
Abstract
Ferroptosis is a type of cell death that depends on iron and reactive oxygen species (ROS). The accumulation of iron and lipid peroxidation primarily initiates oxidative membrane damage during ferroptosis. The core molecular mechanism of ferroptosis includes the regulation of oxidation and the balance between damage and antioxidant defense. Tumor cells usually contain a large amount of H2O2, and ferrous/iron ions will react with excessive H2O2 in cells to produce hydroxyl radicals and induce ferroptosis in tumor cells. Here, we reviewed the latest studies on the regulation of ferroptosis in tumor cells and introduced the tumor-related signaling pathways of ferroptosis. We paid particular attention to the role of noncoding RNA, nanomaterials, the role of drugs, and targeted treatment using ferroptosis drugs for mediating the ferroptosis process in tumor cells. Finally, we discussed the currently unresolved problems and future research directions for ferroptosis in tumor cells and the prospects of this emerging field. Therefore, we have attempted to provide a reference for further understanding of the pathogenesis of ferroptosis and proposed new targets for cancer treatment.
Similar content being viewed by others
Facts
-
Ferroptosis regulates the balance between oxidative damage and antioxidant defense.
-
It is a type of programmed cell death dependent on iron-mediated oxidative damage.
-
Some ncRNAs related to ferroptosis are mainly miRNA, lncRNAs, and circRNAs.
Open questions
-
What is the crosstalk between ferroptosis and other cell death pathways?
-
To what extent does lipid peroxidation induce ferroptosis?
-
Are ferroptosis inducers effective in killing tumor cells in clinical therapy?
-
What is the progress of ferroptosis induced by nanomaterials in tumor therapy?
Introduction
Cell death has long been recognized as a characteristic of malignancy [1, 2]. As early as the 1960s, the concept of programmed cell death (PCD) was introduced. Cell death is regulated by molecular mechanisms to ensure homeostasis and normal development of the body. Imbalance in this regulation leads to the appearance of various pathological symptoms in the body [3, 4]. In the following decades, new forms of cell death, such as apoptosis, necroptosis, pyroptosis, and autophagy were discovered, each with its unique mechanism [5, 6]. Apoptosis refers to cells in certain physiological or pathological conditions, controlled by internal genetic mechanism, in accordance with their own procedures of initiative, physiological death process. It is regulated by apoptosis-related genes, such as Bcl-2, P53, cytochrome C (Cyt C), APAF-1, and the caspase family proteins [7]; Necroptosis is a regulated form of death mediated by RIP1 and RIP3 kinases. Features include early loss of plasma membrane integrity, leakage of cell contents, and swelling of organelles. necroptosis, neither necrosis nor apoptosis, is an alternative modulated cell death model that simulates the characteristics of apoptosis and necrosis [8]. Pyroptosis is programmed cell necrosis mediated by Gasdermin D. It mainly relies on inflammasome to activate the part of the proteins in the caspase family, causing them to cut Gasdermin D protein, activate Gasdermin D protein, and the activated Gasdermin D protein is translocated to the membrane to form holes. The cell expands until its membrane ruptures, causing the release of its contents and triggering an intense inflammatory response [9, 10]. Autophagy plays a key role in cell and tissue homeostasis. Many stimuli, such as nutrient deficiency, oxidative stress, and protein aggregation, can kick-start autophagy. It can be divided into three subtypes: macro-autophagy, microautophagy, and chaperone-mediated autophagy [11].
In 2012, scientists named a new mode of regulated cell death (RCD) induced by the ferrous ion (Fe2+)-dependent accumulation of lipid peroxides as ferroptosis [12,13,14,15,16,17]. The morphological and biochemical characteristics of ferroptosis are distinct from other types of PCDs (e.g., apoptosis, necroptosis, Pyroptosis, and autophagy) [18]. In terms of biochemical characteristics, cells undergoing ferroptosis show an imbalance of oxidation–reduction levels: a significant increase in intracellular reactive oxygen species (ROS) and a significant decrease in NADPH, and therefore lipid antioxidants can inhibit ferroptosis. Cancer cells accumulate high levels of iron and ROS to promote their metabolic activities and growth [19]. Mitochondria are important sites of ROS generation and fatty acid metabolism and provide specific lipid precursors for ferroptosis to occur in the cell. Therefore, mitochondria as iron-rich (iron is necessary for the respiratory chain), ROS generating organelles, are considered as significant locations for ferroptosis. Morphologically, mitochondria undergoing ferroptosis show distinct changes compared to those in normal cells, which can be observed using transmission electron microscopy. It can be clearly seen that the volume of mitochondria in the cell reduces, the boundary shrinks, and the cristae reduce or disappear. The shape of mitochondria changes from a long rod shape to a punctate shape along with a ruptured outer membrane [20,21,22]. Further, during ferroptosis, no apoptotic bodies appear, and characteristic apoptotic proteins such as Caspases, Bax, and Bak are not activated; hence, apoptosis inhibitors cannot prevent ferroptosis [14, 23]. Ferroptosis can be inhibited to a certain extent by necroptosis inhibitors [24]. However, ferroptosis could not be inhibited by inhibitors of apoptosis, pyroptosis, and autophagy, but by iron-chelating agents and antioxidants [25]. Studies have found that different forms of RCD have different molecular mechanisms and modes of death, and there are interactions and influences among various forms of RCD [26]. Ferroptosis involves a series of complex biochemical reactions, gene expression, and signal transduction. It has gained widespread attention and is expected to bring a breakthrough in the treatment of several diseases, including cancer [27,28,29,30].
The antioxidant systems and execution systems of ferroptosis
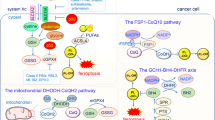
The regulation of ferroptosis is mainly competition between ferroptosis antioxidant defense system and ferroptosis execution system. Ferroptosis antioxidant defense system mainly divided into GPX4-dependent systems (SLC7A11/GSH/GPX4 axis) and GPX4-non-dependent systems (FSP1-CoQ10-NAD(P)H axis, GCH1/BH4 axis, and DHODH/CoQH2 axis) [31, 32] (Fig. 1).

Four ferroptosis defense pathways have been found so far, namely, Cyst(e)ine/GSH/GPX4 axis, FSP1-CoQ10-NAD(P)H axis, GCH1/BH4 axis, and DHODH/CoQH2 axis; ① GPX4 specifically catalyzes the loss of oxidative activity of lipid peroxides through glutathione (GSH)-dependent manner, and protects cells from the threat of ferroptosis is the most classic way; ② as a phospholipid peroxidation inhibitor that does not rely on glutathione, FSP1 converts ubiquinone on the cell membrane into reduced ubiquinol, which can inhibit peroxidation and prevent ferroptosis; ③ the GCH1/BH4 pathway acts as an endogenous antioxidant pathway, GCH1 protects cells from ferroptosis primarily through the antioxidant action of BH4; ④ DHODH resists ferroptosis in mitochondria by regulating the production of dihydroubiquinone in the inner membrane of mitochondria.
SLC7A11/GSH/GPX4 axis
The solute carrier family 7-member 11-glutathione-GPX4(SLC7A11-GSH-GPX4) signaling axis is one of the most classic ferroptosis defense pathways [33,34,35]. The l-cystine/glutamate antiporter SLC7A11 introduces cystine from the extracellular environment, which is transferred to the cytoplasm and converted into cysteine through a reduction reaction that consumes NADPH. Cysteine is subsequently used for glutathione (GSH) biosynthesis (and other biomolecules) and is involved in antioxidant defense mechanism, it is considered to be the rate-limiting precursor of the antioxidant glutathione [36] . GSH is a tripeptide composed of three amino acids (cysteine, glutamate, and glycine), which exists in a reduced (GSH) or an oxidized state (GSSG). These two states are important metabolites that maintain intracellular redox homeostasis and inhibit cell ferroptosis [37, 38]. GPX4 is the core regulatory protein of ferroptosis and has a unique role in inhibiting lipid peroxidation [24, 33]. It can reduce complex lipid peroxides to alcohols or convert free hydrogen peroxide into the water to prevent ferroptosis [33, 39, 40]. It can catalyze the oxidation of GSH to GSSG and peroxidize reduced polyunsaturated fatty acids, thereby protecting the phospholipid bilayer in the cell from oxidative damage and inhibiting ferroptosis. When the GSH-dependent lipid peroxide repair system is damaged, it causes ferroptosis through the fatal accumulation of lipid-reactive oxygen species [20, 41,42,43].
GCH1/BH4 axis
GTP cyclohydrolyse-1 (GCH1) is the rate-limiting enzyme of the 6(R)-l-erythro-5,6,7,8-tetrahydrobiopterin (BH4) complex. Kraft found that the expression of GCH1 triggered the production of potent antioxidant BH4, thus preventing lipid peroxidation [44]; BH4 has powerful antioxidant capacity and can act as a direct antioxidant to protect cells from lipid peroxidation, it can also be used for de novo synthesis of CoQ10, which also protects cells from ferroptosis [45, 46]. The GCH1/BH4 pathway acts as an endogenous antioxidant pathway, GCH1 protects cells from ferroptosis primarily through the antioxidant action of BH4, and is completely independent of GPX4-mediated protection against ferroptosis [44]. GCH1 determines the level of BH4, highlighting a direct correlation between the GCH1/BH4 axis and inhibition of ferroptosis [47,48,49].
FSP1-CoQ10-NAD(P)H axis
The pharmacological targeting of FSP1 has a strong synergistic effect with GPX4 inhibitors, which can trigger the ferroptosis of multiple tumor entities [24, 45]. There is a classic myristoylated motif at the N-terminus of ferroptosis suppressor protein 1 (FSP1), and the existence of this motif indicates that FSP1 may be related to lipid biomolecules. The mutation of the myristoylation site in FSP1 induced ferroptosis proving that FSP1 can only resist ferroptosis in the myristoylated form [46]. Ubiquinone, also known as coenzyme Q (CoQ10), exists in lipid membranes. It helps ATP production in mitochondria, and its reduced form is called ubiquinol. FSP1 converts the ubiquinone on the cell membrane into its reduced form ubiquinol, which can inhibit peroxidation and prevent ferroptosis [50]. NAD(P)H, one of the principal reductants, is produced by the pentose phosphate pathway (PPP) and can be phosphorylated by NAD kinase (NADK) to synthesize NADPH, which inhibits peroxidation damage caused by ferroptosis. The FSP1-CoQ10-NAD(P)H pathway exists as an independent parallel system, compatible with GPX4, and works with glutathione to inhibit phospholipid peroxidation and ferroptosis.
DHODH/CoQH2 axis
Dihydroorotate dehydrogenase (DHODH) is a flavin-dependent enzyme located in the inner membrane of mitochondria. Its main function is to catalyze the fourth step of the pyrimidine biosynthesis pathway. That is, dihydroorotate (DHO) is oxidized to orotate (OA), and at the same time electrons are transferred to ubiquinone in the inner membrane of mitochondria, which is reduced to dihydroubiquinone [51]. Mao et al. reported that DHODH (Dihydroorotate dehydrogenase) coordinates with GPX4 by reducing ubiquitin formation in cancer cells, blocking the action of ferroptosis in mitochondrial intima [52], DHODH inhibitors induce ferroptosis and significantly inhibit tumor growth in solid tumors with low GPX4 expression, and the combination of ferroptosis inducer sulfasalazine and DHODH inhibitors has a good therapeutic effect in solid tumors with high GPX4 expression. DHODH/CoQH2 axis is an ferroptosis inhibitor independent of the classical GPX4 signaling pathway and based on mitochondrial lipid peroxidation of ferroptosis. The findings provide a new strategy for targeting ferroptosis in cancer treatment [52, 53].
The lipid peroxidation of ferroptosis
ACSL protein mainly exists on the endoplasmic reticulum and the outer mitochondrial membrane and is a fatty acid activating enzyme. It is mainly responsible for the conversion of long-chain fatty acids to their active form acyl-CoA for their oxidation and lipid biosynthesis. Specifically, ACSL4 has a preference for long-chain polyunsaturated fatty acids, such as arachidonic acid (AA) or adrenergic acid (Ada) [54, 55], and converts them to arachidonic CoA and adrenal CoA, respectively, which are more likely to be oxidized to form lipid peroxides [56, 57].
Under normal circumstances, the accumulation of AA in cells is much lower than that of other fatty acids. The expression of ACSL4 upregulation is considered a biomarker and contributor to ferroptosis, it can esterify free polyunsaturated fatty acids into membrane phospholipids with the help of lysophosphatidylcholine acyltransferase 3 (LPCAT3). Synthesis of PUFA-PL mediated by LPCAT3 and ACSL4, as well as ALOX- and POR-mediated PUFA-PL peroxidation are necessary for ferroptosis to occur. Exogenous supplementation of AA/Ada (and other long-chain polyunsaturated fatty acids) can make ACSL4 knockout cells sensitive to ferroptosis. Hydroxyl radicals can catalyze the peroxidation of various biological macromolecules in cells, including polyunsaturated fatty acids (PUFA), and it is known that various phospholipid bilayers perform important biological functions in cells. The basic unit of phospholipid is composed of hydrophilic phosphoglycerol and hydrophobic polyunsaturated fatty acid chains. The peroxidation of the PUFA chains leads to the destruction of the phospholipid bilayer membrane structure of the cell enhancing membrane permeability, which ultimately leads to cell death [58, 59].
Iron metabolism
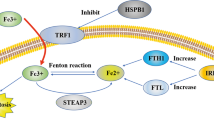
Compared with non-malignant cells, the growth of cancer cells is strongly dependent on the micronutrient iron (ferrum in Latin), which is necessary for the ferroptosis process and can be inhibited by various iron-chelating agents. The excessive iron load may lead to ferroptosis in cancer patients [60,61,62], membrane transferrin receptor 1 (TFR1) can transport Fe3+ ions into cells [63], was also recently identified as a biomarker for ferroptosis [63]. Under the action of DMT1, Fe3+ is converted to Fe2+. Excess iron is stored in ferritin, which includes a ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1) [64, 65]. Therefore, ferritin abundance, especially ferritin heavy chain (FTH1) abundance, is critical for inhibiting ferroptosis [66]. Intracellular unstable iron (Fe2+) can produce a large number of ROS through the Fenton reaction, providing sufficient raw materials for lipid peroxidation, and it is also a cofactor of lipid peroxidase (ALOXs and POR), which determines the activity of these enzymes (Fig. 2).

System XC- obtains cystine from the extracellular environment. The cystine introduced into the cells is converted into cysteine in the cytoplasm through a reduction reaction that consumes NADPH; cysteine is then used for the biosynthesis of glutathione, Glutathione peroxidase 4 (GPX4) can catalyze reduced glutathione (GSH) to oxidized form (GSSG), at the same time, the peroxidized polyunsaturated fatty acids are reduced to protect the phospholipid bilayer in the cell from oxidative damage and inhibit ferroptosis; while the long-chain acyl-CoA synthase 4 (ACSL4) can reduce arachidonic acid and adrenal acid. Polyunsaturated fatty acids (PUFA) are activated to arachidonic CoA and adrenal CoA, which are more likely oxidized to form lipid peroxides and induce ferroptosis; membrane transferrin receptor 1 (transferrin receptor 1, TFR1) can transport trivalent iron ions into the cell, and under the action of divalent metal transporter1 (DMT1), it can convert Fe3+ to Fe2+, and excess iron is stored in ferritin. Nuclear receptor coativator 4 (NCOA4) mediates the degradation of ferritin in autophagosomes, resulting in the release of ferritin-bound iron as free Fe2+ (ferritinophagy), promoting ferroptosis; FSP1 converts ubiquinone to ubiquinol, alleviating persistent oxidative damage to cell membranes and inhibiting ferroptosis.
Tumor-related signaling pathways in ferroptosis
NRF2
The nuclear factor-erythroid factor 2-related factor 2 (NRF2) signaling pathway is an important defense mechanism against ferroptosis, which needs to be activated to exert its antioxidant properties. NRF2 activity is affected by some related regulatory factors during ferroptosis. Subsequently, the activated NRF2 induces and regulates the expression of a series of downstream antioxidant factors. Therefore, in the NRF2 antioxidant regulatory factors category, both activation-related regulatory factors and downstream antioxidant factors or antioxidant systems are included.
A previous study indicated that NRF2 is an important and novel transcriptional regulator of ferroptosis in hepatocellular carcinoma (HCC) cells, and NRF2 activation can inhibit ferroptosis in HCC cells [67,68,69]. First, p62-mediated degradation of KEAP1 contributes to the activation of NRF2 during ferroptosis. Second, the genes quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), and FTH1 regulated by NRF2 inhibit ferroptosis by inhibiting iron metabolism and lipid peroxidation [67]. Inhibition of the p62-KEAP1-NRF2 antioxidant signaling pathway can significantly enhance the anticancer activity of erastin and sorafenib in liver cancer cells in vivo and in vitro [67]. The detailed functional characterization of this pathway may provide references for the treatment of liver cancer. The findings of Chen et al. showed that ARF inhibits the ability of NRF2 to transcriptionally activate its target gene SLC7A11 in vivo and in vitro and regulates the ferroptosis response in a p53-dependent and p53-independent manner. The ARF–NRF2 interaction is essential for ARF to inhibit p53-dependent tumor growth [70, 71]. Therefore, these results reveal a new mechanistic role of ARF–NRF2 interaction in controlling the growth of cancer cells in vivo (Fig. 3).

a p62-mediated degradation of KEAP1 contributes to the activation of NRF2 in ferroptosis. The activated NRF2 enters the nucleus to initiate the transcription of antioxidant enzymes, glutathione redox system, iron metabolism, and other related molecules, alleviating oxidative stress, making tumor cells resistant to ferroptosis. b When NRF2 is degraded by ubiquitination, it cannot enter the nucleus to initiate the transcription of related genes. Second, ARE is a key regulator of NRF2-mediated SLC7A11 activation, and the ARE-NRF2 interaction is a p53-independent enhanced ferroptosis sensitivity approach.
Finally, gene inactivation of tumor suppressor NRF2 makes cancer cells in orthotopic malignant mesothelioma mouse models more sensitive to ferroptosis. The results show the role of cell–cell interaction and intracellular NRF2-YAP signaling pathway in determining ferroptosis and show that malignant mutations in the NRF2-YAP signaling pathway can predict the response of cancer cells to future induction therapy for ferroptosis [72].
p53
Recent studies suggest that p53 plays an important role in controlling metabolism and ferroptosis and that p53 in normal cells can negatively regulate lipid synthesis and glycolysis and positively regulates oxidative phosphorylation and lipid catabolism. In tumor cells, mutant p53 positively regulates lipid synthesis and glycolysis. Therefore, in normal tissues, p53 tends to positively regulate ferroptosis, whereas mutant p53 makes tumor cells more sensitive to ferroptosis.
p53 plays a dual role in regulating ferroptosis. On the one hand, it promotes ferroptosis by inhibiting the expression of SLC7A11 or promoting the expression of metabolism-related genes spermidine/Spermine N1-acetyltransferase 1 (SAT1) and glutaminase 2 (GLS2) [73,74,75,76]. On the other hand, p53 inhibits ferroptosis by inhibiting the activity of dipeptidyl peptidase 4 (DPP4) or inducing the expression of cyclin-dependent kinase inhibitor 1A (CDKN1A/p21) [77], both indicate the importance of p53 as a regulator of metabolism-related genes in ferroptosis (Fig. 4).

a P53 can further promote the upregulation of ALOX15 by promoting the expression of GLS2, PTGS2, and STAT1, and ultimately promote ferroptosis, or p53 can indirectly activate the function of ALOX12 by inhibiting the transcription of SLC7A11, leading to ALOX12-dependent ferroptosis after ROS stress, this pathway mediated by ALOX12 and independent of ACSL4, promotes the peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLS). b P53 can also inhibit the generation of cellular lipid-reactive oxygen species by competitively binding to DPP4 with NOX1; at the same time, the P53-DPP4 complex promotes the expression of SLC7A11 and CDKN1A to inhibit ferroptosis.
However, p53 activation has no evident effect on the function of GPX4, which shows that it does not induce ferroptosis through GPX4 [73]. In addition to the regulation of GPX4, the level of lipid peroxides in cells can also be regulated by lipoxygenase [17, 78]. Knockout of ALOX12 by CRISPR/Cas9 can also inhibit cell ferroptosis. These studies indicate that ALOX12 is necessary for p53-mediated ferroptosis [74].
NUPR1
The stress response gene, nuclear protein 1, transcriptional regulator (NUPR1) is a multifunctional stress-inducing protein, which is produced under a variety of environmental pressures, including oxidative damage and unfolded protein response. Oxidative stress activates NUPR1 by both ER stress (METH [79] and H2O2 [80]) and non-ER stress pathways, NUPR1 regulates ferroptosis via iron metabolism, ROS homeostasis, and the GSH/GPX4 pathway [81, 82]. An increase in intracellular ROS is a major signal that reacts with intracellular iron to produce more active ROS-type hydroxyl radicals (HO•) that trigger ferroptosis [81]. The importance of NUPR1 in maintaining mitochondrial function is now clear. Mitochondrial membranes, like cell membranes, are rich in polyunsaturated fatty acids (PUFAs) and are also susceptible to high levels of ROS [83]. NUPR1 inactivation can regulate mitochondria-related ferroptosis through mitochondrial dysfunction, decreased antioxidant capacity, and increased accumulation of endogenous iron content [84].
As a stress-inducing protein, NUPR1 is overexpressed in various malignant tumors. Sorafenib, an ferroptosis inducer, can induce ferroptosis by inhibiting System Xc-, also increases the expression level of NUPR1 [85]. In addition, other studies have found that various iron inducers, such as Erastin, and RSL3, strongly activate NUPR1, supporting its protective effect on iron [86]. Recently, NUPR1 can transmediate the gene encoding lipocalin 2 (LCN2) to reduce iron-induced oxidative damage and inhibit ferroptosis. Blocking NUPR1-dependent LCN2 expression significantly increased intracellular iron concentration and subsequent oxidative damage, including lipid peroxidation and DNA damage [87]. In general, NUPR1 is critical in regulating mitochondrial-related ferroptosis, either genetic or pharmacological inhibition of NUPR1 presents tumor-killing activity.
Hippo signaling pathway
The Hippo signaling pathway regulator transcription regulator 1 (TAZ) regulates ferroptosis through epithelial membrane protein 1 (EMP1)-NOX4, which means that ferroptosis may be a therapeutic target for renal cell carcinoma and other TAZ-activated tumors [88]. TAZ affects the levels of EMP1 and NOX4 and causes lipid peroxidation and ferroptosis. Therefore, TAZ activation can enhance the sensitivity of ferroptosis and promote ferroptosis. In addition, YAP/TAZ is also regulated by metabolic pathways, which may help explain the important role of glutamine metabolism in ferroptosis [89]. In 1935, Hans Krebs proposed the famous tricarboxylic acid cycle (TCA), pointing out the importance of glutamine metabolism in animals [90]. Tumors can control ROS levels through products produced by the glutamine metabolic pathway, and the most important pathway for glutamine to control ROS is glutathione synthesis [91]. Glutamine is transported to cells through the transporter SLC1A5 (member of solute carrier family 1 neutral amino acid transporter 5), and glutamate is generated under the catalysis of glutaminase (GLS or GLS2) as one of the raw materials for glutathione synthesis [92]. Glutamic acid can be catalyzed by glutamase (GLUD) or transaminase to produce α-ketoglutaric acid, and eventually to acetyl-CoA, which can be used for the direct synthesis of lipids [93]. In addition, TAZ-EMP1-NOX4 and ZEB1-GPX4 may represent two different ways to connect chemotherapy resistance and ferroptosis, and they may play a role in providing resistance in different environments [88].
In epithelial cells, E-cadherin inhibits ferroptosis by mediating the interaction of intracellular NF2 (also called merlin) and Hippo signaling pathway while antagonizing the signal axis mediated by E-cadherin promotes ferroptosis through YAP [72]. The cadherin–Merlin–Hippo–YAP signaling axis often undergoes mutations in cancer. When this signaling pathway is antagonized, the protooncogene transcription coactivator YAP will be upregulated, leading to an increase in the key ferroptosis factors ACSL4 and TFRC, thereby inducing ferroptosis in cancer cells [88].
Epithelial–mesenchymal transition
Both epithelial–mesenchymal transition (EMT) and ferroptosis can be regulated by epigenetics. In tumor cells, the epigenetic reprogramming of EMT makes head and neck cancer (HNC) cells more sensitive to ferroptosis [94]. To obtain more mesenchymal characteristics [95], cells use SIRT1 induction or miR-200 family inhibition, causing metastasis in cancer cells with interstitial properties [96, 97]. Viswanathan et al. [12] reported that in some cancer cells drug resistance occurs mainly because cancer cells are in the “quasi-stable state” with mesenchymal characteristics. In this state, the GPX4 is active in cells, and lipid peroxides are reduced, therefore leading to ferroptosis resistance. After knocking out the ZEB1 gene related to lipid uptake, aggregation, and migration, the sensitivity to ferroptosis and susceptibility to ferroptosis inducers was significantly increased, leading to drug-resistant cancer cell death [78, 98]. Therefore, decreased E-cadherin expression or increased ZEB1 expression leads to increased sensitivity to ferroptosis inducers [72, 99]. EMT regulation of cancer cells is a promising therapeutic strategy to promote the anticancer effect of ferroptosis inducers.
Noncoding RNA and tumor cell ferroptosis
Noncoding RNAs (ncRNAs) can be divided into micro RNAs (miRNA), long noncoding RNAs (lncRNAs), and circular RNAs (circRNA) [4, 100, 101]. They are widely involved in the regulation of gene expression in cells [102,103,104], and play an important role in the occurrence and development of cancer [105, 106]. At present, the most widely studied ncRNAs related to ferroptosis are mainly miRNA, followed by lncRNAs and circRNAs. ncRNA can directly target ferroptosis-related molecules or downstream target genes and proteins in the form of miRNA–mRNA, IncRNA–miRNA, IncRNA–mRNA, circRNA–miRNA, and other regulatory networks to affect ferroptosis in tumor cells. Existing studies have partially revealed the mechanism of ncRNA regulating ferroptosis and other cell death in the context of tumor, and the intricate regulatory network of ncRNA may become the “hub” of various signaling pathways related to different cell death modes.
miRNA and ferroptosis
miRNAs may regulate ferroptosis by affecting the expression of ROS [107]. For example, miR-206 induces ROS accumulation by binding to superoxide dismutase 1 (SOD1) mRNA [108], miR-155 increases ROS production by inducing FOXO3a deficiency [109]. While miR-25 and miR-448-3p reduce ROS levels by targeting nicotinamide adenine dinucleotide phosphate oxidase (NOX) [110, 111], tumor-associated fibroblasts (cancer-associated fibroblasts, CAFs) inhibit the ferroptosis in cancer cells by secreting exosomal miR-522 to target ALOX15 and blocking the accumulation of lipid ROS [112]. miR-9 regulates ferroptosis by targeting glutamate oxaloacetate transaminase 1 (GOT1), then reduce ROS levels in melanoma cells [113]. Polyunsaturated fatty acids (PUFAs), miR-3595 [114], miR-205 [115], and miR-224-5p [116] can reduce the expression of ACSL4. miR-150-5p [117] targets the downregulation of SLC38A1 to inhibit ferroptosis, whereas some miRNAs may promote ferroptosis by targeting SLC7A11. For example, miR-375, miR-27a, and miR-26b can inhibit the transcription of SLC7A11 mRNA [118,119,120]; miR-182–5p and miR-378a-3p negatively regulate the expression of GPX4 and SLC7A11 by directly binding the 3' UTR of GPX4 and SLC7A11 mRNA [121]; and miR-137 directly targets glutamine transport SLC1A5 to regulate ferroptosis negatively in melanoma cells [122].
Some miRNAs can induce ferroptosis by regulating the expression of NRF2. First, miR-7 and miR-200A can easily induce the activation of the NRF2 pathway by inhibiting the expression of Keap1 [123, 124]. Second, miR-101 and miR-455 can be targeted by Cullin-3 (CUL3) [125, 126] to promote the nuclear accumulation of Nrf2. According to existing research results, miRNAs are also involved in iron output, storage, utilization, and absorption. miR-20a and miR-485–3p can reduce iron output by targeting FPN genes [127, 128], miR-200b, and miR-let-7d effectively reducing iron accumulation by inhibiting the expression of FTH, DMT1, and iron response element (IRE) [129, 130]. miR-335 increases iron release, lipid peroxidation, and ROS accumulation by degrading FTH1, and reduces mitochondrial membrane potential (MMP), thereby promoting ferroptosis [131] (Table 1).
LncRNA and ferroptosis
ELAVL1 (ELAV-like RNA-binding protein 1) is highly expressed in many human tumors and regulates eukaryotic gene expression at the post-transcriptional level, which can enhance RNA stability [132, 133]. Wang et al. found that LINC00336 is an oncogene that combines with ELAVL1 to promote tumor cell proliferation, inhibit ferroptosis, and induce tumor formation in an ELAVL1-dependent manner [134]; LINC00618 interacts with lymphoid-specific helicase (LSH) to reduce the expression of SLC7A11, thereby inhibiting ferroptosis [135]. Erastin upregulated the expression of lncRNA GABPB1-AS1, inhibited the translation of GABPB1 and the expression of the peroxidase gene, leading to the accumulation of ROS and MDA, and promoting tumor cell death, suggesting that GABPB1-AS1 may be the key to Erastin-induced ferroptosis [136, 137] (Table 2).
CircRNA and ferroptosis
CircRNAs participate in the ferroptosis process of tumor cells through the competitive endogenous RNA (ceRNA) pathway. For example, CircABCB10 inhibits ferroptosis and apoptosis of rectal cancer cells by regulating the miR-326/CCL5 axis, providing a potential therapeutic target for the treatment of rectal cancer [138]. CircIL4R acts as a miR-541–3p sponge to regulate its target gene Gpx4; thus, circIL4R acts as a tumor promoter and ferroptosis inhibitor in liver cancer through the miR-541–3p/Gpx4 network [139]. Circ-TTBK2 uses sponge miR-761 to target ITGB8 to regulate the proliferation, invasion, and ferroptosis of glioma cells, providing a promising biomarker for the clinical treatment of human glioma [140]. The circular RNA clARS regulates the ferroptosis of HCC through the binding protein ALKBH5 [141]. To date, there have been only a few studies on circRNA in ferroptosis, which is an emerging field that needs to be further explored (Table 3).
In recent years, researchers have detected some ncRNAs associated with ferroptosis in tumor cells. However, the specific mechanism has not been discussed yet, and there are still many obstacles in the clinical treatment of ferroptosis dependent on ncRNAs. The information summarized in our paper is not enough to support the application of ferroptosis inducers in cancer, more ncRNAs identification and further studies are needed. Downstream molecules regulated by ncRNAs, P53, HSPB1, NRF2, and NOX2, key regulators of ferroptosis, are also involved in the regulation of other cell death types [65], suggesting that ferroptosis and other types of cell death are independent and related. Further exploration of the interregulation of various types of cell death can provide insights into the role of regulatory cell death in cancer. Although it has been reported in the artical, ncRNAs may become markers to filter cancer patients who are fit for ferroptosis therapy and become therapeutic targets of ferroptosis inducers [142], there is no mature technique for ncRNA-mediated tumor therapy. To further understand the factors influencing the direction of ferroptosis regulated by these molecules, fully exert the upstream regulatory role of ncRNA in the signaling pathway, promote the pathway of ncRNA promoting ferroptosis in tumor cells, and inhibit its negative regulation of ferroptosis.
Ferroptosis in tumor therapy
Targeted cell death is a common approach in tumor therapy [143]. Since ferroptosis inducers have the possibility of targeting cancer cells specifically, the use of ferroptosis-inducing drugs can improve the antitumor efficacy of these drugs [27]. In the course of chemotherapy or targeted therapy for cancer patients, drug resistance is a key problem to be solved urgently. Viswanathan et al. found that a drug-resistant high mesenchymal state is dependent on a GPX4-regulated lipid peroxidase pathway that protects cells against ferroptosis [78]. Tumor cells may significantly enhance their oxidative stress defense ability by negatively regulating iron ions, thereby achieving drug-resistant survival. By inducing tumor cell ferroptosis, it may be possible to reverse chemotherapy or targeted therapy drug resistance [144,145,146]. At present, ferroptosis inducers used in clinical tumor treatment primarily inhibit the activity of System XC- and GPX4. In addition, some natural products play an important role in inducing ferroptosis in tumor cells (Table 4).
Ferroptosis-inducing drugs used for tumor treatment
Inhibition of the System XC- activity
The main function of System XC- is to transfer glutamate from inside the cell to the outside and transfer the extracellular cystine into the cell, and the transferred cystine is used for the synthesis of glutathione in the cell. When System Xc- is inhibited by ferroptosis inducers, glutathione synthesis is reduced. Subsequently, GPX4 is unable to use glutathione to reduce lipid peroxides, causing cell ferroptosis (Table 5).
Erastin is the first identified compound used to induce ferroptosis. Erastin selectively acts on tumor cells carrying the oncogene RAS; after acting on the tumor cell surface System XC- [112], it directly binds to mitochondrial voltage-dependent anion channel 2 (VDAC2) and induces mitochondrial damage that produces ROS in an NADH-dependent manner to inhibit GSH synthesis. Erastin induces cell death through the RAS–RAF–MEK pathway in some tumor cells expressing ferroptosis activating mutations [147]. In addition, Erastin strongly enhances the effect of wild-type epidermal growth factor receptor cells by inducing ROS-mediated caspase-independent cell death [148].
Sulfasalazine (SAS) also acts on System XC- to inhibit the uptake of cystine [149], causing chronic depletion of glutathione in cells, thereby destroying the redox defense of cells, hindering tumor growth, and inducing tumors ferroptosis [149,150,151,152,153].
Sorafenib induces ferroptosis by targeting the cystine/glutamate anti-transport system Xc- [154,155,156,157], an effect that impinges on cystine uptake, thereby preventing subsequent synthesis of glutathione (GSH), the major intracellular antioxidant, preventing GPX4 activation [158, 159], and can trigger ER stress and ferroptosis.
Inhibition of the GPX4 activity
There are some ferroptosis inducers that block the intracellular antioxidant enzyme GPX4 through endogenous pathways. For example, RSL3, an activator of ferroptosis, does not depend on VDAC2/3 and is selective for tumor cells carrying tumorigenic RAS. Based on affinity chemical proteomics, the chloroacetamide part of the RSL3 structure is essential for its activity. The alkylation of selenocysteine directly inactivates GPX4 and induces lipid peroxidation, thereby inducing ROS production [23, 150, 160]. The other ferroptosis inducer, ML162 [161] and ML210, has the same effect as RSL3 in inducing ferroptosis and inhibiting the activity of GPX4. The ML210 small molecule is likely to exert its inhibitory effect by covalently modifying the selenocysteine of GPX4 [162, 163].
FIN56 was discovered through the modulation map of 56 caspase-independent lethal compounds. Idebenone is the only inhibitor of ferroptosis caused by FIN56 [164]. The oxime group in the structure of FIN56 is essential for the induction of ferroptosis, and the hydrophobicity of the piperidine group affects the potential of ferroptosis. There are two different ways in which FIN56 can induce ferroptosis. First, it uses the activity of acetyl-coenzyme A carboxylase (ACC) to degrade GPX4; second, FIN56 binds and activates squalene synthase (SQS). SQS is an enzyme involved in the synthesis of cholesterol, which leads to the depletion of the endogenous antioxidant Coenzyme Q10 (CoQ10), a non-steroidal metabolite in the glutaric acid pathway. This process enhances the sensitivity of ferroptosis induced by FIN56 [159, 164,165,166].
Glutathione is a tripeptide composed of three amino acids (cysteine, glutamic acid, and glycine). Buthionine sulfoximine (BSO) can selectively inhibit r-glutamylcysteine synthase (R-GCS), thus preventing the synthesis of dipeptides from glutamic acid and cysteine. Therefore, as a rate-limiting enzyme in the synthesis of glutathione (GSH), BSO can inhibit the synthesis of reduced glutathione, reduce the activity of GPX4, and promote ferroptosis [167,168,169,170].
Withaferin A, as a steroid isolated from Withania somnifera, is a natural ferritin inducer for neuroblastoma, dose-dependently either activates the NRF2 pathway through targeting of Kelch-like ECH-associated protein 1 (KEAP1) or aspartate aminotransferase (GPX4) to induce ferroptosis [171, 172].
Other drugs induce ferroptosis
Artemisinin is a natural product of sesquiterpenes and is an effective component of the dried stems and leaves of Artemisia annua in the Compositae family. Studies have found that artemisinin can exert anticancer effects by inducing iron-dependent death of tumor cells [173]. Mechanistic studies have shown that artemisinin can bind to transferrin to enhance its selectivity and cytotoxicity in cancer cells. In addition, artesunate, a derivative of artemisinin, has significant antitumor activity. Artesunate can increase iron concentration by increasing ferritin hydrolysis and inducing ferroptosis through iron dependence [174]. Under the induction of artesunate, the level of lipid peroxidation in hepatocellular carcinoma (SMMC-7721) cells increases, and the use of iron-chelating agent deferoxamine can eliminate the accumulation of intracellular lipids caused by artesunate peroxidation [175, 176]. After the intervention of ovarian cancer cells, the intracellular ROS level increased and the ferroptosis inhibitor (Fer-1) can significantly inhibit artesunate-induced cell death [177].
Statins stand out as promising candidates for the therapeutic induction of ferroptosis in chemoresistant cancer cells [178]. By reducing isopentenic pyrophosphate production in the mevalonate pathway, statins inhibit biosynthesis of selenoproteins such as GPX4 and CoQ10, thereby promoting ferroptosis or selectively inducing regulatory cell death in mesenchymal cells [71]. satins could effectively kill triple-negative breast cancer (TNBC) through induce ferroptosis [179].
Lapatinib is approved for the treatment of ErbB2-positive breast cancer and other cancers that overexpress ErbB2. In particular, it is used as a treatment for patients with advanced or metastatic ErbB2-positive breast cancer [180, 181]. Combining siramesine and lapatinib causes ferroptosis through iron transport disruption leading to increased ROS in breast cancer [182, 183], or targeting GPX4 might be a potential strategy to enhance antitumor effects of lapatinib in NSCLC cells [184].
BAY-87–2243 inhibits complex I (CI) of the mitochondrial respiratory chain then triggers ferroptosis of BRAFV600E melanoma cell lines [185]. On the other hand, In contrast to previously described iron inducers, FINO2 neither inhibits systemic XC -, nor directly targets the reductase GPX4 as erastin and RSL3 do, nor consumes the GPX4 protein as FIN56 does. In contrast, FINO2 both indirectly inhibits the enzyme function of GPX4 and directly oxidizes iron, ultimately leading to extensive lipid peroxidation [186, 187].
In addition to the preclinical drugs associated with ferroptosis mentioned above, some ferroptosis inhibitors are widely used in tumor research. Such as ferrostatin-1 and liproxstatin-1, both inhibitors work by inhibiting lipid peroxides, thereby inhibiting ferroptosis in tumor cells [188, 189]. Vitamin E, a fat-soluble Vitamin, is one of the main antioxidants and also inhibits ferroptosis by inhibiting the production of lipid peroxides [190]. Deferoxamine (DFO) [187], Ciclopirox [191], and Deferiprone [192] inhibit ferroptosis by deplete iron.
We summarize the mechanisms of ferroptosis in reversing drug resistance in preclinical studies. But there is still a long way to go before it can actually be used in patients. At the same time, we also face many challenges: the development of novel ferroptosis-inducing drugs requires consideration of drug toxicity and prevention of off-target effects to avoid other adverse reactions in patients; ferroptosis may be associated with a variety of pathological conditions—including acute kidney injury, tissue deficiency Blood and reperfusion injury and neurodegeneration, etc., in the process of inducing ferroptosis of tumor cells, it is also necessary to avoid other systemic adverse reactions in patients; In addition, since various types of cancer have different sensitivities to ferroptosis, we are temporarily unable to confirm whether the therapeutic strategy of inducing tumor cells ferroptosis in patients is universal? And which drugs are most suitable for clinical treatment? We also need to identify the target patient population most likely to benefit from this strategy.
Nanomaterials used for the treatment of tumor ferroptosis
In recent years, researchers have tried to combine bio-nanotechnology with ferroptosis to develop candidates with a stronger antitumor effect [193, 194]. The delivery of nano-drugs is based on engineering technology. Nanoparticles are used to deliver and control drug release and adjust the intracellular chemical reaction to affect the ROS levels, thereby improving the pharmacokinetic properties of the drug [58, 195, 196]. Nanomaterials can be used to supplement exogenous lipids in tumor cells to increase the accumulation of intracellular lipid peroxides, promote ferroptosis, and achieve the goal of curing cancer [197]. The current emerging nanotherapies mainly focus on inhibiting the expression of GPX4 in tumor cells, increasing the accumulation of ferrous/iron ions in tumor cells, and regulating lipid peroxidation [198,199,200]. This process primarily involves triggering or promoting the Fenton response in tumor cells [198].
Inhibition of GPX4 expression
Currently, nanomaterials can be used to disrupt pathways related to the activity of GPX4 to induce ferroptosis and drive cancer therapy [201, 202]. Some nanomaterials, such as sorafenib, are encapsulated into network-like nanostructures composed of Fe3+ and tannic acid (TA) [201]. Sorafenib is a typical small-molecule System Xc- inhibitor. It inhibits GPX4, leading to tumor-specific ferroptosis, and TA is used to chemically reduce Fe3+ to Fe2+ and continuously supply Fe2+ to maintain the iron redox cycle and maintain the Fenton reaction [201]. Shen et al. by using lactoferrin (LF) and RGD dimer (RGD2)-coupled cisplatin (CDDP) Fe3O4/Gd2O3 hybrid nanoparticles FeGd-HN@Pt@LF/RGD2 successfully combined and delivered Fe2+, Fe3+, and H2O2 (the reactants involved in the Fenton reaction) to the tumor sites. Their local concentration was increased to accelerate the Fenton reaction, significantly improving the efficacy of in situ brain tumor ferroptosis treatment [203]; conversely, targeting GPX4 by nanodrug delivery systems (nano-DDS) of small-molecule inhibitors can overcome the shortcomings of rapid systemic clearance and poor tumor targeting. Similar to the classic ferroptosis inducer Erastin, nano-DDS can inactivate GPX4 by depleting the intracellular GSH substrate. Shuaifei Wang et al. synthesized arginine-rich manganese silicate nanobubbles (AMSNs) to target tumor cells by inducing ferroptosis [202].
Increasing the accumulation of ferrous/iron ions in tumor cells
Iron-based nanomaterials can induce cell ferroptosis and provide an innovative method for cancer treatment. Current “ferroptosis” therapeutic nano preparations are often combined with other treatment methods, resulting in complex nanostructures and multi-metal compositions. Shasha He and others from Nanyang Technological University in Singapore reported the progress on the development of iron-chelated semiconductor multi-composite nanoparticles (SPFeN) under the guidance of photoacoustic (PA) imaging for the treatment of cancer using photothermal “ferroptosis” [204]. Professor Song Yang and Associate Professor Zhu Xiaokang from Southwest University designed a poly-nanosystem Fe3O4-PLGA-Ce6 coated with PLGA, containing iron oxide (Fe3O4) and photosensitizer Ce6, and used it to synergize ferroptosis–photodynamics anticancer treatment. Fe3O4-PLGA-Ce6 nanosystem can dissociate in acidic TME and release ferrous/iron ions and Ce6. Subsequently, the released ferrous/iron ions will react with excess hydrogen peroxide in the cell to produce a Fenton-like reaction generating hydroxyl free radicals (•OH), and induce ferroptosis of tumor cells [205]. In addition, in the xenograft model, ultra-small silica nanoparticles were shown to induce ferroptosis by increasing the transport and accumulation of iron in cells and inhibit tumor growth [206, 207].
Exogenous regulation of lipid peroxidation in tumor cells
Ferroptosis is closely related to the accumulation of lipid hydrogen peroxide in cells, which is mainly derived from PUFAs of membrane phospholipids under oxidative stimulation [144, 208]. Therefore, the use of nano-DDS to exogenously supplement cancer cells with extra polyunsaturated fatty acids to exogenously regulate lipid peroxidation is also an effective strategy to improve the therapeutic effect of ferroptosis-driven cancer treatment [209,210,211]. Zijian Zhou et al. found that in triple-negative breast cancer (TNBC) cells, there is a close relationship between the level of PUFAs in the cell and ferroptosis [211]. Among them, conjugated linolenic acid showed the strongest activity as a ferroptosis inducer. These results provide evidence that PUFAs exert antitumor activity by inducing ferroptosis.
Combining ferroptosis-inducing inducers with bio-nanotechnology for tumor therapy has broad application prospects [212], but there are still many problems to be solved in clinical ferroptosis-based nanotherapy. First of all, the potential toxic and side effects of nanomaterials should be fully studied to ensure the safety of their application in clinical treatment; in addition, there is an urgent need to develop more precise targeted nanomaterials to ensure tumor-specific triggering of ferroptosis while avoiding off-target toxicity to normal tissues; as nanomaterial-mediated ferroptosis may vary greatly between animals and patients, a more complete assessment system is needed in clinical studies.
Conclusions and perspectives
So far, researchers have elucidated some regulatory mechanisms and signal transduction pathways of ferroptosis [213, 214]. Anticancer strategies based on ferroptosis have been widely recognized, and there has been an upsurge in the development of anticancer drugs worldwide. Several natural products have been found to have the potential to be used as ferroptosis anticancer drugs. Therefore, the rational use of these mechanisms in the biomedical field to regulate cell ferroptosis and specifically induce tumor cell ferroptosis during tumor treatment is a promising cancer treatment strategy [215]. Ferroptosis inducers such as Erastin and RSL3 can synergistically induce tumor growth inhibition with anti-PD-L1 antibodies both in vitro and in vivo, and participate in immunotherapies [71]. Wang et al. found that in vitro culture with low cystine and in vivo data show that ferroptosis is involved in T cell-mediated cancer immunity [216]. Which also provoked a review of the relationship between ferroptosis mechanisms and immune system activation [217].
In addition, nanoparticles carrying chemicals or biological materials will provide the possibility to improve the efficacy of existing ferroptosis inducers and to develop new inducers for the treatment of cancer. Some nanoparticles can sensitize effective ferroptosis, produce mild immunogenicity, and improve the response rate of non-inflammatory tumors in cancer immunotherapy [218]. The biomimetic magnetic nanoparticles Fe3O4-SAS@PLT-mediated ferroptosis and immunotherapy is expected to provide great potential in treating tumor metastasis [219]. Typical strategies mainly focus on developing high-performance nanocatalysts and increasing intracellular reactants (such as H2O2 and iron ions) based on the Fenton reaction of nano-DDS [198, 200]. Several nanocatalysts have been developed to initiate the local Fenton response in tumors for cancer treatment. However, the long-term effects of nanoparticles on human health still need to be carefully evaluated.
The antitumor effect of ionizing radiation may be enhanced by triggering ferroptosis and ferroptosis inducers with effective radiosensitizers [71, 220,221,222,223,224]. Due to the non-apoptotic nature of ferroptosis, ferroptosis-based cancer treatments are expected to bypass the shortcomings of traditional therapies mediated by apoptotic pathways [225]. Targeting pathways that can regulate tumor cell ferroptosis is an emerging antitumor strategy because malignant tumor cells usually rely on oncogenic and survival signals, making them particularly vulnerable to ferroptosis. A better understanding of the regulatory mechanism and signaling pathways of ferroptosis and finding ways to help detect and track ferroptosis biomarkers will be an active research area in the next few years [32, 71].
Data availability
All data generated or analyzed during this study are included in this published article.
References
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Cui C, Yang J, Li X, Liu D, Fu L, Wang X. Functions and mechanisms of circular RNAs in cancer radiotherapy and chemotherapy resistance. Mol Cancer. 2020;19:58.
Wu Y, Wang D, Wei F, Xiong F, Zhang S, Gong Z, et al. EBV-miR-BART12 accelerates migration and invasion in EBV-associated cancer cells by targeting tubulin polymerization-promoting protein 1. FASEB J. 2020;34:16205–23.
Wang D, Zeng Z, Zhang S, Xiong F, He B, Wu Y, et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell proliferation through the LOC553103-STMN1 axis. FASEB J. 2020;34:8012–27.
Wang Y, Mo Y, Peng M, Zhang S, Gong Z, Yan Q, et al. The influence of circular RNAs on autophagy and disease progression. Autophagy. 2021:18:240–53.
Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21:678–95.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417.
Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15:199.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–42.
Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top medicinal Chem. 2001;1:497–506.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci: CMLS. 2016;73:2195–209.
Bebber CM, Müller F, Prieto Clemente L, Weber J, von Karstedt S. Ferroptosis in cancer cell biology. Cancers. 2020;12:164.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Yu X, Long YC. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep. 2016;6:30033.
Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the "iron maiden" cell death. Cells. 2020;9:1505.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017;12:558–70.
Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48:1033–43.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat cell Biol. 2014;16:1180–91.
Green DR. The coming decade of cell death research: five riddles. Cell. 2019;177:1094–107.
Snyder AG, Oberst A. The antisocial network: cross talk between cell death programs in host defense. Annu Rev Immunol. 2021;39:77–101.
Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The role of erastin in ferroptosis and its prospects in cancer therapy. OncoTargets Ther. 2020;13:5429–41.
Stockwell BR, Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. 2020;27:365–75.
Wang M, Dai M, Wang D, Tang T, Xiong F, Xiang B, et al. The long noncoding RNA AATBC promotes breast cancer migration and invasion by interacting with YBX1 and activating the YAP1/Hippo signaling pathway. Cancer Lett. 2021;512:60–72.
Zhang Y, Wang D, Peng M, Tang L, Ouyang J, Xiong F, et al. Single-cell RNA sequencing in cancer research. J Exp Clin Cancer Res : CR. 2021;40:81.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12:836–57.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, et al. MYCN-amplified neuroblastoma is addicted to iron and vulnerable to inhibition of the system Xc-/glutathione axis. Cancer Res. 2021;81:1896–1908.
Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun. 2017;8:14844.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2020;12:599–620.
Krummel B, Plotz T, Jorns A, Lenzen S, Mehmeti I. The central role of glutathione peroxidase 4 in the regulation of ferroptosis and its implications for pro-inflammatory cytokine-mediated beta-cell death. Biochimica et Biophysica Acta Mol Basis Dis. 2021;1867:166114.
Wang R, Su Q, Yin H, Wu D, Lv C, Yan Z. Inhibition of SRSF9 enhances the sensitivity of colorectal cancer to erastin-induced ferroptosis by reducing glutathione peroxidase 4 expression. Int J Biochem cell Biol. 2021;134:105948.
Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52.
Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS, Lee HC, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget. 2017;8:114588–602.
Wei F, Wang D, Wei J, Tang N, Tang L, Xiong F, et al. Metabolic crosstalk in the tumor microenvironment regulates antitumor immunosuppression and immunotherapy resisitance. Cell Mol Life Sci : CMLS. 2021;78:173–93.
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Werner ER, Blau N, Thöny B. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochemical J. 2011;438:397–414.
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–60.
Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochemical J. 2000;347:1–16.
Stockwell BR. A powerful cell-protection system prevents cell death by ferroptosis. Nature. 2019;575:597–8.
Martínez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature. 2020;585:288–92.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Wang F, Min J. DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduct Target Ther. 2021;6:244.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Wei X, Yi X, Zhu XH, Jiang DS. Posttranslational modifications in ferroptosis. Oxid Med Cell Longev. 2020;2020:8832043.
Cui Y, Zhang Y, Zhao X, Shao L, Liu G, Sun C, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immunity. 2021;93:312–21.
Tian X, Li S, Ge G. Apatinib promotes ferroptosis in colorectal cancer cells by targeting ELOVL6/ACSL4 signaling. Cancer Manag Res. 2021;13:1333–42.
Li L, Sun S, Tan L, Wang Y, Wang L, Zhang Z, et al. Polystyrene nanoparticles reduced ROS and inhibited ferroptosis by triggering lysosome stress and TFEB nucleus translocation in a size-dependent manner. Nano Lett. 2019;19:7781–92.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Tang B, Zhu J, Li J, Fan K, Gao Y, Cheng S, et al. The ferroptosis and iron-metabolism signature robustly predicts clinical diagnosis, prognosis and immune microenvironment for hepatocellular carcinoma. Cell Commun Signal: CCS. 2020;18:174.
Liu H, Gao L, Xie T, Li J, Zhai TS, Xu Y. Identification and validation of a prognostic signature for prostate cancer based on ferroptosis-related genes. Front Oncol. 2021;11:623313.
Ma J, Hu X, Yao Y, Wu L, Sheng C, Chen K, et al. Characterization of two ferroptosis subtypes with distinct immune infiltration and gender difference in gastric cancer. Front Nutr. 2021;8:756193.
Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30:3411–e3417.
Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55–63.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Mumbauer S, Pascual J, Kolotuev I, Hamaratoglu F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019;15:e1008396.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Liu Y, Tao S, Liao L, Li Y, Li H, Li Z, et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun. 2020;11:348.
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426.
Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68:224–e224.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402–6.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–91.
Zhang Z, Guo M, Shen M, Kong D, Zhang F, Shao J, et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020;36:101619.
Galluzzi L, Bravo-San Pedro JM, Kroemer G. Ferroptosis in p53-dependent oncosuppression and organismal homeostasis. Cell Death Differ. 2015;22:1237–8.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Xu X, Huang E, Tai Y, Zhao X, Chen X, Chen C, et al. Nupr1 modulates methamphetamine-induced dopaminergic neuronal apoptosis and autophagy through CHOP-Trib3-mediated endoplasmic reticulum stress signaling pathway. Front Mol Neurosci. 2017;10:203.
Okamoto A, Iwamoto Y, Maru Y. Oxidative stress-responsive transcription factor ATF3 potentially mediates diabetic angiopathy. Mol Cell Biol. 2006;26:1087–97.
Huang C, Santofimia-Castaño P, Iovanna J. NUPR1: a critical regulator of the antioxidant system. Cancers. 2021;13:3670.
Deng HF, Yue LX, Wang NN, Zhou YQ, Zhou W, Liu X, et al. Mitochondrial iron overload-mediated inhibition of Nrf2-HO-1/GPX4 assisted ALI-induced nephrotoxicity. Front Pharmacol. 2020;11:624529.
Sullivan EM, Pennington ER, Green WD, Beck MA, Brown DA, Shaikh SR. Mechanisms by which dietary fatty acids regulate mitochondrial structure-function in health and disease. Adv Nutr. 2018;9:247–62.
Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med. 2018;117:45–57.
Emma MR, Iovanna JL, Bachvarov D, Puleio R, Loria GR, Augello G, et al. NUPR1, a new target in liver cancer: implication in controlling cell growth, migration, invasion and sorafenib resistance. Cell Death Dis. 2016;7:e2269.
Lin X, Ping J, Wen Y, Wu Y. The mechanism of ferroptosis and applications in tumor treatment. Front Pharmacol. 2020;11:1061.
Liu J, Song X, Kuang F, Zhang Q, Xie Y, Kang R, et al. NUPR1 is a critical repressor of ferroptosis. Nat Commun. 2021;12:647.
Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The Hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28:2501–e2504.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–24.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–34.
Mullard A. Cancer metabolism pipeline breaks new ground. Nat Rev Drug Discov. 2016;15:735–7.
Huang W, Choi W, Chen Y, Zhang Q, Deng H, He W, et al. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013;23:724–7.
Lee J, You JH, Kim MS, Roh JL. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 2020;37:101697.
Galle E, Thienpont B, Cappuyns S, Venken T, Busschaert P, Van Haele M, et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin Epigenet. 2020;12:27.
Drápela S, Bouchal J, Jolly MK, Culig Z, Souček K. ZEB1: a critical regulator of cell plasticity, DNA damage response, and therapy resistance. Front Mol Biosci. 2020;7:36.
Song N, Jing W, Li C, Bai M, Cheng Y, Li H, et al. ZEB1 inhibition sensitizes cells to the ATR inhibitor VE-821 by abrogating epithelial-mesenchymal transition and enhancing DNA damage. Cell Cycle. 2018;17:595–604.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Chen P, Li X, Zhang R, Liu S, Xiang Y, Zhang M, et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics. 2020;10:5107–19.
Wu P, Mo Y, Peng M, Tang T, Zhong Y, Deng X, et al. Emerging role of tumor-related functional peptides encoded by lncRNA and circRNA. Mol Cancer. 2020;19:22.
Fan CM, Wang JP, Tang YY, Zhao J, He SY, Xiong F, et al. circMAN1A2 could serve as a novel serum biomarker for malignant tumors. Cancer Sci. 2019;110:2180–8.
Klingenberg M, Matsuda A, Diederichs S, Patel T. Non-coding RNA in hepatocellular carcinoma: Mechanisms, biomarkers and therapeutic targets. J Hepatol. 2017;67:603–18.
Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiological Rev. 2016;96:1297–325.
Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019;179:1033–55.
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31.
Chen J, Wang R, Zhang K, Chen LB. Long non-coding RNAs in non-small cell lung cancer as biomarkers and therapeutic targets. J Cell Mol Med. 2014;18:2425–36.
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34.
Zhang Y, Zheng S, Geng Y, Xue J, Wang Z, Xie X, et al. MicroRNA profiling of atrial fibrillation in canines: miR-206 modulates intrinsic cardiac autonomic nerve remodeling by regulating SOD1. PLoS ONE. 2015;10:e0122674.
Wang P, Zhu CF, Ma MZ, Chen G, Song M, Zeng ZL, et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget. 2015;6:21148–58.
Varga ZV, Kupai K, Szűcs G, Gáspár R, Pálóczi J, Faragó N, et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol. 2013;62:111–21.
Kyrychenko S, Kyrychenko V, Badr MA, Ikeda Y, Sadoshima J, Shirokova N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovascular Res. 2015;108:324–34.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19:43.
Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, et al. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol Carcinogenesis. 2018;57:1566–76.
Wu X, Zhi F, Lun W, Deng Q, Zhang W. Baicalin inhibits PDGF-BB-induced hepatic stellate cell proliferation, apoptosis, invasion, migration and activation via the miR-3595/ACSL4 axis. Int J Mol Med. 2018;41:1992–2002.
Cui M, Xiao Z, Sun B, Wang Y, Zheng M, Ye L, et al. Involvement of cholesterol in hepatitis B virus X protein-induced abnormal lipid metabolism of hepatoma cells via up-regulating miR-205-targeted ACSL4. Biochemical Biophysical Res Commun. 2014;445:651–5.
Peng Y, Xiang H, Chen C, Zheng R, Chai J, Peng J, et al. MiR-224 impairs adipocyte early differentiation and regulates fatty acid metabolism. Int J Biochem Cell Biol. 2013;45:1585–93.
Yang Y, Tai W, Lu N, Li T, Liu Y, Wu W, et al. lncRNA ZFAS1 promotes lung fibroblast-to-myofibroblast transition and ferroptosis via functioning as a ceRNA through miR-150-5p/SLC38A1 axis. Aging. 2020;12:9085–102.
Wu Y, Sun X, Song B, Qiu X, Zhao J. MiR-375/SLC7A11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med. 2017;6:1686–97.
Drayton RM, Dudziec E, Peter S, Bertz S, Hartmann A, Bryant HE, et al. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res. 2014;20:1990–2000.
Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX, et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 2011;585:1363–7.
Ding C, Ding X, Zheng J, Wang B, Li Y, Xiang H, et al. miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death Dis. 2020;11:929.
Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25:1457–72.
Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic Biol Med. 2015;89:548–56.
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286:40725–33.
Kim JH, Lee KS, Lee DK, Kim J, Kwak SN, Ha KS, et al. Hypoxia-responsive microRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxid Redox Signal. 2014;21:2469–82.
Xu D, Zhu H, Wang C, Zhu X, Liu G, Chen C, et al. microRNA-455 targets cullin 3 to activate Nrf2 signaling and protect human osteoblasts from hydrogen peroxide. Oncotarget. 2017;8:59225–34.
Babu KR, Muckenthaler MU. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J Mol Med. 2016;94:347–59.
Sangokoya C, Doss JF, Chi JT. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013;9:e1003408.
Shpyleva SI, Tryndyak VP, Kovalchuk O, Starlard-Davenport A, Chekhun VF, Beland FA, et al. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res Treat. 2011;126:63–71.
Andolfo I, De Falco L, Asci R, Russo R, Colucci S, Gorrese M, et al. Regulation of divalent metal transporter 1 (DMT1) non-IRE isoform by the microRNA Let-7d in erythroid cells. Haematologica. 2010;95:1244–52.
Li X, Si W, Li Z, Tian Y, Liu X, Ye S, et al. miR‑335 promotes ferroptosis by targeting ferritin heavy chain 1 in in vivo and in vitro models of Parkinson's disease. Int J Mol Med. 2021;47;1–12.
Filippova N, Nabors LB. ELAVL1 role in cell fusion and tunneling membrane nanotube formations with implication to treat glioma heterogeneity. Cancers. 2020;12:3069.
Schultz CW, Preet R, Dhir T, Dixon DA, Brody JR. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip Rev RNA. 2020;11:e1581.
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019;26:2329–43.
Wang Z, Chen X, Liu N, Shi Y, Liu Y, Ouyang L, et al. A nuclear long non-coding RNA LINC00618 accelerates ferroptosis in a manner dependent upon apoptosis. Mol Ther : J Am Soc Gene Ther. 2021;29:263–74.
Qi W, Li Z, Xia L, Dai J, Zhang Q, Wu C, et al. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci Rep. 2019;9:16185.
Yuan J, Liu Z, Song R. Antisense lncRNA As-SLC7A11 suppresses epithelial ovarian cancer progression mainly by targeting SLC7A11. Die Pharmazie. 2017;72:402–7.
Xian ZY, Hu B, Wang T, Cai JL, Zeng JY, Zou Q, et al. CircABCB10 silencing inhibits the cell ferroptosis and apoptosis by regulating the miR-326/CCL5 axis in rectal cancer. Neoplasma. 2020;67:1063–73.
Xu Q, Zhou L, Yang G, Meng F, Wan Y, Wang L, et al. CircIL4R facilitates the tumorigenesis and inhibits ferroptosis in hepatocellular carcinoma by regulating the miR-541-3p/GPX4 axis. Cell Biol Int. 2020;44:2344–56.
Zhang HY, Zhang BW, Zhang ZB, Deng QJ. Circular RNA TTBK2 regulates cell proliferation, invasion and ferroptosis via miR-761/ITGB8 axis in glioma. Eur Rev Med Pharmacol Sci. 2020;24:2585–2600.
Liu Z, Wang Q, Wang X, Xu Z, Wei X, Li J. Circular RNA cIARS regulates ferroptosis in HCC cells through interacting with RNA binding protein ALKBH5. Cell Death Discov. 2020;6:72.
Zhang X, Wang L, Li H, Zhang L, Zheng X, Cheng W. Crosstalk between noncoding RNAs and ferroptosis: new dawn for overcoming cancer progression. Cell Death Dis. 2020;11:580.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21:47.
Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–14.
Sun Y, Qiao Y, Liu Y, Zhou J, Wang X, Zheng H, et al. ent-Kaurane diterpenoids induce apoptosis and ferroptosis through targeting redox resetting to overcome cisplatin resistance. Redox Biol. 2021;43:101977.
Du J, Wang X, Li Y, Ren X, Zhou Y, Hu W, et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 2021;12:705.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8.
Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS, et al. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin Cancer Res. 2013;19:845–54.
Yang J, Zhou Y, Xie S, Wang J, Li Z, Chen L, et al. Metformin induces ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J Exp Clin Cancer Res: CR. 2021;40:206.
Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021;42:101928.
Li C, Liu J, Hou W, Kang R, Tang D. STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front Cell Dev Biol. 2021;9:698679.
Liu N, Zhang J, Yin M, Liu H, Zhang X, Li J, et al. Inhibition of xCT suppresses the efficacy of anti-PD-1/L1 melanoma treatment through exosomal PD-L1-induced macrophage M2 polarization. Mol Ther: J Am Soc Gene Ther. 2021;29:2321–34.
Wang K, Zhang Z, Tsai HI, Liu Y, Gao J, Wang M, et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021;28:1222–36.
Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA, et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol. 2021;17:665–74.
Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med. 2021;13:e14351.
Zhang X, Yu K, Ma L, Qian Z, Tian X, Miao Y, et al. Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics. 2021;11:5650–74.
Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y, et al. Targeting SLC3A2 subunit of system X(C)(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021;168:25–43.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr Biol : CB. 2018;28:2388–e2385.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med. 2018;129:454–62.
Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497–506.
Bi J, Yang S, Li L, Dai Q, Borcherding N, Wagner BA, et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis. 2019;10:682.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Jia M, Qin D, Zhao C, Chai L, Yu Z, Wang W, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol. 2020;21:727–35.
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51:575–e574.
Broquist HP. Buthionine sulfoximine, an experimental tool to induce glutathione deficiency: elucidation of glutathione and ascorbate in their role as antioxidants. Nutr Rev. 1992;50(Pt 1):110–1.
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–89.
Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell. 2020;181:1188–1188.e1181.
Zhang J, Yang J, Lin C, Liu W, Huo Y, Yang M, et al. Endoplasmic reticulum stress-dependent expression of ERO1L promotes aerobic glycolysis in pancreatic cancer. Theranostics. 2020;10:8400–14.
Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Investig. 2018;128:3341–55.
Logie E, Novo CP, Driesen A, Van Vlierberghe P, Vanden Berghe W. Phosphocatalytic kinome activity profiling of apoptotic and ferroptotic agents in multiple myeloma cells. Int J Mol Sci. 2021;22:12731.
Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27:242–54.
Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381:165–75.
Kong Z, Liu R, Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomedicine Pharmacother = Biomedecine pharmacotherapie. 2019;109:2043–53.
Li ZJ, Dai HQ, Huang XW, Feng J, Deng JH, Wang ZX, et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta pharmacologica Sin. 2021;42:301–10.
Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10:67.
Chen JJ, Galluzzi L. Fighting resilient cancers with iron. Trends Cell Biol. 2018;28:77–78.
Yao X, Xie R, Cao Y, Tang J, Men Y, Peng H, et al. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J Nanobiotechnol. 2021;19:311.
Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–9.
Li H, Li L, Xue C, Huang R, Hu A, An X, et al. A novel ferroptosis-related gene signature predicts overall survival of breast cancer patients. Biology. 2021;10:151.
Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307.
Ma S, Dielschneider RF, Henson ES, Xiao W, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE. 2017;12:e0182921.
Ni J, Chen K, Zhang J, Zhang X. Inhibition of GPX4 or mTOR overcomes resistance to Lapatinib via promoting ferroptosis in NSCLC cells. Biochemical Biophysical Res Commun. 2021;567:154–60.
Basit F, van Oppen LM, Schöckel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716.
Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–15.
Zheng Q, Li P, Zhou X, Qiang Y, Fan J, Lin Y, et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics. 2021;11:8674–91.
Sun Y, Lu D, Yin Y, Song J, Liu Y, Hao W, et al. PTENα functions as an immune suppressor and promotes immune resistance in PTEN-mutant cancer. Nat Commun. 2021;12:5147.
Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther. 2020;5:51.
Han X, Duan X, Liu Z, Long Y, Liu C, Zhou J, et al. ZEB1 directly inhibits GPX4 transcription contributing to ROS accumulation in breast cancer cells. Breast Cancer Res Treat. 2021;188:329–42.
Wei D, Ke YQ, Duan P, Zhou L, Wang CY, Cao P. MicroRNA-302a-3p induces ferroptosis of non-small cell lung cancer cells via targeting ferroportin. Free Radic Res. 2021;55:821–30.
Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. 2018;46:441–e448.
Lin H, Chen Y, Shi J. Nanoparticle-triggered in situ catalytic chemical reactions for tumour-specific therapy. Chem Soc Rev. 2018;47:1938–58.
Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X. Emerging strategies of cancer therapy based on ferroptosis. Adv Mater. 2018;30:e1704007.
Wang M, Chen Y, Cai W, Feng H, Du T, Liu W, et al. In situ self-assembling Au-DNA complexes for targeted cancer bioimaging and inhibition. Proc Natl Acad Sci USA. 2020;117:308–16.
Wang M, Zhao J, Jiang H, Wang X. Tumor-targeted nano-delivery system of therapeutic RNA. Mater Horizons. 2022;9:1111–40.
Shan X, Li S, Sun B, Chen Q, Sun J, He Z, et al. Ferroptosis-driven nanotherapeutics for cancer treatment. J Controlled Release. 2020;319:322–32.
Qian X, Zhang J, Gu Z, Chen Y. Nanocatalysts-augmented Fenton chemical reaction for nanocatalytic tumor therapy. Biomaterials. 2019;211:1–13.
Wang L, Huo M, Chen Y, Shi J. Iron-engineered mesoporous silica nanocatalyst with biodegradable and catalytic framework for tumor-specific therapy. Biomaterials. 2018;163:1–13.
Huo M, Wang L, Chen Y, Shi J. Tumor-selective catalytic nanomedicine by nanocatalyst delivery. Nat Commun. 2017;8:357.
Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano. 2018;12:12181–92.
Wang S, Li F, Qiao R, Hu X, Liao H, Chen L, et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano. 2018;12:12380–92.
Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano. 2018;12:11355–65.
He S, Jiang Y, Li J, Pu K. Semiconducting polycomplex nanoparticles for photothermal ferrotherapy of cancer. Angew Chem. 2020;59:10633–8.
Chen Q, Ma X, Xie L, Chen W, Xu Z, Song E, et al. Iron-based nanoparticles for MR imaging-guided ferroptosis in combination with photodynamic therapy to enhance cancer treatment. Nanoscale. 2021;13:4855–70.
Tang H, Chen D, Li C, Zheng C, Wu X, Zhang Y, et al. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J Pharmaceutics. 2019;572:118782.
Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–85.
Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830–49.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–4975.
Ou W, Mulik RS, Anwar A, McDonald JG, He X, Corbin IR. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic Biol Med. 2017;112:597–607.
Zhou Z, Song J, Tian R, Yang Z, Yu G, Lin L, et al. Activatable singlet oxygen generation from lipid hydroperoxide nanoparticles for cancer therapy. Angew Chem. 2017;56:6492–6.
Luo L, Wang H, Tian W, Li X, Zhu Z, Huang R, et al. Targeting ferroptosis-based cancer therapy using nanomaterials: strategies and applications. Theranostics. 2021;11:9937–52.
Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21:648–57.
Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–66.
Wang D, Tang L, Wu Y, Fan C, Zhang S, Xiang B, et al. Abnormal X chromosome inactivation and tumor development. Cell Mol Life Sci: CMLS. 2020;77:2949–58.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13:110.
Wang M, Zhao J, Xiong H, Lu H. Reviews XWJCC. Advance of nano anticancer therapies targeted on tumor-associated macrophages. Coordination Chem Rev. 2021;446:214126.
Jiang Q, Wang K, Zhang X, Ouyang B, Liu H, Pang Z, et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small. 2020;16:e2001704.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15:469–84.
Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–43.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69:423–34.
Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res: CR. 2018;37:87.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31:e1904197.
Acknowledgements
This study was supported by grants from Natural Science Foundation of Hunan Province of China (2021JJ31127) and we thank Editage for their assistance with language editing.
Author information
Authors and Affiliations
Contributions
DW wrote the article. LT, YJZ, and GLG collected the related papers. YZM, PW, LYL, and SCZ participated in the design of the review. QJY, SSZ, XJJ, and XYD provided the idea. BX, XYL, LS, and FYW initiated the study. MZ, QJL, and CG revised the study. ZYZ, ZJG, and WX finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Boris Zhivotovsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, D., Tang, L., Zhang, Y. et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis 13, 544 (2022). https://doi.org/10.1038/s41419-022-04927-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-04927-1
This article is cited by
-
Unlocking ferroptosis in prostate cancer — the road to novel therapies and imaging markers
Nature Reviews Urology (2024)
-
Metformin and long non-coding RNAs in breast cancer
Journal of Translational Medicine (2023)
-
Ferroptosis in tumors and its relationship to other programmed cell death: role of non-coding RNAs
Journal of Translational Medicine (2023)
-
The mechanism of ferroptosis and its related diseases
Molecular Biomedicine (2023)
-
Ferroptosis as a potential target for cancer therapy
Cell Death & Disease (2023)
