
Abstract
Pancreatic cancer (PC) is one of the leading causes of cancer-related death worldwide due to delayed diagnosis and limited treatments. More than 90% of all pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC). Extensive communication between tumour cells and other cell types in the tumour microenvironment have been identified which regulate cancer hallmarks during pancreatic tumorigenesis via secretory factors and extracellular vesicles (EVs). The EV-capsuled factors not only facilitate tumour growth locally, but also enter circulation and reach distant organs to construct a pre-metastatic niche. In this review, we delineate the key factors in pancreatic ductal adenocarcinoma derived EVs that mediate different tumour processes. Also, we highlight the factors that are related to the crosstalk with cancer stem cells/cancer-initiating cells (CSC/CIC), the subpopulation of cancer cells that can efficiently metastasize and resist currently used chemotherapies. Lastly, we discuss the potential of EV-capsuled factors in early diagnosis and antitumour therapeutic strategies.
Similar content being viewed by others


Facts
-
PDAC-derived exosomes show heterogeneity in their cargos.
-
PDAC-derived exosomes distinctly regulate angiogenesis, cancer-associated thrombosis, immune evasion, stromal reprogramming, chemoresistance, metastasis and tumour growth during PDAC tumorigenesis.
-
CSC/CIC-derived exosomes transmit signals for efficient metastasis and for creating a pre-metastatic niche in the distant organ.
-
Around 10–20% of identified exosomal proteins are transcription factors and nucleotide-binding factors.
Open questions
-
How do EVs mediate the pivotal crosstalk in the microenvironment to regulate the main hallmarks of tumorigenesis?
-
How do EVs modulate distant organs that will home metastases?
-
Can EVs mediate intercellular epigenetic regulation between cells and different cell types through transmitting transcription factors and nucleotide-binding factors to recipient cells?
-
Which EV cargo proteins or nucleic acids can be used as reliable markers for early detection and prognosis of pancreatic cancers from blood or other sources?
-
Will a combination of current biomarkers and the exosomal proteins provide better sensitivity and specificity of early cancer detection?
-
Can exosome-based therapies improve therapeutic outcomes in pancreatic cancer?
Pancreatic ductal adenocarcinoma (PDAC) and extracellular vesicles (EVs)
Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer, remains one of the deadliest cancers in the world. This is due to its poor diagnosis at early stages, high risk of metastasis, and limited response to treatments, including surgery and chemotherapy [1]. Surgical resection is the potential curative therapy and chemotherapy drugs serve as the first-line treatment for patients. Nevertheless, surgical resection is usually followed by cancer recurrence, and chemotherapy resistance commonly appears in patients with rapid tumour progression [1, 2]. It is reported that the 5-year survival rate for PDAC patients made up <10% and the median survival time is 5–6 months [1].
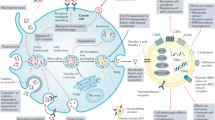
Tumour progression is driven by the communication between tumour cells and the surrounding cells in the microenvironment, as well as the distant organs. It can be mediated by direct cell–cell contacts, secretory proteins, and extracellular vesicle (EVs). EV is defined as a lipid bilayer particle that is naturally secreted by cells into extracellular microenvironment without replication capability. EVs display in a wide range of size, biogenesis, biochemical composition and biological functions, including cell–cell communication, immune response modulation, and disease progression [3]. Based on the particle sizes and origins, EVs are generally divided into three groups: exosomes (diameter from 30 to 150 nm), microvesicles (microparticles or ectosome, diameter from 50 to 1000 nm), and apoptotic bodies (diameter from 500- over 1000 nm) [4]. Though EV release is a normal process, an increasing rate in their release and change in cargos such as DNA, RNA, miRNA and proteins, can favour cancer development. Over the course of tumorigenesis, exosomes are generated from the tumour cells as well as from the surrounding stromal cells, such as cancer-associated fibroblasts (CAFs), tumour-associated macrophages (TAMs) and bone marrow mesenchymal stem cell (BMSCs). These EVs deliver cargos to different recipients which facilitates tumour progression and tumour niche construction, through regulating angiogenesis, cellular metabolism, metastasis, cell survival, immune regulation and therapeutic resistance (Fig. 1).

A PDAC-derived EVs contain various cargos that selectively modulate the surrounding cells types. B Different cell types exist in PDAC microenvironment and produce EVs with various contents that regulate PDAC progression. PDAC pancreatic ductal adenocarcinoma, PSC pancreatic stellate cell, TAM tumour-associated macrophage, CAF cancer-associated fibroblast, BM-MSC bone marrow-derived mesenchymal stem cell, NK natural killer cell, DC dendritic cell.
We summarise the current understanding of the role of EVs in pancreatic tumour progression and discuss the potential strategy of using EVs in cancer treatment (Table 1).
PDAC-derived EVs
PDAC-derived EVs regulate angiogenesis
Communication between tumour cells and endothelial cells leads to new blood vessel formation that supports tumour growth. PDAC-derived EVs induce HUVEC cell proliferation and mobility, as well as influence their proangiogenic factor secretion [5,6,7]. Tetraspanins are a family of proteins with four transmembrane domains that are abundant in membranes of organelles and in exosomes. Two tetraspanins, CD151 and tetraspanin 8 (also known as CO-029 and TM4SF3 in human, and D6.1A in rats), are upregulated in PDAC and support tumour progression. Overexpression of D6.1A in PDAC promotes angiogenesis in vivo through transmitting PDAC-derived exosomal D6.1A to endothelial cells and consequently induces the expression of angiogenic factors such as VEGFR and CD31 [8]. CD151 selectively affects angiogenesis [9, 10]. Assembly defects during skin wound healing and tubular formation are observed in cd151 knockout and tspan8 knockout mice, and even more severe effects in the double knockout mice [9]. Interestingly these impaired phenomena in knockout mice can be mitigated by Tspan8/CD151-competent serum exosomes, through stimulating GPCR and RTK-dependent angiogenesis which ultimately contribute to tumour progression in vivo [9]. This suggests a potential application of joint Tspan8 and CD151 blockage in cancer therapeutics.
PDAC cell-derived exosomal miR-27a promotes angiogenesis through upregulating the expression of angiogenesis factors VEGF and VEGFR, and migration-related factors MMP2 and MMP9 in tumour cells [11]. Another PDAC-derived exosomal protein, ANXA1, positively regulates endothelial tubular formation [12] and fibroblast and endothelial cell mobility [13]. Similarly, PK-45H cell-derived exosomes promote in vitro HUVEC tube formation via phosphorylation of Akt and Erk signalling pathway and dynamin-dependent endocytosis [14]. Tumour-derived exosomes also increase permeability of endothelial monolayer cells via transferring circ-IARS into recipient cells and activating their miR122/RhoA signalling, thereby leading to upregulation of F-actin and downregulation of ZO-1 and facilitating tumour metastasis [15]. Depletion of myoferlin influences exosome production in PDAC and alleviates exosome entry into HUVEC, decreasing proliferation and migration of endothelial cells [16].
A recent study showed that DUSP2/VEGF-C axis in PDAC mediated lymphangiogenesis and tumour invasion [17]. Inhibition of DUSP2 increased VEGF-C expression in PDAC-derived exosomes [17]. These VEGF-C-containing exosomes promoted proliferation of lymph endothelial cells and lymphovascular invasion in the orthotopic PDAC mouse model [17].
PDAC-derived EVs deliver tissue factor (TF) promoting cancer-associated thrombosis
Cancer-associated thrombosis is the most common complication in patients that causes high mortality. It shows a higher association with certain cancer types, such as PDAC, lung cancer, brain cancer, kidney cancer, lymphoma and ovarian cancer, while the reasons remain unclear. Tissue factor (TF) is an initiator of the coagulation cascade and is essential for haemostasis. Under normal condition, TF is not expressed by the cells that directly contact with blood, except a low level in CD14+ monocytes, to prevent inappropriate activation of coagulation in the absence of vascular injury. In cancers, TF is expressed by tumour cells as an extracellular secretory factor or EV cargo. PDAC-derived EVs carrying TF can trigger venous thrombosis in mice [18] and foster prothrombotic activity in assistance of phospholipids [19, 20]. This prothrombotic activity can occur in platelet-dependent manner [21,22,23], and cooperate with neutrophils that release neutrophil extracellular trap (NET) [24]. NETs serve as a scaffold for platelet adhesion and subsequently initiate coagulation. On the other hand, Stark and colleagues showed that initiation of thrombosis was a platelet- and leucocyte-independent mechanism which required phosphatidylethanolamine-dependent TF+EVs from host PDAC cells [19].
PDAC-derived exosomes regulate pancreatic functions
High prevalence of PDAC-associated diabetes is a hallmark of PDAC progression. Many patients report dramatic weight loss and diabetes indicating that tumour cells disrupt normal pancreatic metabolism. The PDAC-derived exosomal adrenomedullin (AM) induces p38 and ERK1/2 MAPK-mediated lipidosis in adipocytes [25], and inhibits insulin secretion by β-cells [26]. Also, ER stress markers, ROS and NO are upregulated in β-cells upon PDAC-derived exosomal AM treatment [26]. Mechanically, AM interacts with its receptor, ADMRs, to initiate the dysfunction of the recipient cells, and blockage of ARMRs abrogates these effects. An independent study showed that lipidosis and glucose intake inhibition caused by PDAC-derived exosomes is PI3K/Akt/FoxO1 signalling dependent, which eventually develops insulin resistance in skeleton muscle cells [27]. PDAC-derived exosomes suppress the synthesis of glucose-dependent insulinotropic peptide (GIP) and glucagon-like peptide-1 (GLP-1) by downregulating the proprotein convertase subtilisin/kexin type 1/3 (PCSK1/3) [28]. Insulin resistance in skeleton muscle cells together with impaired GIP and GLP-1 are involved in pathogenesis of type 2 diabetes mellitus. Further investigations in the relationship between PDAC and systemic metabolism will help to discover potential targets for treating metabolic disorders and improving the diagnosis/treatment in PDAC.
PDAC-derived exosomes in immune regulation
An imbalance of immune cells in PDAC microenvironment leads to an immunosuppressive and protumorigenic phenotype. These include immunosuppressive Treg lymphocytes, M2 polarized TAM and myeloid-derived suppressor cells (MDSCs) that prevail over immune effector CD8+ T cells, dendritic cells (DCs) and M1 polarized TAM in the tumour microenvironment, blood and lymphoid organs. PDAC-associated immune cell alterations concur with immune escaping, hence favouring tumour progression. Lymphocyte lineage alterations with Treg cell accumulation appear to occur early, while myeloid lineage with MDSCs accumulation occur later during PDAC development. Such a Treg cell expansion is mediated by PDAC-derived EVs through upregulating the expression of FOXO transcription factors and nuclear translocation in FOXP3+ Treg cells [29].
PDAC-derived exosomes cause an imbalance of immune cells via expanding monocytic myeloid-derived suppressor cells (mMDSCs; CD11b+CD14+HLA-DR−), while reducing DCs (CD11b+CD14−HLA-DR+) and granulocytic MDSCs (gMDSCs; CD11b+CD14−HLA-DR−) [30]. These exosomes increase intracellular calcium trafficking in a SMAD4-dependent manner in mMDSCs and induce aerobic glycolysis in a SMAD4-independent manner in PBMCs [30]. An increase in CD14+HLA-DRlo/neg monocyte population was confirmed in PDAC patients by tumour-derived exosomes that involves an alteration in STAT3 signalling, induction of arginase expression and ROS generation [31]. Also, tumour-derived exosomes can regulate monocytes via CD44H, a transmembrane protein on monocytes, that initiates phagocytosis and chemokine production, namely CCL5 and CCL4, via STAT3-mediated signalling [32].
Using rat PDAC-derived exosomes, Zöller et al. showed that these exosomes enter all leukocyte subpopulations and strengthen cytotoxic T cell and NK cell activity [33]. In line with these findings, PDAC exosomes enriched in heat shock protein 70 (Hsp70) stimulate NK cell migration and trigger their cytotoxicity against tumour cells via releasing granzyme B [34]. By contrast, PDAC-derived EVs have also been reported to attenuate NK cell cytotoxicity by downregulating the expression of NKG2D, CD107a, TNFα and INFϒ, as well as impairing their glucose uptake [35]. PDAC patient serum-derived EVs are TGFβ1 enriched and able to induce SMAD2/3-dependent signalling in NK cells, suggesting a potential distant immunosuppression by PDAC-derived exosomes [35]. Similarly, saliva exosomes from pancreatic tumour-bearing mice suppress natural killer (NK) cell cytotoxic capability against tumour cells [36]. PDAC-derived exosomes activate p38 MAPK in T lymphocytes leading to ER stress-mediated apoptosis and ultimately immunosuppression [37].
PDAC-derived exosomal miR-203 is transferable to DCs and decreases their toll-like receptor 4 (TLR4) expression, TNFα and IL-12 secretion [38]. A further report showed a reduction of regulatory factor X-associated protein (RFXAP) in DCs by PDAC-secreted exosomal miR-212-3p, resulting in decreased MHC II expression and induced immune tolerance [39].
PDAC-derived exosomes deliver arachidonic acid (AA) to non-polarised THP-1 macrophages (M0) to induce a M2-like phenotype (CD14hiCD163hiCD206hi) and increase a panel of pro-tumoral factors, including VEGF, MCP-1, IL-6, IL-1β, MMP-9 and TNFα [40]. KRAS is another extracellular molecule transferred by PDAC-derived exosomes to macrophages to cause M2-like phenotype switch via STAT3 dependent fatty acid oxidation [41].
B-cell-associated immune response is reported in many cancers including PDAC. An accumulation of B-cell subpopulation has been observed during early pancreatic neoplasia [42] and in HIF-1α depleted PDAC model [43], with a positive correlation to pancreatic tumorigenesis. Bruton tyrosine kinase, a key B-cell and macrophage kinase [44], and PD-1/PD-L1 pathway [45], suppress T-cell-dependent antitumour immune responses in PDAC, indicating potential mechanisms of B cells in favour of tumour progression. Although these murine studies have indicated a pro-tumour role for B cells, the clinical data are controversial. For instance, higher B-cell infiltration within PDAC tumours related to shorter survival time is reported in some studies [45, 46], whereas others state differently [47,48,49]. The detailed roles of B cells in PDAC need further investigations. B-cell-associated autoimmune response is evidenced by the production of autoantibodies against tumour-associated antigens (TAAs). Capello et al. have described a panel of TAAs, such as LGALSBP3, PKM2, HSPA8, KRT17, KRT16, ACTB, H2AF, KRT5 and JUP, which was enriched on the surface of PDAC plasma-derived exosomes vs healthy donors [50]. Moreover, the exosomal antigens induce and bind to autoantibodies in PDAC patients, indicating PDAC-derived exosomes inhibit complement-dependent cytotoxicity towards tumour cells [50]. This suggests a decoy-like function of PDAC-derived exosomes by binding to circulating autoantibodies in patient serum and attenuating B-cell-associated antitumour responses, hence favouring an immune escape.
PDAC-derived exosomes deliver chemoresistance
Chemoresistance happens during long-term exposure to chemotherapeutic drugs. As it usually occurs rapidly and comprehensively, an efficient mechanism such as exosomes has been proposed to spread the signals in tumour cells. In this case, exosomal cargos from chemoresistant cells can regulate gene expression in the chemosensitive recipient cells to support their survival or anti-apoptotic capacity against chemotherapy. Gemcitabine is a primary cancer drug that can change tumour cell contents including their derived exosomes. miR-155 is a well-studied onco-miRNA that has been found to be overexpressed in most PDAC cases and to correlate with a poor survival of PDAC patients. Upregulation of miR-155 in PDAC cells and exosomes by gemcitabine can be delivered to other PDAC cells to protect against gemcitabine-induced cell death in vitro and in NOD/SCID mice [51, 52]. Deoxycytidine kinase (DCK) is one of miR-155 targets. Upon chemotherapy, DCK converts gemcitabine into an active form which incorporates into DNA and inhibits DNA synthesis, consequently leading to tumour cell apoptosis. Downregulation of DCK by miR-155 halts this process and supports PDAC cell survival [51, 52]. TP53INP1, a pro-apoptotic stress-induced p53 target gene, was also decreased in PDAC cells upon the uptake of exosomal miR-155 [51, 52]. Hence, upregulation of miR-155 represents a protection mechanism by tumours against chemotherapy and an indicator of poor survival. EphA2 protein is another example. Gemcitabine-resistant PDAC cells secrete EphA2 enriched exosomes that can be transmitted into gemcitabine-sensitive PDAC cells to support their survival [53]. Both miR-210 and exosomal miR-210 expression is upregulated in PDAC by gemcitabine and the latter is also transferrable from a chemoresistant CD133+CD44+ PDAC cell population to a chemosensitive PDAC cell population [54]. Exosomal miR-210 promotes tumour growth in nude mice via activating mTOR signalling [54].
PDAC-derived exosomes promote invasion and metastasis
The exosomes collected from advanced PDAC patient serum enhance PDAC cell migration and proliferation in vitro, increase CD44 and PPP2R1A expression and decrease TP53 expression in recipient PDAC cells [55]. Asparaginyl endopeptidase (AEP), highly abundant in PDAC patient sera, shows positive effects on PDAC cell invasion [56]. Such an invasive ability is transferrable between tumour cells. In vitro studies using paired PDAC cell lines show that metastatic ability is able to be transferred to their less motile counterparts through exosomes cargos, such as miR-125b-5p [57], ZIP4 [58], circular-RNA-PDE8A [59], miR-222 [60] or lncRNA-Sox2ot [61,62,63].
Long-distant metastasis niche
Liver metastases are reported in up to 80% of PDAC patients and lung metastases occur in 45% of PDAC patients [64]. Pre-treating the mouse with PDAC-derived exosomes has been shown to enhance tumour growth and tumour metastasis [63]. Costa-Silva and colleagues showed that pre-injection of PDAC-derived exosomes in mice ‘educated’ Kupffer cells to enhance the metastatic burden in liver [65]. These PDAC-derived exosomes fused with Kupffer cells and changed their ECM components by increasing fibronectin and decreasing vitronectin and tenascin C [65]. It also induced liver fibrosis signalling molecules, such as CTGF and TGFβ, and recruited bone marrow-derived F4/80 macrophages to the liver pre-metastatic niche [65]. In their study, also migration inhibitory factor (MIF) was identified in exosomes for such liver pre-metastatic niche construction [65]. Tumour-derived exosomes deliver cargos into macrophages via CD36, a lipid receptor, to create a pre-metastatic niche in mouse model [66].
Emmanouilidi et al. revealed that oncogenic exosomes contain factors known to regulate the pre-metastatic niche (S100A4, F3, ITGβ5, ANXA1), invasion (PODXL, ITGA3) and metastasis (LAMP1, ST14) [67]. Protein Kinase D1 (PRKD1) [68] and p53 [69] are reported in PDAC-derived exosomes for lung metastases. Reduced expression of PRKD1 in PDAC increases their exosome secretion and promotes lung metastasis in mice [68]. Mutant p53-expressing exosomes from tumour cells can transfer invasive/migratory ability to other tumour cells as well as regulate lung ECM to facilitate their invasion [69]. Hypoxia regulates tumour mobility through exosomal cargos. Wang et al. showed that hypoxic exosomes derived from PDAC activate macrophages to the M2 polarity, which in turn promoted tumour metastasis to lung [70]. The exosomal miR-301a is upregulated by hypoxia-inducible factors (HIF) and transmitted into macrophages for their activation via stimulating PI3K signalling [70]. Earlier, Zöller’s group revealed both CD44v6 and soluble matrix were necessary for lung pre-metastatic niche formation [71]. Similarly, exosomal CD44v6/complement C1q binding protein (C1QBP) also activates hepatic satellite cell for liver metastasis and liver fibrosis in mice [72].
PDAC-derived exosomes in tumour growth and progression
Pancreatic cancer cell-derived exosomes show potential to support malignant cell transformation compared to normal pancreatic cell-derived exosomes [73]. Verince’s group reported that PDAC-derived EVs induced mitochondria-dependent apoptotic pathway in tumour cells through PTEN and GSK-3β activation [74], and downregulation of Hes-1 expression, a Notch-1 signalling pathway mediator [75]. In these studies, the decrease in Hes-1 expression by presenilin blockage resulted in PTEN and GSK-3β activation [74], whereas inhibiting either PTEN or GSK-3β activation increases Hes-1 expression which counteracts their effects on inhibiting tumour cell proliferation [75]. This suggests a complexity in signalling crosstalk induced by EVs, whereas the effects may depend on where the vesicles anchor and trigger the intracellular signalling in recipient cells. Another study has identified a group of long non-coding RNAs (lncRNAs), including NONHSAT105177, capable of mediating drug inhibition on PDAC proliferation via cancer-derived exosomes [76]. Overexpressing NONHSAT105177 significantly downregulates cholesterol pathway genes, and inhibits PDAC cell proliferation, migration, and the epithelial–mesenchymal transition (EMT) both in vitro and in vivo [76]. Exosomal lncRNA 01133 (LINC01133) [77], has-circ-0000069 [78] and Survivin [79] have also been recently reported in tumour progression and may impact treatment.
EV-mediated crosstalk between tumour cells and stroma
Between tumour cells and pancreatic stellate cells (PSCs)
PSCs play a pivotal role in pancreatic fibrogenesis to develop a conditioned microenvironment for tumour growth. PDAC-derived exosomes stimulate proliferation, migration and profibrogenic activity of PSCs, potentially via transforming growth factor β1 (TGFβ) and tumour necrosis factor (TNF) that mediate ERK and AKT activation [80]. miR-1246 and miR-1290 are abundantly expressed in PDAC-derived exosomes and induce α-smooth muscle actin (ACTA2) and fibrosis-related genes in PSCs to support tumour growth [81]. PDAC-derived exosomal Lin28B protein chemoattracts PSC migration, which in turn supports tumour progression in mice [82]. Co-incubation of PSC-derived exosomes with PDAC cells induces their chemokine gene expression, such as CXCL1, CXCL2 and CCL20, and stimulates PDAC proliferation and migration [83]. Also, hypoxia regulates PSC-derived exosomal miRNAs in favour of tumour progression and invasion [84]. By contrast, a stroma specific miR-145 can be delivered in exosomes to tumour cells that induces apoptosis and suppresses tumour cell growth [85]. Furthermore, PSC-derived exosomal miR-5703 inhibits PDAC proliferation via targeting CMTM4 expression [86], which in turn suppressed PI3K/Akt pathway through downregulating PAK4 [86].
Between tumour cells and CAFs
PDAC-derived microvesicles containing miR-155 promote conversion of normal fibroblasts to CAFs by downregulating TP53INP1 expression and upregulating the expression of a-SMA, fibroblast activation protein (FAP), and fibroblast growth factor 2 [81, 87]. These profibrogenic activities eventually facilitate tumour spreading. Furthermore, CAFs are innately drug resistant and able to transfer such ability to their neighbouring tumour cells via exosomes [88, 89]. CAF-derived exosomes support PDAC cell growth upon gemcitabine treatment in vitro [88]. miR-106b carried in CAF-derived exosomes blocks apoptosis through targeting TP53INP1 in PDAC cells, though the detailed mechanism is still unclear [89]. Metabolically, CAF-derived exosomes inhibit mitochondrial oxidative phosphorylation in tumour cells, thereby increasing glycolysis and glutamine-dependent reductive carboxylation to support tumour growth [90]. In hypoxic and lipid starvation condition, like a tumour microenvironment, co-culture of CAFs with macrophages facilitates ANXA6/LRP/TSP1 complex formation in CAFs [91]. This complex is subsequently delivered in exosomes to PDAC cells enhancing their migration ability and tumour progression [91]. Manipulating ANXA6 expression in CAFs in a co-culture with PDAC cells correlates with liver metastasis in xenografts, suggesting a potential of ANXA6+ EVs as a biomarker for PDAC aggressiveness [91]. In addition, a number of oncogenic miRNAs (miR-10a-5p, miR-92a-3p, miR-181a-5p, miR-191–5p, and miR-92b-3p) have been identified in CAF-derived EVs that support tumour migration [92]. In contrast, the exosomal miRNA-520b derived from normal fibroblasts, inhibits PDAC cell proliferation, invasion, migration and stimulates apoptosis, suggesting its therapeutic potential in PDAC treatment [93].
Between tumour cells and TAMs
A higher expression level of exosomal Ezrin protein is observed in PDAC patients and pancreatic ductal cell lines [94]. Enriched exosomal Ezrin protein can regulate M2 macrophage polarity which consequently leads to tumour metastasis [94]. Interestingly, exosome containing Ezrin promotes M2 marker expression and cytokine production while direct incubation of Ezrin with macrophages showed no statistical significance. Injection of patient-derived exosomes that expressed Ezrin before and post tumour injection into mouse models facilitated tumour metastasis to liver. This suggests that specific exosomal proteins derived from tumour cells could regulate stromal cells in the tumour microenvironment to support tumour spreading.
TAMs-derived exosomes support PDAC cell survival in response to gemcitabine via interfering with gemcitabine activation [95]. Chitinase 3-like-1 (CHI3L1) and fibronectin (FN1) are two abundant proteins in macrophage-derived EVs that contribute to gemcitabine resistance [96]. In addition, the uptake of TAM-derived exosomal miR-365 by PDAC cells increases the expression of cytidine deaminase (CDA), an enzyme that deaminates gemcitabine, and excretes gemcitabine out of the cells [95]. miR-365 has been previously reported to directly target SHC1 and BAX to block apoptosis in PDAC upon gemcitabine treatment, indicating that the miR-365/SHC1/BAX axis modulates survival of PDAC cells [97]. Exosomal miR-365 from M2 macrophages not only regulates chemoresistance but it also promotes PDAC development through suppressing B-cell translocation gene 2 (BTG2) expression and activating FAK/AKT pathway in tumour cells [98]. Together, M2-derived exosomal miR-365 exerts distinct functions in tumour cells to support cancer progression.
M2 macrophages secrete exosomes that regulate gene expression which promote PDAC migration, invasion and angiogenesis [99]. miR-501-3p supports tumour formation and liver metastasis in nude mice via downregulating TGFBR3 in recipient PDAC cells while upregulating TGFBR1 and TGFBR2 [99]. Blocking miR-501-3p reduces tubular formation, tumour formation and metastasis to liver, as well as downregulates stemness-related genes, such as CD133, OCT4 and NANOG [99]. Constrained lncRNA-SBF2-AS1 in M2 macrophage-derived exosomes increases miR-122-5p expression to reduce XIAP expression in tumour cells leading to tumour suppression [100].
Other immune cell-derived exosomes suppress PDAC progression
There is also crosstalk between PDAC cells and other stromal cells. miR-3607-3p is enriched in the NK cell-derived EVs while a low level of miR-3607-3p correlates with poor diagnosis in PDAC patients [101]. Exosomal miR-3607-3p from NK cells targets IL-26, and thereby inhibit proliferation, migration and invasion of PDAC cells. miR-1231 derived from Bone marrow mesenchymal stem cells (BM-MSCs) negatively correlates with PDAC tumour progression and its downregulation shortens cell cycle progression and enhances PDAC cell migration/invasion [102]. Ding et al. applied exosomes from human umbilical cord mesenchymal stromal cells (hucMSCs) to deliver exogenous miR-145-5p to pancreatic cells and inhibited cell proliferation and invasion, while inducing cell cycle arrest and apoptosis in vitro and in vivo [103]. Exosomal miR-126-3p derived from primary BM-MSC inhibited PDAC cell proliferation, migration, invasion, and promoted their apoptosis [104]. Exosomal miR-143 [105] and miR-124 [106] from BM-MSCs have shown similar suppression of PDAC progression.
Exosomes and cancer stem cell/cancer-initiating cell (CSC/CIC)
A cancer stem cell (CSC), alternatively cancer-initiating cell (CIC) or tumorigenic cell, is defined as a cell within a tumour that possesses the capacity to self-renew and divide asymmetrically to cause the heterogeneous lineages of cancer cells that constitute the bulk of the tumour [107]. CSCs represent a side-population in the tumour which exerts stem cell-like characteristics such as entering a quiescent state, slow cell cycling, expressing stemness markers and stemness transcription factors. These together make CSCs responsible for the maintenance and proliferation of the tumour, enhance metastatic characteristics and resistance to chemo- and radiotherapy, which ultimately lead to cancer relapse. Though the specificity remains controversial, several CSC markers have been reported in PDACs, including CD133, CD24, CD44, CXCR4, EpCAM, ABCG2, c-Met, ALDH-1 and Nestin [108].
Increasing evidence suggests that exosomes derived from CSCs promote survival, EMT and motility in tumour cells, and angiogenesis in endothelial cells (Fig. 2). These ultimately promote tumour progression. Wang et al. reported Tspan8, CD44v6 and α6β4 as biomarkers for PDAC CIC [109]. As mentioned previously, Tspan8/CD151 exosomes contribute to tumour progression by stimulating angiogenesis in vivo [10]. In addition, PDAC-derived exosomal CD151 and Tspan8 induce matrix remodelling in recipient cells via exosomal tetraspanin–integrin and tetraspanin–protease associations, which ultimately promote the metastatic phenotype [110]. Similarly, CD151 and Tspan8 defects cause reduction of exosome uptake and therefore weaken their biological functions in recipients.

PDAC cell-derived EVs contain cargos such as CD44v6, C1P, Tspan8 and CD151, while CSC/CIC-derived EVs carry factors including Tspan8, CD151, miR210 and miR-195-5p that regulate biological functions in the counterparts. This includes upregulation of stemness and survival genes in the recipient cells that provide resistance to radiation and chemotherapies, stimulate protein secretion for stromal reprogramming and angiogenesis, and the switching to metastatic phenotype for metastasizing to distant sites.
The same group also reported that CD44 variant isoform v6 (CD44v6), a known CIC marker and pro-metastatic molecule, contributes to PDAC migration and invasion [111]. Genetically knocking down CD44v6 concomitantly decreased the expression of CD151 and Tspan8, whereas loss of CD44v6 and Tspan8 lead to decreased invasiveness and lower protease expression. CD44v6 mediates exosome uptake by recipients while its deficiency can be reversed by CD44v6 wild-type exosomes and even enhanced by the exosomes derived from spheroids or holoclonal cells (a colony-forming stem cell that has a higher growth potential and does not contain differentiated cells). Furthermore, the PDAC CIC-derived exosomes promote various stem cell-like features, including apoptosis-resistance, in vivo tumour growth and progression, and inducing EMT gene expressions in CD44v6kd/Tspan8kd non-CIC cells via initiating receptor tyrosine kinase (RTK) signalling [112]. Recently, Jiang and colleagues reported that exosomal miR-194-5p derived from dying tumour cells support ALDH1A1+ cell repopulation upon radiotherapy [113]. The exosomal miR-194-5p supports ALDH1A1+ cell survival via downregulating E2F3 expression and inducing G1/S arrest. It also triggers DNA damage response for DNA repair in irradiated ALDH1A1+ cells and facilitates their survival. In addition, treating with exosomal miR-194-5p recovers the expression of HMGA2 in irradiated ALDH1A1+ cells which promotes cell stemness and cancer progression.
An independent study showed that highly invasive PDAC cells secrete lncRNA-Sox2ot in exosomes which promote recipient PDAC epithelial–mesenchymal transition (EMT) and stem cell-like properties via regulating Sox2 expression [61]. PDAC cells secrete ceramide-1-phosphate (C1P)-containing exosomes that selectively stimulate CIC migration and adhesion on fibronectin [114]. On the other hand, exosomal miR-21-5p derived from M2 macrophage stimulates CSC/CIC differentiation through targeting KLF3 [115].
CSC-derived exosomes are believed to contribute to the communication in tumour microenvironment as well as long distant communication to other organs to promote tumour progression. Given the CSC biological characteristics, the CSC-derived exosomes can support tumour mass through delivering self-renewal capacity to one another and educating the recipients to regenerate multiple malignant cell types, as well as providing a niche for tumour growth. Moreover, CSCs can also transfer their material via exosome secretion to promote chemoresistance in other cancer cell subtypes [63].
Exosomes and epigenetic regulation
Many studies show that exosomes regulate gene expression in receipt cells via delivering miRNAs and lncRNAs, whereas only a few have so far discussed a possible role of transcriptional regulators as exosomal cargo. PDAC related exosomal proteomic studies indicate that a minor proportion of identified proteins functionally associate with transcriptional regulation or translation. Between 10 and 20% of total identified exosomal proteins are usually residing in the cell nucleus, ranging in function from transcription or translation activity and nucleotide/DNA/RNA binding [116,117,118]. TP53, for instance, a widely known transcription regulator that is commonly mutated in late stage of PDAC, is present in plasma-derived exosomes from PDAC patients [119]. This suggests that the exosomes may be able to influence recipients’ behaviours not only with RNA-type cargo, but with proteins that directly regulate gene expression. In line with this hypothesis, a previous review indicates a similar proportion of identified proteins in human brain tumour or medulloblastoma cell line-derived exosomes has some role in transcriptional and/or translational activities [120]. Whether exosomes transport transcription factors or epigenetic regulators that could directly impact chromatin architecture and gene expression in recipients is still an open question. Since this has been suggested in other disease contexts it will be interesting to discover if such mechanisms are relevant for pancreatic cancer.
Exosomes and early detection of PDAC
One of the main factors that causes the high mortality rate in PDAC is the lack of early detection markers. CA19-9 antigen, found about 35 years ago, is the most common indicator that is currently used for PDAC monitoring [121]. CA19-9 is regularly expressed on cells of the pancreatic-biliary system with low concentrations in healthy serum and elevated levels in cancer patients, particularly in PDAC. Although being routinely used in clinic, serum CA19-9 as a biomarker has its limitations, for example, the concentration can be interfered by other diseases, such as cholangitis, pancreatitis or obstructive jaundice, hepatic, and pancreatic cysts. Hence, other biomarkers are needed whether in combination with serum CA19-9 or separately to provide better diagnosis. A number of exosomal cargos including proteins, DNA, miRNAs, lncRNA, as well as exosomal membrane lipids, for instance, a well-known phosphatidylserine (PS) highly expressed on tumour cells and present in tumour-derived exosomes, have been proposed as potential biomarkers for PDAC [122].
Glypican-1 is upregulated in PDAC and breast cancer. Its elevated expression in circulating exosomes of pancreatic patients suggests its suitability as a biomarker for detection of early PDAC. Higher level of secretory and exosomal glypican-1 from serum is correlating with poor prognosis and tumour burden of PDAC [123, 124], suggesting its potential as a biomarker for early detection [125, 126] and prognosis [127]. A recent clinical case study reported that the high level of exosomal glypican-1 detected in a patient only showed progression one year after its detection [128]. Combining exosomal glypican-1 and serum CA19-9 may provide better diagnosis [129, 130]. Contrary, Lucien et al. reported they could not distinguish PDAC from benign pancreatic diseases by exosomal glypican 1 and 2 [131]. Other candidates have been reported, such as MIF [132], EphA2-EVs [133, 134] and EGFR [135, 136], epidermal growth factor receptor pathway substrate 8 (Eps8) [137], CKAP4, a DKK1 receptor [138], DCBLD2 [139] and ALPPL2 [140] which are overexpressed in circulating exosomes and can serve as biomarkers for PDAC. Exosomal c-Met and PD-L1 expression correlate with shorter postoperative survival time and serve as a negative prognostic indicator in combination with detection of CA19-9 [141]. A higher glycosylation level of CD133 in ascite-derived exosomes could indicate advanced PDAC [142]. PDAC-derived exosomal EpCAM shows an increasing expression during treatment and hence represents a useful prognostic factor [143]. As described above, tissue factor (TF) has been reported in PDAC-derived EVs and leads to venous thrombosis via interacting with endothelial cells in patients. The TF+ EVs are reported as biomarkers for survival of cancer patients and venous thrombosis in PDAC patients [144, 145]. Mutations on KRAS and TP53 have been well studied in PDAC and show important roles in pancreatic tumorigenesis. Both DNA and protein of mutant KRAS and TP53 are detected in circulating exosomes of patients, suggesting a potential of being biomarkers for PDAC [146,147,148].
miRNA and long non-coding RNAs (lncRNAs) are also detected in exosomes. These nucleic acids are more resistant to RNA degrading enzymes than those present in free circulation, which allows for a higher detection sensitivity and specificity. For example, expression profiling of lncRNAs that revealed high upregulation in liver cancer (HULC) was also found highly expressed and induced by TGFβ in PDAC serum exosomes compared to healthy individuals or Intraductal papillary mucinous neoplasm (IPMN) patients [149, 150]. Numerous findings suggest novel biomarkers for early PDAC detection using different miRNA profiling from circulating exosomes (Table 2) [151,152,153,154,155,156,157,158,159]. The expression of serum exosomal miR-23b-3p shows correlation with serum CA19-9 level in PDAC patients [160]. Exosomal miR-10b and miR-30c signature is stated to be superior to exosomal GPC1 or plasma CA19-9 levels for PDAC diagnosis and differentiating between PDAC and CP [161]. Other exosome sources including saliva [162], pancreatic juice [163] and urine exosomes [164] are also studied to generate exosomal miRNAs profiling for PDAC detection (Fig. 3).

EVs isolated from biofluid, including saliva, peripheral blood serum or plasma, pancreatic juice, and urine, provide specific exosomal protein and miRNA profiling that can distinguish PDAC patients and healthy donors. Combination of novel exosomal factors (e.g. exosomal glypican-1) and known PDAC markers, such as CA19-9 from serum, provide new strategies for earlier and more reliable PDAC diagnosis.
Challenges of exosome-based therapies
One long-standing strategy of cancer treatments is targeting secretory proteins and their receptors that are involved in tumorigenesis. As exosomes also play an orchestral role in tumour microenvironment, targeting exosomes provides a new approach in cancer therapies. Especially, exosomes provide protection to those cargos from antagonist treatment and enter recipient cells bypassing receptors, which can escape from current therapies and eventually cause tumour relapse. This might explain why a combination of drugs targeting both secretory proteins and their receptors increase patient survival in the first instance, but the disease still progresses later. One exosome-based strategy can be interfering exosome biogenesis and uptake. Exosome biogenesis can go through ESCRT-dependent and independent mechanisms, and inhibition of both mechanisms has shown significant suppression of cancer invasion and progression. Secondly, specific surface proteins on tumour-derived EVs which are responsible for exosome uptake regulation can be targeted. CD47, for example, is an integrin transmembrane molecule that shields exosomes from phagocytosis and facilitates micropinocytosis [165]. While exosomes have been largely described as tumorigenesis promoters, it is conceivable that exosomes also possess antitumor functions and act to restrain disease progression. Exosomes loaded with interferon-γ from dendritic cells facilitate anticancer immune responses in NK and T cells [166]. Also, constrained lncRNA SBF2-AS1 in M2 macrophage-derived exosomes inhibit PDAC progression [100]. Exosomes from NKs [101] and BM-MSCs [102,103,104,105,106] exert tumour suppression abilities too. This suggests an additional route in line with immune cell therapy against cancers. Intriguingly, an increasing number of studies are investigating drug-loaded exosomes as a cancer therapy. Gemcitabine and paclitaxel loaded exosomes enhanced anticancer efficacy of therapeutics [167, 168]. This suggests that exosomes can serve as vesicles since drugs incorporate during exosome biogenesis. Liposome-based drug delivery has been one of the earliest efforts. Nevertheless, the drug loading capability, the release of payload, and antitumor efficacies are varied in donor cell types [169]. Therefore, these major obstructions remain to revolutionize therapeutic advances.
Conclusion
The evidence included in this review demonstrates that exosomes impact multiple hallmarks of PDAC progression, revealing their contribution to niche construction, metastasis, immune regulation, chemoresistance and potential for diagnosis, prognosis and therapy. However, heterogenous characteristics of exosomes make accurate exosome isolation and precise detection of exosomes still challenging. Understanding how exosomes deliver signals and how this reshapes and reprograms target cells into more malignant phenotypes provide important insight into PDAC detection and treatment.
References
Lan B, Zeng S, Grutzmann R, Pilarsky C. The role of exosomes in pancreatic cancer. Int J Mol Sci. 2019;20:4332.
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85.
Bandu R, Oh JW, Kim KP. Mass spectrometry-based proteome profiling of extracellular vesicles and their roles in cancer biology. Exp Mol Med. 2019;51:30.
Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
Baj-Krzyworzeka M, Mytar B, Weglarczyk K, Szatanek R, Kijowski J, Siedlar M. Protumorogenic potential of pancreatic adenocarcinoma-derived extracellular vesicles. Folia Biol. 2020;66:104–10.
Han W, Sulidankazha Q, Nie X, Yilidan R, Len K. Pancreatic cancer cells-derived exosomal long non-coding RNA CCAT1/microRNA-138-5p/HMGA1 axis promotes tumor angiogenesis. Life Sci. 2021;278:119495.
Guo Z, Wang X, Yang Y, Chen W, Zhang K, Teng B, et al. Hypoxic tumor-derived exosomal long noncoding RNA UCA1 promotes angiogenesis via miR-96-5p/AMOTL2 in pancreatic cancer. Mol Ther Nucleic Acids. 2020;22:179–95.
Gesierich S, Berezovskiy I, Ryschich E, Zoller M. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res. 2006;66:7083–94.
Zhao K, Erb U, Hackert T, Zoller M, Yue S. Distorted leukocyte migration, angiogenesis, wound repair and metastasis in Tspan8 and Tspan8/CD151 double knockout mice indicate complementary activities of Tspan8 and CD51. Biochim Biophys Acta Mol Cell Res. 2018;1865:379–91.
Zhao K, Wang Z, Hackert T, Pitzer C, Zoller M. Tspan8 and Tspan8/CD151 knockout mice unravel the contribution of tumor and host exosomes to tumor progression. J Exp Clin Cancer Res. 2018;37:312.
Shang D, Xie C, Hu J, Tan J, Yuan Y, Liu Z, et al. Pancreatic cancer cell-derived exosomal microRNA-27a promotes angiogenesis of human microvascular endothelial cells in pancreatic cancer via BTG2. J Cell Mol Med. 2020;24:588–604.
Pessolano E, Belvedere R, Bizzarro V, Franco P, Marco I, Porta A, et al. Annexin A1 may induce pancreatic cancer progression as a key player of extracellular vesicles effects as evidenced in the in vitro MIA PaCa-2 model system. Int J Mol Sci. 2018;19:3878.
Novizio N, Belvedere R, Pessolano E, Tosco A, Porta A, Perretti M, et al. Annexin A1 released in extracellular vesicles by pancreatic cancer cells activates components of the tumor microenvironment, through interaction with the formyl-peptide receptors. Cells. 2020;9:2719.
Chiba M, Kubota S, Sato K, Monzen S. Exosomes released from pancreatic cancer cells enhance angiogenic activities via dynamin-dependent endocytosis in endothelial cells in vitro. Sci Rep. 2018;8:11972.
Li J, Li Z, Jiang P, Peng M, Zhang X, Chen K, et al. Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J Exp Clin Cancer Res. 2018;37:177.
Blomme A, Fahmy K, Peulen O, Costanza B, Fontaine M, Struman I, et al. Myoferlin is a novel exosomal protein and functional regulator of cancer-derived exosomes. Oncotarget. 2016;7:83669–83.
Wang CA, Chang IH, Hou PC, Tai YJ, Li WN, Hsu PL, et al. DUSP2 regulates extracellular vesicle-VEGF-C secretion and pancreatic cancer early dissemination. J Extracell Vesicles. 2020;9:1746529.
Wang JG, Geddings JE, Aleman MM, Cardenas JC, Chantrathammachart P, Williams JC, et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood. 2012;119:5543–52.
Stark K, Schubert I, Joshi U, Kilani B, Hoseinpour P, Thakur M, et al. Distinct pathogenesis of pancreatic cancer microvesicle-associated venous thrombosis identifies new antithrombotic targets in vivo. Arterioscler Thromb Vasc Biol. 2018;38:772–86.
Yates KR, Welsh J, Echrish HH, Greenman J, Maraveyas A, Madden LA. Pancreatic cancer cell and microparticle procoagulant surface characterization: involvement of membrane-expressed tissue factor, phosphatidylserine and phosphatidylethanolamine. Blood Coagul Fibrinolysis. 2011;22:680–7.
Geddings JE, Hisada Y, Boulaftali Y, Getz TM, Whelihan M, Fuentes R, et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J Thromb Haemost. 2016;14:153–66.
Mezouar S, Darbousset R, Dignat-George F, Panicot-Dubois L, Dubois C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J Cancer. 2015;136:462–75.
Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med. 2009;206:1913–27.
Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, et al. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J Thromb Haemost. 2015;13:1310–9.
Sagar G, Sah RP, Javeed N, Dutta SK, Smyrk TC, Lau JS, et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut. 2016;65:1165–74.
Javeed N, Sagar G, Dutta SK, Smyrk TC, Lau JS, Bhattacharya S, et al. Pancreatic cancer-derived exosomes cause paraneoplastic beta-cell dysfunction. Clin Cancer Res. 2015;21:1722–33.
Wang L, Zhang B, Zheng W, Kang M, Chen Q, Qin W, et al. Exosomes derived from pancreatic cancer cells induce insulin resistance in C2C12 myotube cells through the PI3K/Akt/FoxO1 pathway. Sci Rep. 2017;7:5384.
Zhang Y, Huang S, Li P, Chen Q, Li Y, Zhou Y, et al. Pancreatic cancer-derived exosomes suppress the production of GIP and GLP-1 from STC-1cells in vitro by down-regulating the PCSK1/3. Cancer Lett. 2018;431:190–200.
Shen T, Jia S, Ding G, Ping D, Zhou L, Zhou S, et al. BxPC-3-derived small extracellular vesicles induce FOXP3+ Treg through ATM-AMPK-Sirtuins-mediated FOXOs nuclear translocations. iScience. 2020;23:101431.
Basso D, Gnatta E, Padoan A, Fogar P, Furlanello S, Aita A, et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget. 2017;8:84928–44.
Javeed N, Gustafson MP, Dutta SK, Lin Y, Bamlet WR, Oberg AL, et al. Immunosuppressive CD14(+)HLA-DR(lo/neg) monocytes are elevated in pancreatic cancer and “primed” by tumor-derived exosomes. Oncoimmunology. 2017;6:e1252013.
Baj-Krzyworzeka M, Weglarczyk K, Szatanek R, Mytar B, Baran J, Siedlar M. The role of CD44H molecule in the interactions between human monocytes and pancreatic adenocarcinoma-derived microvesicles. Folia Histochem Cytobiol. 2019;57:28–34.
Zech D, Rana S, Buchler MW, Zoller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun Signal. 2012;10:37.
Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005;65:5238–47.
Zhao J, Schlosser HA, Wang Z, Qin J, Li J, Popp F, et al. Tumor-derived extracellular vesicles inhibit natural killer cell function in pancreatic cancer. Cancers. 2019;11:874.
Katsiougiannis S, Chia D, Kim Y, Singh RP, Wong DT. Saliva exosomes from pancreatic tumor-bearing mice modulate NK cell phenotype and antitumor cytotoxicity. FASEB J. 2017;31:998–1010.
Shen T, Huang Z, Shi C, Pu X, Xu X, Wu Z, et al. Pancreatic cancer-derived exosomes induce apoptosis of T lymphocytes through the p38 MAPK-mediated endoplasmic reticulum stress. FASEB J. 2020;34:8442–58.
Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014;292:65–9.
Ding G, Zhou L, Qian Y, Fu M, Chen J, Chen J, et al. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget. 2015;6:29877–88.
Linton SS, Abraham T, Liao J, Clawson GA, Butler PJ, Fox T, et al. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PLoS ONE. 2018;13:e0206759.
Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069–83.
Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Disco. 2016;6:247–55.
Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, et al. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Disco. 2016;6:256–69.
Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas. Cancer Cancer Discov. 2016;6:270–85.
Tong DN, Guan J, Sun JH, Zhao CY, Chen SG, Zhang ZY, et al. Characterization of B cell-mediated PD-1/PD-L1 interaction in pancreatic cancer patients. Clin Exp Pharm Physiol. 2020;47:1342–9.
Takahashi R, Macchini M, Sunagawa M, Jiang Z, Tanaka T, Valenti G, et al. Interleukin-1beta-induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte-mediated immune suppression. Gut. 2021;70:330–41.
Spear S, Candido JB, McDermott JR, Ghirelli C, Maniati E, Beers SA, et al. Discrepancies in the tumor microenvironment of spontaneous and orthotopic murine models of pancreatic cancer uncover a new immunostimulatory phenotype for B cells. Front Immunol. 2019;10:542.
D’Angelo A, Sobhani N, Roviello G, Bagby S, Bonazza D, Bottin C, et al. Tumour infiltrating lymphocytes and immune-related genes as predictors of outcome in pancreatic adenocarcinoma. PLoS ONE. 2019;14:e0219566.
Brunner M, Maier K, Rummele P, Jacobsen A, Merkel S, Benard A, et al. Upregulation of CD20 positive B-cells and B-cell aggregates in the tumor infiltration zone is associated with better survival of patients with pancreatic ductal adenocarcinoma. Int J Mol Sci. 2020;21:1779.
Capello M, Vykoukal JV, Katayama H, Bantis LE, Wang H, Kundnani DL, et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat Commun. 2019;10:254.
Patel GK, Khan MA, Bhardwaj A, Srivastava SK, Zubair H, Patton MC, et al. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer. 2017;116:609–19.
Mikamori M, Yamada D, Eguchi H, Hasegawa S, Kishimoto T, Tomimaru Y, et al. MicroRNA-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci Rep. 2017;7:42339.
Fan J, Wei Q, Koay EJ, Liu Y, Ning B, Bernard PW, et al. Chemoresistance transmission via exosome-mediated EphA2 transfer in pancreatic cancer. Theranostics. 2018;8:5986–94.
Yang Z, Zhao N, Cui J, Wu H, Xiong J, Peng T. Exosomes derived from cancer stem cells of gemcitabine-resistant pancreatic cancer cells enhance drug resistance by delivering miR-210. Cell Oncol. 2020;43:123–36.
An M, Zhu J, Wu J, Cuneo KC, Lubman DM. Circulating microvesicles from pancreatic cancer accelerate the migration and proliferation of PANC-1 cells. J Proteome Res. 2018;17:1690–9.
Yan Q, Yuan WB, Sun X, Zhang MJ, Cen F, Zhou SY, et al. Asparaginyl endopeptidase enhances pancreatic ductal adenocarcinoma cell invasion in an exosome-dependent manner and correlates with poor prognosis. Int J Oncol. 2018;52:1651–60.
Wu M, Tan X, Liu P, Yang Y, Huang Y, Liu X, et al. Role of exosomal microRNA-125b-5p in conferring the metastatic phenotype among pancreatic cancer cells with different potential of metastasis. Life Sci. 2020;255:117857.
Jin H, Liu P, Wu Y, Meng X, Wu M, Han J, et al. Exosomal zinc transporter ZIP4 promotes cancer growth and is a novel diagnostic biomarker for pancreatic cancer. Cancer Sci. 2018;109:2946–56.
Li Z, Yanfang W, Li J, Jiang P, Peng T, Chen K, et al. Tumor-released exosomal circular RNA PDE8A promotes invasive growth via the miR-338/MACC1/MET pathway in pancreatic cancer. Cancer Lett. 2018;432:237–50.
Li Z, Tao Y, Wang X, Jiang P, Li J, Peng M, et al. Tumor-secreted exosomal miR-222 promotes tumor progression via regulating P27 expression and re-localization in pancreatic cancer. Cell Physiol Biochem. 2018;51:610–29.
Li Z, Jiang P, Li J, Peng M, Zhao X, Zhang X, et al. Tumor-derived exosomal lnc-Sox2ot promotes EMT and stemness by acting as a ceRNA in pancreatic ductal adenocarcinoma. Oncogene. 2018;37:3822–38.
Chiba M, Kubota S, Sakai A, Monzen S. Celltocell communication via extracellular vesicles among human pancreatic cancer cells derived from the same patient. Mol Med Rep. 2018;18:3989–96.
Yu Z, Zhao S, Ren L, Wang L, Chen Z, Hoffman RM, et al. Pancreatic cancer-derived exosomes promote tumor metastasis and liver pre-metastatic niche formation. Oncotarget. 2017;8:63461–83.
Houg DS, Bijlsma MF. The hepatic pre-metastatic niche in pancreatic ductal adenocarcinoma. Mol Cancer. 2018;17:95.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–26.
Pfeiler S, Thakur M, Grunauer P, Megens RTA, Joshi U, Coletti R, et al. CD36-triggered cell invasion and persistent tissue colonization by tumor microvesicles during metastasis. FASEB J. 2019;33:1860–72.
Emmanouilidi A, Paladin D, Greening DW, Falasca M. Oncogenic and non-malignant pancreatic exosome cargo reveal distinct expression of oncogenic and prognostic factors involved in tumor invasion and metastasis. Proteomics. 2019;19:e1800158.
Armacki M, Polaschek S, Waldenmaier M, Morawe M, Ruhland C, Schmid R, et al. Protein kinase D1, reduced in human pancreatic tumors, increases secretion of small extracellular vesicles from cancer cells that promote metastasis to lung in mice. Gastroenterology. 2020;159:1019–35 e22.
Novo D, Heath N, Mitchell L, Caligiuri G, MacFarlane A, Reijmer D, et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat Commun. 2018;9:5069.
Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kgamma to promote pancreatic cancer metastasis. Cancer Res. 2018;78:4586–98.
Jung T, Castellana D, Klingbeil P, Cuesta Hernandez I, Vitacolonna M, Orlicky DJ, et al. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 2009;11:1093–105.
Xie Z, Gao Y, Ho C, Li L, Jin C, Wang X, et al. Exosome-delivered CD44v6/C1QBP complex drives pancreatic cancer liver metastasis by promoting fibrotic liver microenvironment. Gut. 2021;gutjnl-2020-323014.
Stefanius K, Servage K, de Souza Santos M, Gray HF, Toombs JE, Chimalapati S, et al. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. Elife. 2019;8:e40226.
Ristorcelli E, Beraud E, Verrando P, Villard C, Lafitte D, Sbarra V, et al. Human tumor nanoparticles induce apoptosis of pancreatic cancer cells. FASEB J. 2008;22:3358–69.
Ristorcelli E, Beraud E, Mathieu S, Lombardo D, Verine A. Essential role of Notch signaling in apoptosis of human pancreatic tumoral cells mediated by exosomal nanoparticles. Int J Cancer. 2009;125:1016–26.
Wang X, Li H, Lu X, Wen C, Huo Z, Shi M, et al. Melittin-induced long non-coding RNA NONHSAT105177 inhibits proliferation and migration of pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9:940.
Liu Y, Tang T, Yang X, Qin P, Wang P, Zhang H, et al. Tumor-derived exosomal long noncoding RNA LINC01133, regulated by Periostin, contributes to pancreatic ductal adenocarcinoma epithelial-mesenchymal transition through the Wnt/beta-catenin pathway by silencing AXIN2. Oncogene 2021;40:3164–79.
Ye Z, Zhu Z, Xie J, Feng Z, Li Y, Xu X, et al. Hsa_circ_0000069 knockdown inhibits tumorigenesis and exosomes with downregulated hsa_circ_0000069 suppress malignant transformation via inhibition of STIL in pancreatic cancer. Int J Nanomed. 2020;15:9859–73.
Chang WH, Nguyen TT, Hsu CH, Bryant KL, Kim HJ, Ying H, et al. KRAS-dependent cancer cells promote survival by producing exosomes enriched in Survivin. Cancer Lett. 2021;517:66–77.
Masamune A, Yoshida N, Hamada S, Takikawa T, Nabeshima T, Shimosegawa T. Exosomes derived from pancreatic cancer cells induce activation and profibrogenic activities in pancreatic stellate cells. Biochem Biophys Res Commun. 2018;495:71–7.
Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, et al. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015;106:1362–9.
Zhang YF, Zhou YZ, Zhang B, Huang SF, Li PP, He XM, et al. Pancreatic cancer-derived exosomes promoted pancreatic stellate cells recruitment by pancreatic cancer. J Cancer. 2019;10:4397–407.
Takikawa T, Masamune A, Yoshida N, Hamada S, Kogure T, Shimosegawa T. Exosomes derived from pancreatic stellate cells: microRNA signature and effects on pancreatic cancer cells. Pancreas. 2017;46:19–27.
Cao W, Zeng Z, He Z, Lei S. Hypoxic pancreatic stellate cell-derived exosomal mirnas promote proliferation and invasion of pancreatic cancer through the PTEN/AKT pathway. Aging. 2021;13:7120–32.
Han S, Gonzalo DH, Feely M, Rinaldi C, Belsare S, Zhai H, et al. Stroma-derived extracellular vesicles deliver tumor-suppressive miRNAs to pancreatic cancer cells. Oncotarget. 2018;9:5764–77.
Li M, Guo H, Wang Q, Chen K, Marko K, Tian X, et al. Pancreatic stellate cells derived exosomal miR-5703 promotes pancreatic cancer by downregulating CMTM4 and activating PI3K/Akt pathway. Cancer Lett. 2020;490:20–30.
Prieto-Garcia E, Diaz-Garcia CV, Agudo-Lopez A, Pardo-Marques V, Garcia-Consuegra I, Asensio-Pena S, et al. Tumor-stromal interactions in a co-culture model of human pancreatic adenocarcinoma cells and fibroblasts and their connection with tumor spread. Biomedicines. 2021;9:364.
Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770–8.
Fang Y, Zhou W, Rong Y, Kuang T, Xu X, Wu W, et al. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp Cell Res. 2019;383:111543.
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016;5:e10250.
Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. 2016;126:4140–56.
Kong F, Li L, Wang G, Deng X, Li Z, Kong X. VDR signaling inhibits cancer-associated-fibroblasts’ release of exosomal miR-10a-5p and limits their supportive effects on pancreatic cancer cells. Gut. 2019;68:950–1.
Shi H, Li H, Zhen T, Dong Y, Pei X, Zhang X. The potential therapeutic role of exosomal microRNA-520b derived from normal fibroblasts in pancreatic cancer. Mol Ther Nucleic Acids. 2020;20:373–84.
Chang YT, Peng HY, Hu CM, Huang SC, Tien SC, Jeng YM. Pancreatic cancer-derived small extracellular vesical Ezrin regulates macrophage polarization and promotes metastasis. Am J Cancer Res. 2020;10:12–37.
Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben David G, et al. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Cancer Res. 2018;78:5287–99.
Xavier CPR, Castro I, Caires HR, Ferreira D, Cavadas B, Pereira L, et al. Chitinase 3-like-1 and fibronectin in the cargo of extracellular vesicles shed by human macrophages influence pancreatic cancer cellular response to gemcitabine. Cancer Lett. 2021;501:210–23.
Hamada S, Masamune A, Miura S, Satoh K, Shimosegawa T. MiR-365 induces gemcitabine resistance in pancreatic cancer cells by targeting the adaptor protein SHC1 and pro-apoptotic regulator BAX. Cell Signal. 2014;26:179–85.
Li X, Xu H, Yi J, Dong C, Zhang H, Wang Z, et al. miR-365 secreted from M2 macrophage-derived extracellular vesicles promotes pancreatic ductal adenocarcinoma progression through the BTG2/FAK/AKT axis. J Cell Mol Med. 2021;25:4671–83.
Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, et al. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-beta signaling pathway. J Exp Clin Cancer Res. 2019;38:310.
Yin Z, Zhou Y, Ma T, Chen S, Shi N, Zou Y, et al. Down-regulated lncRNA SBF2-AS1 in M2 macrophage-derived exosomes elevates miR-122-5p to restrict XIAP, thereby limiting pancreatic cancer development. J Cell Mol Med. 2020;24:5028–38.
Sun H, Shi K, Qi K, Kong H, Zhang J, Dai S, et al. Natural killer cell-derived exosomal miR-3607-3p inhibits pancreatic cancer progression by targeting IL-26. Front Immunol. 2019;10:2819.
Shang S, Wang J, Chen S, Tian R, Zeng H, Wang L, et al. Exosomal miRNA-1231 derived from bone marrow mesenchymal stem cells inhibits the activity of pancreatic cancer. Cancer Med. 2019;8:7728–40.
Ding Y, Cao F, Sun H, Wang Y, Liu S, Wu Y, et al. Exosomes derived from human umbilical cord mesenchymal stromal cells deliver exogenous miR-145-5p to inhibit pancreatic ductal adenocarcinoma progression. Cancer Lett. 2019;442:351–61.
Wu DM, Wen X, Han XR, Wang S, Wang YJ, Shen M, et al. Bone marrow mesenchymal stem cell-derived exosomal microRNA-126-3p inhibits pancreatic cancer development by targeting ADAM9. Mol Ther Nucleic Acids. 2019;16:229–45.
Wang B, Xu Y, Wei Y, Lv L, Liu N, Lin R, et al. Human mesenchymal stem cell-derived exosomal microRNA-143 promotes apoptosis and suppresses cell growth in pancreatic cancer via target gene regulation. Front Genet. 2021;12:581694.
Xu Y, Liu N, Wei Y, Zhou D, Lin R, Wang X, et al. Anticancer effects of miR-124 delivered by BM-MSC derived exosomes on cell proliferation, epithelial mesenchymal transition, and chemotherapy sensitivity of pancreatic cancer cells. Aging. 2020;12:19660–76.
Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44.
Ishiwata T, Matsuda Y, Yoshimura H, Sasaki N, Ishiwata S, Ishikawa N, et al. Pancreatic cancer stem cells: features and detection methods. Pathol Oncol Res. 2018;24:797–805.
Wang H, Rana S, Giese N, Buchler MW, Zoller M. Tspan8, CD44v6 and alpha6beta4 are biomarkers of migrating pancreatic cancer-initiating cells. Int J Cancer. 2013;133:416–26.
Yue S, Mu W, Erb U, Zoller M. The tetraspanins CD151 and Tspan8 are essential exosome components for the crosstalk between cancer initiating cells and their surrounding. Oncotarget. 2015;6:2366–84.
Wang Z, von Au A, Schnolzer M, Hackert T, Zoller M. CD44v6-competent tumor exosomes promote motility, invasion and cancer-initiating cell marker expression in pancreatic and colorectal cancer cells. Oncotarget. 2016;7:55409–36.
Wang Z, Sun H, Provaznik J, Hackert T, Zoller M. Pancreatic cancer-initiating cell exosome message transfer into noncancer-initiating cells: the importance of CD44v6 in reprogramming. J Exp Clin Cancer Res. 2019;38:132.
Jiang MJ, Chen YY, Dai JJ, Gu DN, Mei Z, Liu FR, et al. Dying tumor cell-derived exosomal miR-194-5p potentiates survival and repopulation of tumor repopulating cells upon radiotherapy in pancreatic cancer. Mol Cancer. 2020;19:68.
Kuc N, Doermann A, Shirey C, Lee DD, Lowe CW, Awasthi N, et al. Pancreatic ductal adenocarcinoma cell secreted extracellular vesicles containing ceramide-1-phosphate promote pancreatic cancer stem cell motility. Biochem Pharm. 2018;156:458–66.
Chang J, Li H, Zhu Z, Mei P, Hu W, Xiong X, et al. microRNA-21-5p from M2 macrophage-derived extracellular vesicles promotes the differentiation and activity of pancreatic cancer stem cells by mediating KLF3. Cell Biol Toxicol. 2021. https://doi.org/10.1007/s10565-021-09597-x.
Klein-Scory S, Tehrani MM, Eilert-Micus C, Adamczyk KA, Wojtalewicz N, Schnolzer M, et al. New insights in the composition of extracellular vesicles from pancreatic cancer cells: implications for biomarkers and functions. Proteome Sci. 2014;12:50.
Han S, Huo Z, Nguyen K, Zhu F, Underwood PW, Basso KBG, et al. The proteome of pancreatic cancer-derived exosomes reveals signatures rich in key signaling pathways. Proteomics. 2019;19:e1800394.
Servage KA, Stefanius K, Gray HF, Orth K. Proteomic profiling of small extracellular vesicles secreted by human pancreatic cancer cells implicated in cellular transformation. Sci Rep. 2020;10:7713.
Fahrmann JF, Mao X, Irajizad E, Katayama H, Capello M, Tanaka I, et al. Plasma-derived extracellular vesicles convey protein signatures that reflect pathophysiology in lung and pancreatic adenocarcinomas. Cancers. 2020;12:1147.
Ung TH, Madsen HJ, Hellwinkel JE, Lencioni AM, Graner MW. Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 2014;105:1384–92.
Herlyn M, Blaszczyk M, Bennicelli J, Sears HF, Ernst C, Ross AH, et al. Selection of monoclonal antibodies detecting serodiagnostic human tumor markers. J Immunol Methods. 1985;80:107–16.
Sharma R, Huang X, Brekken RA, Schroit AJ. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br J Cancer. 2017;117:545–52.
Zhou CY, Dong YP, Sun X, Sui X, Zhu H, Zhao YQ, et al. High levels of serum glypican-1 indicate poor prognosis in pancreatic ductal adenocarcinoma. Cancer Med. 2018;7:5525–33.
Frampton AE, Prado MM, Lopez-Jimenez E, Fajardo-Puerta AB, Jawad ZAR, Lawton P, et al. Glypican-1 is enriched in circulating-exosomes in pancreatic cancer and correlates with tumor burden. Oncotarget. 2018;9:19006–13.
Herreros-Villanueva M, Bujanda L. Glypican-1 in exosomes as biomarker for early detection of pancreatic cancer. Ann Transl Med. 2016;4:64.
Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–82.
Qian JY, Tan YL, Zhang Y, Yang YF, Li XQ. Prognostic value of glypican-1 for patients with advanced pancreatic cancer following regional intra-arterial chemotherapy. Oncol Lett. 2018;16:1253–8.
Moutinho-Ribeiro P, Costa-Moreira P, Adem B, Batista I, Almeida M, Barroca H, et al. Exosomal glypican-1 for risk stratification of pancreatic cystic lesions: A case of pathological progression in the absence of any suspicious imaging finding. Pancreatology. 2020;20:571–5.
Buscail E, Chauvet A, Quincy P, Degrandi O, Buscail C, Lamrissi I, et al. CD63-GPC1-positive exosomes coupled with CA19-9 offer good diagnostic potential for resectable pancreatic ductal adenocarcinoma. Transl Oncol. 2019;12:1395–403.
Xiao D, Dong Z, Zhen L, Xia G, Huang X, Wang T, et al. Combined exosomal GPC1, CD82, and serum CA19-9 as multiplex targets: a specific, sensitive, and reproducible detection panel for the diagnosis of pancreatic cancer. Mol Cancer Res. 2020;18:300–10.
Lucien F, Lac V, Billadeau DD, Borgida A, Gallinger S, Leong HS. Glypican-1 and glycoprotein 2 bearing extracellular vesicles do not discern pancreatic cancer from benign pancreatic diseases. Oncotarget. 2019;10:1045–55.
Matsushita H, Yang YM, Pandol SJ, Seki E. Exosome migration inhibitory factor as a marker and therapeutic target for pancreatic cancer. Gastroenterology. 2016;150:1033–5.
Liang K, Liu F, Fan J, Sun D, Liu C, Lyon CJ, et al. Nanoplasmonic quantification of tumor-derived extracellular vesicles in plasma microsamples for diagnosis and treatment monitoring. Nat Biomed Eng. 2017;1:0021.
Wei Q, Wei L, Zhang J, Li Z, Feng H, Ren L. EphA2enriched exosomes promote cell migration and are a potential diagnostic serum marker in pancreatic cancer. Mol Med Rep. 2020;22:2941–7.
Adamczyk KA, Klein-Scory S, Tehrani MM, Warnken U, Schmiegel W, Schnolzer M, et al. Characterization of soluble and exosomal forms of the EGFR released from pancreatic cancer cells. Life Sci. 2011;89:304–12.
Arscott WT, Camphausen KA. EGFR isoforms in exosomes as a novel method for biomarker discovery in pancreatic cancer. Biomark Med. 2011;5:821.
Ohshima K, Hatakeyama K, Kanto K, Ide T, Watanabe Y, Moromizato S, et al. Comparative proteomic analysis identifies exosomal Eps8 protein as a potential metastatic biomarker for pancreatic cancer. Oncol Rep. 2019;41:1019–34.
Kimura H, Yamamoto H, Harada T, Fumoto K, Osugi Y, Sada R, et al. CKAP4, a DKK1 receptor, is a biomarker in exosomes derived from pancreatic cancer and a molecular target for therapy. Clin Cancer Res. 2019;25:1936–47.
Feng Z, Li K, Wu Y, Peng C. Transcriptomic profiling identifies DCBLD2 as a diagnostic and prognostic biomarker in pancreatic ductal adenocarcinoma. Front Mol Biosci. 2021;8:659168.
Shin HS, Jung SB, Park S, Dua P, Lee DK. ALPPL2 is a potential diagnostic biomarker for pancreatic cancer-derived extracellular vesicles. Mol Ther Methods Clin Dev. 2019;15:204–10.
Lux A, Kahlert C, Grutzmann R, Pilarsky C. c-Met and PD-L1 on circulating exosomes as diagnostic and prognostic markers for pancreatic cancer. Int J Mol Sci. 2019;20:3305.
Sakaue T, Koga H, Iwamoto H, Nakamura T, Ikezono Y, Abe M, et al. Glycosylation of ascites-derived exosomal CD133: a potential prognostic biomarker in patients with advanced pancreatic cancer. Med Mol Morphol. 2019;52:198–208.
Giampieri R, Piva F, Occhipinti G, Bittoni A, Righetti A, Pagliaretta S, et al. Clinical impact of different exosomes’ protein expression in pancreatic ductal carcinoma patients treated with standard first line palliative chemotherapy. PLoS ONE. 2019;14:e0215990.
Hisada Y, Mackman N. Cancer cell-derived tissue factor-positive extracellular vesicles: biomarkers of thrombosis and survival. Curr Opin Hematol. 2019;26:349–56.
Ender F, Freund A, Quecke T, Steidel C, Zamzow P, von Bubnoff N, et al. Tissue factor activity on microvesicles from cancer patients. J Cancer Res Clin Oncol. 2020;146:467–75.
Allenson K, Castillo J, San Lucas FA, Scelo G, Kim DU, Bernard V, et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017;28:741–7.
Yang S, Che SP, Kurywchak P, Tavormina JL, Gansmo LB, Correa de Sampaio P, et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol Ther. 2017;18:158–65.
Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289:3869–75.
Takahashi K, Ota Y, Kogure T, Suzuki Y, Iwamoto H, Yamakita K, et al. Circulating extracellular vesicle-encapsulated HULC is a potential biomarker for human pancreatic cancer. Cancer Sci. 2020;111:98–111.
Kumar SR, Kimchi ET, Manjunath Y, Gajagowni S, Stuckel AJ. RNA cargos in extracellular vesicles derived from blood serum in pancreas associated conditions. Sci Rep. 2020;10:2800.
Madhavan B, Yue S, Galli U, Rana S, Gross W, Muller M, et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Cancer. 2015;136:2616–27.
Komatsu S, Ichikawa D, Takeshita H, Morimura R, Hirajima S, Tsujiura M, et al. Circulating miR-18a: a sensitive cancer screening biomarker in human cancer. Vivo. 2014;28:293–7.
Bartsch DK, Gercke N, Strauch K, Wieboldt R, Matthai E, Wagner V, et al. The combination of MiRNA-196b, LCN2, and TIMP1 is a potential set of circulating biomarkers for screening individuals at risk for familial pancreatic cancer. J Clin Med. 2018;7:295.
Zou X, Wei J, Huang Z, Zhou X, Lu Z, Zhu W, et al. Identification of a six-miRNA panel in serum benefiting pancreatic cancer diagnosis. Cancer Med. 2019;8:2810–22.
Pu X, Ding G, Wu M, Zhou S, Jia S, Cao L. Elevated expression of exosomal microRNA-21 as a potential biomarker for the early diagnosis of pancreatic cancer using a tethered cationic lipoplex nanoparticle biochip. Oncol Lett. 2020;19:2062–70.
Takahasi K, Iinuma H, Wada K, Minezaki S, Kawamura S, Kainuma M, et al. Usefulness of exosome-encapsulated microRNA-451a as a minimally invasive biomarker for prediction of recurrence and prognosis in pancreatic ductal adenocarcinoma. J Hepatobiliary Pancreat Sci. 2018;25:155–61.
Kawamura S, Iinuma H, Wada K, Takahashi K, Minezaki S, Kainuma M, et al. Exosome-encapsulated microRNA-4525, microRNA-451a and microRNA-21 in portal vein blood is a high-sensitive liquid biomarker for the selection of high-risk pancreatic ductal adenocarcinoma patients. J Hepatobiliary Pancreat Sci. 2019;26:63–72.
Goto T, Fujiya M, Konishi H, Sasajima J, Fujibayashi S. Hayashi A, et al. An elevated expression of serum exosomal microRNA-191, -21, -451a of pancreatic neoplasm is considered to be efficient diagnostic marker. BMC Cancer. 2018;18:116.
Ko J, Bhagwat N, Black T, Yee SS, Na YJ, Fisher S, et al. miRNA profiling of magnetic nanopore-isolated extracellular vesicles for the diagnosis of pancreatic cancer. Cancer Res. 2018;78:3688–97.
Chen D, Wu X, Xia M, Wu F, Ding J, Jiao Y, et al. Upregulated exosomic miR23b3p plays regulatory roles in the progression of pancreatic cancer. Oncol Rep. 2017;38:2182–8.
Lai X, Wang M, McElyea SD, Sherman S, House M, Korc M. A microRNA signature in circulating exosomes is superior to exosomal glypican-1 levels for diagnosing pancreatic cancer. Cancer Lett. 2017;393:86–93.
Machida T, Tomofuji T, Maruyama T, Yoneda T, Ekuni D, Azuma T, et al. miR1246 and miR4644 in salivary exosome as potential biomarkers for pancreatobiliary tract cancer. Oncol Rep. 2016;36:2375–81.
Nakamura S, Sadakari Y, Ohtsuka T, Okayama T, Nakashima Y, Gotoh Y, et al. Pancreatic juice exosomal microRNAs as biomarkers for detection of pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2019;26:2104–11.
Yoshizawa N, Sugimoto K, Tameda M, Inagaki Y, Ikejiri M, Inoue H, et al. miR-3940-5p/miR-8069 ratio in urine exosomes is a novel diagnostic biomarker for pancreatic ductal adenocarcinoma. Oncol Lett. 2020;19:2677–84.
Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503.
Rao Q, Zuo B, Lu Z, Gao X, You A, Wu C, et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64:456–72.
Li YJ, Wu JY, Wang JM, Hu XB, Cai JX, Xiang DX. Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomater. 2020;101:519–30.
Cocce V, Franze S, Brini AT, Gianni AB, Pascucci L, Ciusani E, et al. In vitro anticancer activity of extracellular vesicles (EVs) secreted by gingival mesenchymal stromal cells primed with paclitaxel. Pharmaceutics. 2019;11:61.
Kanchanapally R, Deshmukh SK, Chavva SR, Tyagi N, Srivastava SK, Patel GK, et al. Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: a comparative analysis. Int J Nanomed. 2019;14:531–41.
Acknowledgements
We are grateful to Dr. Lucy Elphick for the manuscript proofreading.
Funding
This work in S.P.’s laboratory was supported by a Cancer Research UK Career Development Fellowship, Grant ID C59392/A25064. CHC is a Daphne Jackson Fellow funded by The Kennedy Trust for Rheumatology Research, Grant ID 349/106.
Author information
Authors and Affiliations
Contributions
CHC and SP generated the idea for the article. CHC performed the literature search and analysis. CHC and SP performed writing, review and revision of the article. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This article did not require ethical approval.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Stephen Tait
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, CH., Pauklin, S. Extracellular vesicles in pancreatic cancer progression and therapies. Cell Death Dis 12, 973 (2021). https://doi.org/10.1038/s41419-021-04258-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04258-7
This article is cited by
-
Cancer-derived small extracellular vesicles: emerging biomarkers and therapies for pancreatic ductal adenocarcinoma diagnosis/prognosis and treatment
Journal of Nanobiotechnology (2022)
-
Exosomal DNAJB11 promotes the development of pancreatic cancer by modulating the EGFR/MAPK pathway
Cellular & Molecular Biology Letters (2022)
