaling in apoptosis. Oncogene 27, 6245–6251 (2008).
Jinesh, G. G., Chunduru, S. & Kamat, A. M. Smac mimetic enables the anticancer action of BCG-stimulated neutrophils through TNF-alpha but not through TRAIL and FasL. J. Leukoc. Biol. 92, 233–244 (2012).
Jinesh, G. G. & Kamat, A. M. Redirecting neutrophils against bladder cancer cells by BCG and Smac mimetic combination. Oncoimmunology 1, 1161–1162 (2012).
Eguchi, Y., Shimizu, S. & Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57, 1835–1840 (1997).
Tsujimoto, Y. Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 4, 429–434 (1997).
McCurrach, M. E., Connor, T. M., Knudson, C. M., Korsmeyer, S. J. & Lowe, S. W. bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc. Natl Acad. Sci. USA 94, 2345–2349 (1997).
Gao, G. & Dou, Q. P. N-terminal cleavage of Bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes Bcl-2-independent cytochrome C release and apoptotic cell death. J. Cell. Biochem. 80, 53–72 (2001).
Rosen, K. et al. Downregulation of the pro-apoptotic protein Bak is required for the ras-induced transformation of intestinal epithelial cells. Curr. Biol. 8, 1331–1334 (1998).
Finlay, C. A., Hinds, P. W. & Levine, A. J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 57, 1083–1093 (1989).
Tecleab, A., Zhang, X. & Sebti, S. M. Ral GTPase down-regulation stabilizes and reactivates p53 to inhibit malignant transformation. J. Biol. Chem. 289, 31296–31309 (2014).
Lane, D. P. Cell immortalization and transformation by the p53 gene. Nature 312, 596–597 (1984).
Fischer, M. Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017).
Sachet, M., Liang, Y. Y. & Oehler, R. The immune response to secondary necrotic cells. Apoptosis 22, 1189–1204 (2017).
Hardwick J. M. & Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 5, a008722 (2013).
Marash, L. & Kimchi, A. DAP5 and IRES-mediated translation during programmed cell death. Cell Death Differ. 12, 554–562 (2005).
Lee, E. K. et al. A Smac mimetic augments the response of urothelial cancer cells to gemcitabine and cisplatin. Cancer Biol. Ther. 14, 812–822 (2013).
Metwalli, A. R. et al. Smac mimetic reverses resistance to TRAIL and chemotherapy in human urothelial cancer cells. Cancer Biol. Ther. 10, 885–892 (2010).
Boon, K. et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 20, 1383–1393 (2001).
Casinelli, G. et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Disco. 2, 16082 (2016).
Hipp, N. I. et al. MYCN and survivin cooperatively contribute to malignant transformation of fibroblasts. Carcinogenesis 35, 479–488 (2014).
Jinesh, G. G., Lee, E. K., Tran, J. & Kamat, A. M. Lenalidomide augments the efficacy of bacillus Calmette-Guerin (BCG) immunotherapy in vivo. Urol. Oncol. 31, 1676–1682 (2013).
Fay, J. et al. Reactive oxygen species induce expression of vascular endothelial growth factor in chondrocytes and human articular cartilage explants. Arthritis Res. Ther. 8, R189 (2006).
Ushio-Fukai, M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal 9, 731–739 (2007).
Fleming, A. M., Ding, Y. & Burrows, C. J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl Acad. Sci. USA 114, 2604–2609 (2017).
Shih, S. C., Mullen, A., Abrams, K., Mukhopadhyay, D. & Claffey, K. P. Role of protein kinase C isoforms in phorbol ester-induced vascular endothelial growth factor expression in human glioblastoma cells. J. Biol. Chem. 274, 15407–15414 (1999).
Tao, S. et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 74, 7430–7441 (2014).
Mylroie, H. et al. PKCepsilon-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc Res. 106, 509–519 (2015).
Loboda, A., Damulewicz, M., Pyza, E., Jozkowicz, A. & Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol. Life Sci. 73, 3221–3247 (2016).
Gupta, S., Kass, G. E. N., Szegezdi, E. & Joseph, B. The mitochondrial death pathway: a promising therapeutic target in diseases. J. Cell. Mol. Med. 13, 1004–1033 (2009).
Holash, J. et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc. Natl Acad. Sci. USA 99, 11393–11398 (2002).
Jiang, L., Wang, P., Chen, L. & Chen, H. Down-regulation of FoxM1 by thiostrepton or small interfering RNA inhibits proliferation, transformation ability and angiogenesis, and induces apoptosis of nasopharyngeal carcinoma cells. Int J. Clin. Exp. Pathol. 7, 5450–5460 (2014).
Kwok, J. M. et al. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 7, 2022–2032 (2008).
Bai, X. et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 278, 35501–35507 (2003).
Kaushik, G. et al. Honokiol affects melanoma cell growth by targeting the AMP-activated protein kinase signaling pathway. Am. J. Surg. 208, 995–1002 (2014).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Lindsay, C. R. & Blackhall, F. H. Direct Ras G12C inhibitors: crossing the rubicon. Br. J. Cancer 121, 197–198 (2019).
Sakamoto, K., Masutani, T. & Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 10, 21671 (2020).
Ries, S. et al. Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell 103, 321–330 (2000).
Halaschek-Wiener, J., Wacheck, V., Kloog, Y. & Jansen, B. Ras inhibition leads to transcriptional activation of p53 and down-regulation of Mdm2: two mechanisms that cooperatively increase p53 function in colon cancer cells. Cell Signal 16, 1319–1327 (2004).
Lee, S. J., Jung, Y. S., Lee, S. H., Chung, H. Y. & Park, B. J. Isolation of a chemical inhibitor against K-Ras-induced p53 suppression through natural compound screening. Int J. Oncol. 34, 1637–1643 (2009).
Khanal, P. et al. 5’-Nitro-indirubinoxime inhibits epidermal growth factor- and phorbol ester-induced AP-1 activity and cell transformation through inhibition of phosphorylation of Pin1. Mol. Carcinog. 50, 961–971 (2011).
Lee, N. Y. et al. Fucoidan from Laminaria cichorioides inhibits AP-1 transactivation and cell transformation in the mouse epidermal JB6 cells. Mol. Carcinog. 47, 629–637 (2008).
Kang, N. J. et al. Equol, a metabolite of the soybean isoflavone daidzein, inhibits neoplastic cell transformation by targeting the MEK/ERK/p90RSK/activator protein-1 pathway. J. Biol. Chem. 282, 32856–32866 (2007).
Chao, D. C. et al. Evodiamine inhibits 12-O-tetradecanoylphorbol-13-acetate-induced activator protein 1 transactivation and cell transformation in human hepatocytes. Phytother. Res. 25, 1018–1023 (2011).
Kim, E. Y., Kim, A., Kim, S. K. & Chang, Y. S. AZD6244 inhibits cisplatin-induced ERK1/2 activation and potentiates cisplatin-associated cytotoxicity in K-ras G12D preclinical models. Cancer Lett. 358, 85–91 (2015).
Liu, X. et al. Rapamycin inhibits Akt-mediated oncogenic transformation and tumor growth. Anticancer Res. 24, 2697–2704 (2004).
Roe, J. S. et al. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 22, 395–405 (2006).
Zanotto-Filho, A. et al. The curry spice curcumin selectively inhibits cancer cells growth in vitro and in preclinical model of glioblastoma. J. Nutr. Biochem. 23, 591–601 (2012).
Fornari, F. et al. In hepatocellular carcinoma miR-519d is up-regulated by p53 and DNA hypomethylation and targets CDKN1A/p21, PTEN, AKT3 and TIMP2. J. Pathol. 227, 275–285 (2012).
Jinesh, G. G. et al. Regulation of MYO18B mRNA by a network of C19MC miRNA-520G, IFN-gamma, CEBPB, p53 and bFGF in hepatocellular carcinoma. Sci. Rep. 10, 12371 (2020).
Jinesh, G. G., Flores, E. R. & Brohl, A. S. Chromosome 19 miRNA cluster and CEBPB expression specifically mark and potentially drive triple negative breast cancers. PLoS ONE 13, e0206008 (2018).
Acknowledgements
Not applicable.
Funding
This research did not receive any specific grant from any funding agencies including the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All authors contributed equally. All authors read and agree to the contents of this manuscript.
Corresponding authors
Ethics declarations
Ethics statement
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by I. Lavrik
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Godwin, I., Anto, N.P., Bava, S.V. et al. Targeting K-Ras and apoptosis-driven cellular transformation in cancer. Cell Death Discov. 7, 80 (2021). https://doi.org/10.1038/s41420-021-00457-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-021-00457-5
This article is cited by
-
Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis
Signal Transduction and Targeted Therapy (2022)
-
Mutant p53s and chromosome 19 microRNA cluster overexpression regulate cancer testis antigen expression and cellular transformation in hepatocellular carcinoma
Scientific Reports (2021)
