
Abstract
Gene fusions are thought to be driver mutations in multiple cancers and are an important factor for poor patient prognosis. Most of them appear in specific cancers, thus satisfactory strategies can be developed for the precise treatment of these types of cancer. Currently, there are few targeted drugs to treat gynecologic tumors, and patients with gynecologic cancer often have a poor prognosis because of tumor progression or recurrence. With the application of massively parallel sequencing, a large number of fusion genes have been discovered in gynecologic tumors, and some fusions have been confirmed to be involved in the biological process of tumor progression. To this end, the present article reviews the current research status of all confirmed fusion genes in gynecologic tumors, including their rearrangement mechanism and frequency in ovarian cancer, endometrial cancer, endometrial stromal sarcoma, and other types of uterine tumors. We also describe the mechanisms by which fusion genes are generated and their oncogenic mechanism. Finally, we discuss the prospect of fusion genes as therapeutic targets in gynecologic tumors.
Similar content being viewed by others


Facts
-
Fusion genes are cancer-specific and considered to be the driving events of cancer.
-
Chromosome instability and genome reassembly are the structural basis for fusion genes.
-
Cancer-related exposure factors are closely related to the occurrence of fusion genes.
-
Fusion genes change the biological behavior of cancer cells through their molecular functions.
Open questions
-
What is the cause of fusion genes?
-
How fusion genes are involved in tumorigenesis and what are the molecular mechanisms?
-
What are the characteristics of recurrent fusions in gynecological tumors?
-
Are there carcinogenic fusions in gynecological tumors?
-
What is the potential of fusion genes in gynecological tumors as therapeutic targets?
Background
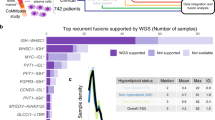
Fusion genes can be defined as new genes that are formed by chromosome breakage and re-splicing at the genome level [1]. Generally, at the genome level, the fusion gene may be expressed; however, if the promoter region or other important elements are destroyed, it may not be expressed. In 1973, researchers first discovered the rearrangement of chromosomes 9 and 22 in chronic myeloid leukemia (CML) and the rearrangement of chromosomes 8 and 21 in acute myeloid leukemia (AML) through chromosome banding technology [2, 3]. Subsequently, researchers carried out cytogenetic analysis of other hematological tumors, and discovered a variety of cancer-characteristic gene rearrangements. For example, t(8;14)(q24; q32), t(2;8)(p11;q24), and t(8;22)(q24;q11) in Burkitt’s lymphoma [4, 5]; t(4;11)(q21;q23) in acute lymphoblastic leukemia (ALL) [6]; t (15;17) (q22;q21) in acute promyelocytic leukemia (APL) [7]; and t(14;18)(q32;q21) in follicular lymphoma [8]. In recent years, deep sequencing technology has been used widely used, and more cancer-related fusion genes have been characterized. Currently, the identification of fusion genes can be based on whole-genome sequencing (WGS), transcriptome sequencing (RNA-seq), or a combination of the two technologies [9, 10]. Fusion genes identified using WGS alone can be determined to be caused by the rearrangement at the genome level; however, if there is no transcriptome sequencing data, it is impossible to accurately determine whether the new fusion gene is expressed or its expression level. Fusion genes identified by RNA-seq alone can be determined to be expressed (Fig. 1a) [11], but it cannot be completely determined whether this is caused by a genomic mutation or RNA fusion that occurs after the transcription of different genes. Therefore, combining the technology of WGS and RNA-seq can obtain more accurate results. For further verification and analysis, methods such as designing specific primers for PCR or real-time fluorescent quantitative PCR followed by DNA gel electrophoresis can be adopted.

The pie chart shows the number of gene fusions in different types of cancer (a). The data comes from the Tumorfusion database. The circos diagram shows the fusion gene on the chromosome in gynecological tumors (b), (c). Bioinformatic analysis was performed using the OmicStudio tools at https://www.omicstudio.cn/tool.
Gene fusion is considered an important driving event for multiple cancers [12]. Some cancer-specific fusion events can be used as diagnostic markers or therapeutic targets, and have achieved good results. In recent years, a large number of fusion events have been described in gynecologic tumors (Fig. 1b, c) [13]; however, their potential roles are not fully understood. Moreover, gene fusion profiles vary in different types of gynecologic tumors. Understanding their occurrence and carcinogenic mechanism will help to identify new therapeutic targets for gynecologic tumors. Therefore, the present article summarizes and classifies the research progress on fusion genes in gynecologic tumors.
Generation mechanism of fusion genes
The structural basis of fusion genes
Chromosome rearrangements and extensive mutations in the genome are significant features of cancer [14, 15]. Fusion events are usually caused by chromosomal rearrangements, including translocations, inversions, and deletions [16, 17], and some fusions are caused by cis or trans splicing of adjacent genes transcripts [18, 19]. In the past, it was generally believed that chromosome instability and chromosome rearrangement were the basis of gene fusion, accompanied by breakage at certain specific sites. When the breakpoint is located in the intron region or the exon boundary, a fusion transcript of the complete exon can be retained; however, when the breakpoint is located inside an exon, the corresponding transcript may be destroyed, leading to changes in gene expression profiles [20].
Physical, chemical, and biological exposure factors induce fusion genes
Exposure to physical, chemical, and biological factors can cause mutations in the genome and induce gene fusion (Fig. 2). After the Chernobyl nuclear leak, many studies reported that radiation exposure has a strong correlation with gene mutation and gene fusion in thyroid cancer. The frequency of RET fusions in radiation-induced papillary thyroid carcinoma (PTC) is very high, ranging from 35 to 80% [21,22,23]. A meta-analysis of the distribution of NCOA4-RET, including 2395 cases of radioactive and sporadic PTC, found that radiation exposure caused an increased risk of RET/PTC, although this association was limited to the NCOA4-RET subtype in the Western population [24]. In addition, the ETV6-NTRK3 fusion was found in 14.5% of PTC cases caused by the Chernobyl incident, and this fusion is also considered to correlate strongly with radiation exposure [25]. A study on the relationship between ionizing radiation with RET fusion in lung adenocarcinoma found that 201T human lung cells exposed to 1 Gy of gamma rays induced RET fusion, and RET rearrangement was also found in 2 of 37 cases of radiation exposure [26]. Another study on the frequency of gene fusion in lung cancer showed that patients exposed to tobacco and coal had the highest gene fusion frequency, and ALK fusion and total gene rearrangement were closely related to these exposures [27]. A genomic study of small cell lung cancer with complex tobacco exposure identified the tandem replication of CHD7 exons 3–8 and a cell line with PVT1-CHD7 fusion [28]. In a study of the relationship between insecticides and cancer-related gene damage, the ETV6-RUNX1 fusion was detected in peripheral blood mononuclear cells (PBMCs) that were exposed acutely to permethrin. Exposure to permethrin could induce the fusion of ETV6-RUNX1 and IGH-BCL2 in the K562 cell line, while malathion can induce the fusion of KMT2A-AFF1 and ETV6-RUNX1 [29]. In squamous cell carcinoma of the head, the EGFR-PPARGC1A fusion is associated with long-term sunlight exposure [30]. Hyperglycemia can induce IGFBP2, which increases the frequency of gene fusion, along with a decrease in PKC DNA levels, suggesting that they are mediated by changes in the rate of double-strand break repair. By contrast, IGF1 and EGF are induced under insulin conditions, which reduces the incidence of gene fusion [31]. Some apoptotic signals, such as serum starvation, etoposide, and salicylic acid, can induce TEL (ETV6) gene disruption and fusion in immature B lymphocytes. The TEL-AML1 fusion is one of the most common genetic mutations in childhood acute lymphoblastic leukemia [32]. It is generally believed that prostate cancer is related to male androgen levels. In-depth studies have found that androgen signaling can induce the TMPRSS2 and ERG genes at the genome level, and cells exposed to gamma-rays experience DNA double-strand breaks, thereby promoting TMPRSS2-ERG gene fusion [33]. Interestingly, the expression of the TMPRSS2-ERG fusion gene changed the chemical and radio reactivity of androgen-independent prostate cancer cells [34].

Here, we summarize the fusion genes and their exposure factors in several common cancers, including nuclear radiation and RET fusions; ionizing radiation, tobacco, coal and RETfusions, ALK fusions; insecticides, permethrin, malathion and ETV6-RUNX1, IGH-BCL2, KMN2A-AFF1; long-term sunlight exposure and EGFR-PDARGC1A; serum starvation, etoposide, salicylic acid, and TEL-AML1; male androgen and TMPRSS1-ERG; The relationship between fusion frequency and hyperglycemia/insulin conditions.
In summary, stimulation by physical, chemical, and biological factors might be crucial conditions for the generation of genomic mutations and gene fusion. Understanding the effects of these exposure factors will help to explore the mechanism of fusion genes in tumorigenesis and provide protective strategies in cancer prevention and treatment.
General mechanism of fusion genes in cancer
Gene fusions can result in the production of new fusion transcripts or fusion proteins. Some fusion products play a key driving role in cancer, such as BCR/ABL and AML1/ETO fusions in hematological malignancies [35, 36]. In recent years, many potential molecular functions of oncogenic fusion proteins have been discovered. Here, we summarize the carcinogenic mechanisms of several fusion genes (Fig. 3), including: destroying protein functional domains, affecting the function of protein complexes, changing molecular subcellular localization, obtaining active or strong promoters, evading regulation by microRNAs (miRNAs), and upregulation of downstream effectors.

MLL fusions lost its functional domain which catalyze H3K4 methylation in Leukemia tumorigenesis (a). SS18-SSX fusion protein cause the lost of BAF47 subunit of BAF complex in synovial sarcoma (b). MAN2A1-FER fusion allows the FER molecule to be located to the Golgi apparatus, thereby activating its tyrosine kinase activity (c). Fusion genes allow some transcription factors or kinases to acquire strong promoters, thereby activating downstream genes (d). FGFR3-TACC3 fusion avoids the regulation of miR199a in tumorigenesis (e). Some fusion proteins act as effectors to activate the target enhancers or genes (f).
Loss of protein functional domains
Some gene fusions result in the loss of part of the coding exons during the rearrangement process, depending on the location of the breakpoint. The loss of important structural domains will cause functional defects (Fig. 3a). The MLL gene encodes a DNA binding protein containing a SET domain, which has H3K4 methyltransferase activity [37]. The MLL gene can regulate positively the expression of a series of downstream HOX genes in hematopoietic stem cells through its H3K4 methyltransferase activity. The fused MLL gene produces a truncated MLL protein lacking the SET domain, resulting in its inability to catalyze H3K4 methylation, which drives the occurrence of leukemia.
Effects on the function of protein complexes
Functional protein complexes are common, and structural integrity is crucial for their normal function. Repeated chromosomal rearrangement is an important sign of synovial sarcoma. The SS18-SSX fusion produced by t(X;18) has been confirmed to be a carcinogenic fusion in synovial sarcoma (Fig. 3b) [38]. SS18 encodes a subunit of the SWI/SNF (BAF) complex and this gene is often rearranged in synovial sarcoma. The SS18-SSX fusion protein competes with the wild-type SS18 protein for binding to the BAF complex, resulting in the loss of the BAF47 subunit (a tumor suppressor) from the complex, which promoting the expression of downstream SOX2 and other genes [39]. In addition, the synergy of the SS18-SSX fusion protein and SWI/SNF complex plays an important role in chromatin structure regulation and histone modification, which jointly promote the tumorigenesis and development of synovial sarcoma [40].
Change in molecular subcellular location
The correct subcellular location of a protein is a significant factor for signal transduction and protein molecular function. Some partner genes that express proteins with specific subcellular localization sequences can play a role by changing the localization of the fusion protein (Fig. 3c). MAN2A1-FER is a common recurrent fusion in liver cancer, esophageal adenocarcinoma, and non-small cell lung cancer, and has carcinogenic effects [41, 42]. The fused MAN2A1 protein retains its signal peptide, resulting in the MAN2A1-FER fusion protein being located in the Golgi apparatus, which results in the ectopic activation FER. The FER-related tyrosine kinase activity of the fusion protein is almost four times that of wild-type FER, which activates downstream signaling molecules significantly, such as BRAF, MEK, and AKT.
Acquisition of an active or strong promoter
The promoter is an important element of gene expression, and its activity is a key factor in determining gene transcription efficiency. It has been reported that some genes acquire active promoters or cis-regulatory elements through fusion events to achieve higher expression levels (Fig. 3d) [43]. For example, the TMPRSS2-ERG fusion is a carcinogenic fusion event in prostate cancer. The fusion allows the ERG gene to share clusters of regulatory elements (COREs) from the TMPRSS2 gene, including its promoter. ERG is as a key transcription factor that maintains the expression of genes required for cancer cell proliferation and metabolism. The EML4-ALK fusion is a successful target for the treatment of non-small cell lung cancer. The fused ALK acquires an active promoter and dimerization site from the EML4 gene, resulting in constitutively activated ALK kinase [44]. Therefore, obtaining a strong promoter is an important mechanism by which oncogenic transcription factors or kinases drive cancer.
Avoiding miRNA regulation
MicroRNAs are small non-coding RNAs with a length of 18–22 nt. miRNAs target and bind to the 3′ untranslated region (UTR) of their target gene mRNAs, leading to mRNA degradation or translational silencing, which play negative roles in the regulation of gene expression (Fig. 3e) [45]. It has been reported that FGFR3 is the target gene of miR-99a and is negatively regulated by miR-99a. In FGFR3-TACC3 fusion-positive gliomas, the fused FGFR3 loses its 3′ UTR region; therefore, it is no longer negatively regulated by miR-99a, which increases the FGFR3 tyrosine kinase signal, thereby promoting tumor progression [46].
Upregulating downstream effectors
The enhancer region is important to regulate gene transcription activity, and the silencing or activation of an enhancer is usually mediated by certain transcriptional activation or repressor factors. For example, the transcriptional activator STAT5 activates the transcription of target genes (such as MYC and BCL2) by binding to their enhancer regions and co-exists with enhancer markers such as BRD4, P300, and H3K27ac. The NUP214-ABL fusion is considered to be a key factor in inducing acute T-cell leukemia (Fig. 3f). The fused protein can activate STAT5 through phosphorylation, and activated STAT5 can increase the transcriptional activity of its target genes significantly [47, 48]. In AML, the RUNXI-ETO fusion protein is considered to be an oncogenic transcription factor (Fig. 3f). The RUNX1-ETO fusion protein replaces the wild-type RUNX1 protein and binds to the open chromatin region upstream of the CCND2 transcription start site (TSS), resulting in the abnormal activation of CCND2, which drives the occurrence of AML [49].
Current research status of fusion genes in gynecologic tumors
The research progress into gene fusions in various types of gynecologic tumors varies. There have been more studies related to ovarian cancer and uterine sarcoma, and fewer studies related to cervical cancer and endometrial cancer. In this section, we describe gene fusions in various types of gynecologic tumors, including the frequency of fusion genes, the method of fusions, the relationship between the genes and cancer, and the mechanism of oncogenic fusion (Table 1). All fusion genes have been confirmed in the relevant literature. The identification methods of fusion genes include RT-PCR, FISH or Sanger sequencing.
Fusion genes in ovarian cancer
Among gynecologic tumors, ovarian cancer is a highly malignant tumor that is prone to metastasis and recurrence. Approximately 70–80% of patients with advanced ovarian cancer experience recurrence within 5 years [50, 51]. The fusion genes that have been identified in ovarian cancer are PCMTD1-CCNL2, ANXA5-CCNA2, CCN4-NRG4, SLC25A40-ABCB1, DPP9-PPP6R3, MAN2A1-FER, CDKN2D-WDFY2, BCAM-AKT2, and FHL2-GLI2. These fusion genes involve cyclin family genes, multi-drug resistance related MDR/TAP subfamily genes, tyrosine kinase family genes, and AKT signaling pathway-related genes [52].
The frequency of the PCMTD1-CCNL2 fusion in endometrioid ovarian cancer is 22% (4/18). It is formed by the rearrangement of the PCMTD1 gene on chromosome 8 and the CCNL2 gene on chromosome 1. The upstream part of the fusion junction is exon 3 of PCMTD1, and the downstream part is exon 6 of CCNL2. The parental gene CCNL2 encodes a cyclin family protein that can interact with a variety of proteins to induce cell cycle arrest and apoptosis in lung cancer and mouse embryonic cancer cells [53, 54]. Interestingly, the other parent gene, PCMTD1, encodes a member of the methyltransferase superfamily, which has methyltransferase activity. Unfortunately, no studies have confirmed that this gene fusion affects the occurrence or development of ovarian cancer or other cancers.
The SLC25A40-ABCB1 fusion is the most common rearrangement involving ABCB1, occurring in 15.7% (20/108) of cases of high-grade serous ovarian cancer [55]. Most of the breakpoints of ABCB1 fusions occur in the intron 1 region, and the fusion includes exon 2 and the following sequence. ABCB1 fusion transcription promoters are mostly replaced by partner gene promoters. ABCB1 fusion-positive tumor tissues are usually accompanied by upregulation of ABCB1 expression, suggesting that gene fusion might allow ABCB1 to escape negative regulation and obtain a strong promoter. The SLC25A40 gene is commonly expressed in tissues and has not been shown to be related to tumors. MDR1, the protein encoded by ABCB1, is a member of the MDR/TAP subfamily, which mediates the efflux of chemotherapeutic drugs and is associated with multidrug resistance in tumors [42, 56].
Although the frequency of MAN2A1-FER fusion in ovarian cancer is only 1.7% (1/60), it appears in a variety of tumors. The frequency of MAN2A1-FER is higher in esophageal adenocarcinoma (25.9%), liver cancer (15.7%), and non-small cell lung cancer (16.8%) [42]. The MAN2A1-FER fusion is formed by the cis splicing of exons 1–13 of MAN2A1 and exons 1–6 of FER on chromosome 5. The parental gene MAN2A1 encodes a glycosyl hydrolase located in the Golgi apparatus that catalyzes the final hydrolysis step in the maturation pathway of asparagine-linked oligosaccharides (N-glycans). MAN2A1 has been shown to be involved in the immune regulation of tumors, and inhibition of MAN2A1 can enhance the tumor’s immune response to anti-PD-L1 drugs [57]. The encoded product of FER is a member of the receptor tyrosine kinase family, which regulates cell adhesion and is related to the epithelial-mesenchymal transition process of tumors [58]. Mechanistically, the MAN2A1-FER fusion protein retains the signal peptide of MAN2A1, which results in the fusion protein being located in the Golgi apparatus, significantly increasing the activity of the fused FER tyrosine kinase [42].
The CDKN2D-WDFY2 fusion is cancer-specific and has only been identified in ovarian cancer. The fusion is formed by the rearrangement of CDKN2D on chromosome 19 and WDFY2 on chromosome 13, including exon 1 of CDKN2D and exons 3–12 of WDFY2, with a fusion frequency of 20% (12/60) [59]. CDKN2D encodes a cell cycle regulator that is involved in the DNA repair process. WDFY2 encodes a protein containing two WD domains and an FYVE zinc finger region, which can specifically target AKT2, leading to decreased phosphorylation of downstream molecules of AKT (such as BAD and FOX3A) [60]. Studies have shown that overexpression of WDFY2 can inhibit the biological behavior of prostate cancer by affecting the AKT pathway [61]. The CDKN2D-WDFY2 fusion resulted in the loss of wild-type WDFY2 protein expression, and the truncated WDFY2 protein was expressed at the same time. The fused WDFY2 protein lacks the AKT-binding domain and results in increased downstream BAD and FOX3A expression.
The BCAM-AKT2 fusion is a specific rearrangement in high-grade serous ovarian cancer. The fusion frequency is 7% (4/60). The fusion gene comprises exons 1–13 of BCAM and exons 5 and the following sequence of AKT2 on chromosome 19. BCAM encodes a receptor for the extracellular matrix protein laminin, which mediates the adhesion of red blood cells [62] and is related to the metastasis of colorectal cancer [63,64,65]. AKT2 is a known oncogene encoding a serine/threonine kinase subfamily protein that can phosphorylate a variety of proteins. Mechanistically, the BCAM-AKT2 fusion protein uses the membrane localization domain of BCAM to guide the fused AKT2 to the membrane where it is activated by phosphorylation [64, 66].
The FHL2-GLI2 fusion is unique to sclerosing stromal tumors of the ovary (SST), with a fusion frequency of 65% (17/26). It consists of exons 1–5 of FHL2 and exons 8–12 GLI2 on chromosome 2 [67]. FHL2 has been reported to play a role in a variety of tumors, participating in epithelial-mesenchymal transition and stabilizing EGFR [68, 69]. The encoded product of GLI2 is a member of the transcription factor of the Gli family, which participates in the Sonic hedgehog (Shh) signaling pathway and promotes tumor progression [70, 71].
Although the TMEM123-MMP27, ZBTB46-WFDC13, PLXNB1-PRKAR2A, and other fusions have been reported in only a few cases of ovarian cancer, it is interesting that the above fusions increase the expression of the 3′ end gene [72] 5. In addition, a study reported that the expression levels of 48 genes located near the fusion gene were upregulated significantly, which also indicated that fusion gene remodeling of the cancer transcriptome is a complicated process [73].
Fusion genes in endometrial cancer
The pathogenesis of endometrial cancer is still unclear. Studies have shown that mutations in some genes or pathways are important driving events of uterine corpus endometrial carcinoma (UCEC), such as the mutation of the P53 gene, the PIK3CA pathway, the KRAS gene, and overexpression of the HER2 gene [74]. In recent years, with the development of deep sequencing technology, a large number of fusion events have been discovered (see Table 1), among which CPQ-PRKDC and TSNAX-DISC1 are two representative fusions [75]. CPQ-PRKDC occurs at a low frequency in endometrial cancer (2.5%, 3/122), and is formed by the rearrangement of exons 1–2 of CPQ and exons 80–87 of PRKDC on chromosome 8 [76]. The TSNAX-DISC1 fusion is not a gene-level rearrangement. It is formed by the splicing of TSNAX and DISC1 transcripts and occurs with a frequency of 71.2% (123/176) in UCEC. Unfortunately, there is no research to show whether these two fusions play a role in cancer.
Fusion genes in cervical cancer
Chronic persistent infection of high-risk human papillomavirus (HPV) is the main cause of cervical cancer, and more than 90% of cases are accompanied by HPV virus infection [77]. However, only 1% of women infected with high-risk HPV eventually develop into cervical cancer. This suggests that there may be other carcinogenic factors besides HPV infections, such as gene mutations and chromosome rearrangement. We found 454 fusions in the Tumorfusion database (Fig. 1a) [11], but only the FGFR3-TACC3 fusion has been confirmed [78]. This fusion is formed by the cis splicing of two adjacent genes on the P16 arm of chromosome 4, including exons 1–17 of FGFR3 and exons 4–16 or 6–16 of TACC3, with an incidence of 1.9% (2/103). FGFR3 encodes a member of the fibroblast growth factor receptor (FGFR) family and contains a tyrosine kinase domain. TACC3 encodes a member of the acidic Escherichia coli chain protein family, which plays a role in the differentiation and growth of certain cancer cells. In glioma, FGFR3 is regulated negatively by miR-99a, while in cervical cancer, FGFR3-TACC3, which has lost the FGFR3 3′ UTR region, has been proven to be a carcinogenic fusion that activates the MAPK pathway by increasing its FGFR3 signal [79]. In addition, the FGFR3-TACC3 fusion protein can replace the EGFR-ERK signaling pathway in tumors, mediating the drug bypass resistance mechanism [80]. This suggests that FGFR3 fusions can fully exert their carcinogenic effects by escaping the negative regulation of miR-99a [81, 82]. Studies have reported that FGFR3-TACC3 is a common fusion in many tumors, and the fusion frequency is similar in various cancers [83]. The FGFR3-TACC3 fusion is a clear oncogene, and its targeted inhibition has achieved good results in other cancers.
Fusion genes in endometrial stromal sarcoma (ESS)
The cause of ESS is unclear; however, it has a high recurrence rate and poor prognosis. It is generally believed that chromosomal rearrangement is closely related to the occurrence of ESS [84]. The instability of chromosomes 6, 7, 10, and 17 in ESS is the cause of some fusions, such as PHF1, JAZF1, EPC1, YWHAE, and MBTD1-CXorf67 fusions.
EPC1 fusion is a rare event in ESS, being only reported in a few cases. Its partner genes include SUZ12, BCOR, and PHF1. EPC1-PHF1 has been reported very early and is associated with the morphology and clinical features of low-grade ESS, which is produced by t (6;10) (p21; p11). By contrast, EPC1-SUZ12 and EPC1-BCOR are more prone to occur in aggressive high-grade ESS, corresponding to t (10;17) (p11; q11) and t (10; x) (p11; p11), respectively [85, 86]. EPC1 is an oncogene that can act as both a transcriptional activator and a repressor, and is related to apoptosis and DNA repair [87, 88]. For example, the EPC1 promoter physically combines with E2F1 to activate transcriptional activity, thereby inducing anti-apoptosis-related genes and promoting cancer growth. Interestingly, the three partner genes are all related to transcriptional regulation [89,90,91], suggesting that the fusion of EPC1 and its partner genes allows them to be physically close, which might promote its participation in transcriptional regulation.
PHF1 fusions mainly include MEAF6-PHF1, EPC1-PHF1, BRD8-PHF1, EPC2-PHF1, and JAZF1-PHF1 [92,93,94]. The PHF1 gene is located on chromosome 6 (6p21), JAZF1 is located at 7p15, EPC2 is located at 2q23, and EPC2 is located at 10p11. The rearrangement of EPC1 or MEAF6 from 1p34 is a recurrent fusion in ESS. The encoded product of PHF1 is a polycomb group protein, which forms a PRC2 complex with, for example, MTF2 and PHF19 to mediate the methylation of histone H3K27 [95]. The PRC2 complex is a methyltransferase that is commonly dysregulated in human cancers. Overexpression of PRC2 is a significant sign of poor prognosis in human cancer [96]. Similar to the EPC1 fusion, most of the PHF1 fusion partner genes are also involved in transcriptional regulation, which suggests that fusion genes might be a mechanism of transcription disorders in ESS.
YWHAE-FAM22A/B fusion is a rearrangement of chromosome 17 arm p13 with chromosome 10 arms q23 and q22, including exons 1–5 of YWHAE and exons 2–7 of FAM22A, with a frequency of 26–58% in ESS [97, 98]. Compared with JAZF1 fusions, YWHAE fusions ESS tend to be associated with higher disease stages and more frequent recurrences, and have diagnostic specificity for high-grade ESS. The encoded product of YWHAE is 14-3-3ε, which belongs to the 14-3-3 protein family, and mediates signal transduction by binding to proteins containing phosphoserine residues. For example, the combination of 14-3-3ε and FBX4 promotes the dimerization of FBX4 and promotes its E3 ligase activity [99]. In prostate cancer, the combination of 14-3-3ε and APAF-1 inhibits cytochrome c from activating the downstream caspase and protects the survival of cancer cells [100]. FAM22A/B encodes a protein with a nuclear localization signal; however, its role in cancer is unclear. The YWHAE-FAM22A/B fusion retains the nuclear localization sequence of FAM22A/B and maintains the complete 14-3-3ε domain. YWHAE, which is directed to the nucleus from the cytoplasm, activates the expression of a series of downstream genes, such as CCND1 and CEBPA [98].
JAZF1-SUZ12 is a carcinogenic fusion related to low-grade ESS. Its fusion frequency is reported to be 75% in endometrial stromal nodule (ESN), 50% in low grade (LG)-ESS, and 15% in high grade (HG)-ESS, and can be used to distinguish LG-ESS from HG-ESS [101]. This fusion is the result of a rearrangement between p15 of the arm of chromosome 7 and q21 of the arm of chromosome 17, which contains exons 1–3 of JAZF1 and exons 2–16 of SUZ12. JAZF1 encodes a nuclear protein with a zinc finger structure. This protein binds to the orphan nuclear receptor TAK1 and acts as a transcriptional regulator [102], which can play a role in tumor suppression or cancer promotion [103, 104]. SUZ12 encodes an important component of the PRC2 complex, which mediates the modification of H3K9 and H3K27 methylation, and is related to chromatin remodeling and gene silencing [105]. The JAZF1-SUZ12 fusion protein in ESS replaces the wild-type SUZ12 protein, which changes the structure of the PRC2 complex and inhibits its H3K27 methylation activity, thus increasing the expression of downstream genes HOXA9 and WNT11 [106].
The fusion frequency of MBTD1-CXorf67 in ESS is 7% (1/14). The MBTD1-CXorf67 fusion is composed of exons 1–16 of MBTD1 and exon 1 of CXorf67 [107]. The result of the MBTD1-CXorf67 fusion in ESS increased the expression of CXorf67 by 5–9 times. Although it is not clear whether CXorf67 has an oncogene effect in ESS, studies have confirmed that CXorf67 affects the DNA repair pathway of homologous recombination in the ependymoma and blocks the methyltransferase activity of EZH2, which plays an important role in glioma tumorigenesis.
Fusion genes in other types of uterine tumors
Uterine inflammatory myofibroblastoma (IMT) is a rare mesenchymal tumor with low-grade malignancy, the recurrence of which can be effectively avoided by complete surgical resection [108]. IMT is usually accompanied by ALK expression and ALK fusion. Common gene fusions in IMT include IGFBP5-ALK, THBS1-ALK, FN1-ALK, and TIMP3-ALK. ALK fusion is considered a diagnostic indicator of IMT [109]. A study found that 10 of 11 IMT tumors contained ALK rearrangements, among which IGFBP5-ALK represented 27% (3/11), THBS1-ALK represented 27% (3/11), FN1-ALK represented 18% (2/11), and TIMP3-ALK represented 9% (1/11) [110]. Another study detected ALK gene fusions in 14 cases of IMT, which indicated that the incidence of ALK fusions in IMT is extremely high [109]. ALK encodes a well-known receptor tyrosine kinase. The ALK fusion partner usually produces oncogenic constitutive tyrosine kinase activity by promoting the polymerization and autophosphorylation of ALK [111].
Uterine tumors similar to ovarian sex cord stromal tumors (UTROSCT) are rare uterine stromal tumors of unknown etiology. Although most of them are benign, some of them recur [112]. Chromosome rearrangement has recently been reported in UTROSCT cases, where ESR1 fusion and GREB1 fusion are common, including ESR1-NCOA2, ESR1-GREB1, GREB1-NCOA2, GREB1-NR4A3, GREB1-SS18, GREB1-NCOA1, and GREB1-CTNNB1. In a recent study, four cases of UTROSCT with GREB1 rearrangements were identified, including GREB1-NCOA2, GREB1-NR4A3, GREB1-SS18, and GREB1-NCOA1. UTROSCTs with these rearrangements were found to be more aggressive than those with ESR1 rearrangements [113]. GREB1 is an early estrogen-responsive gene in the estrogen receptor regulatory pathway. It is believed to stimulate the proliferation of breast, ovarian, and prostate cancer cells [114], and mediate resistance to tamoxifen [115]. NCOA1 and NCOA2 are common partner gene and their encoded proteins act as transcriptional co-activators of steroid and nuclear hormone receptors. They contain nuclear localization signals and bHLH and PAS domains. ESR1 encodes a transcription factor that activates estrogen receptors and ligands, and is considered to be a driving event for breast and endometrial cancer [116, 117]. These reports suggest that fusion genes of the estrogen regulatory pathway have potential carcinogenic effects in UTROSCT.
The prospect of gene fusions as therapeutic targets in gynecologic tumors
For more than half a century, gene fusion has been regarded as a key driving event in cancer. The results of gene fusion can change the expression pattern of the original gene. Some fusion genes can activate protein functions or produce new chimeric proteins, and can participate in the occurrence and development of cancers by changing signaling pathways, affecting protein interaction, changing subcellular localizations, or activating neighboring genes. Chromosome rearrangement and gene fusions are valuable in the diagnosis or classification of cancers [16]. Studies also suggested that gene fusion can be used as a method to monitor the residual small lesions after treatment [118]. Many cancers characterized by fusion genes have a poor prognosis; therefore, targeting specific oncogenic fusion proteins might bring unexpected results. In recent years, targeted drug therapy for gene fusion-positive cancers has significantly improved the prognosis of patients with cancer. For example, tyrosine kinase inhibitors have achieved satisfactory results in a variety of cancers. With the development of deep sequencing technology, scientists have discovered that fusion genes not only exist in sarcomas and hematological malignancies, but also exist in large amounts in some solid tumors [119, 120]. Although most of the fusion events seem to occur accidentally and have no pathogenic characteristics, there are some valuable fusions among the few commonly recurring fusions.
Some of the fusions described in this article have potential value for the management of gynecologic tumors. For example, the JAZF1-SUZ12 fusion related to low-grade ESS and the YWHAE-FAM22 fusion related to high-grade ESS have been confirmed to play a role in the progression of ESS [98, 106]. The CDKN2D-WDFY2 fusion had a detection rate of 20% in 60 cases of ovarian cancer, and it is not found in normal ovarian and fallopian tube tissues [59]. WDFY2 is an important regulator of AKT phosphorylation, and thus might represent a promising target in the diagnosis and treatment of ovarian cancer. The MAN2A1-FER fusion in ovarian cancer can significantly enhance tyrosine kinase activity. In vivo experiments have also confirmed that the fusion promotes tumor progression. Therefore, the application of tyrosine kinase inhibitors to MAN2A1-FER fusion-positive cancers might be a valuable choice [42]. In cervical cancer, glioma, and other tumors, FGFR3-TACC3 is a recurrent and carcinogenic fusion [46, 79], and is sensitive to FGFR inhibitors [83]; therefore, FGFR3-TACC3 fusion protein inhibitors have the potential to treat fusion-positive cancers. At present, the targeted drugs used in gynecologic tumors mainly include VEGF inhibitors, EGF inhibitors, and PARP inhibitors. Although these drugs have achieved certain results, the rates of recurrence and metastasis are still high.
Conclusions
A full understanding of the frequency, associated pathways, and carcinogenic mechanisms of fusion genes in gynecologic tumors have great prospects to identify valuable oncogenic fusions and potential targets.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.
References
Hahn Y, Bera TK, Gehlhaus K, Kirsch IR, Pastan IH, Lee B. Finding fusion genes resulting from chromosome rearrangement by analyzing the expressed sequence databases. Proc Natl Acad Sci USA. 2004;101:13257–61.
Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–3.
Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973;16:109–12.
Zech L, Haglund U, Nilsson K, Klein G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. Int J Cancer. 1976;17:47–56.
Berger R, Bernheim A, Weh HJ, Flandrin G, Daniel MT, Brouet JC, et al. A new translocation in Burkitt’s tumor cells. Hum Genet. 1979;53:111–2.
Oshimura M, Freeman AI, Sandberg AA. Chromosomes and causation of human cancer and leukemia. XXVI. Binding studies in acute lymphoblastic leukemia (ALL). Cancer. 1977;40:1161–72.
Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;1:549–50.
Fukuhara S, Rowley JD, Variakojis D, Golomb HM. Chromosome abnormalities in poorly differentiated lymphocytic lymphoma. Cancer Res. 1979;39:3119–28.
McPherson A, Wu C, Hajirasouliha I, Hormozdiari F, Hach F, Lapuk A, et al. Comrad: detection of expressed rearrangements by integrated analysis of RNA-Seq and low coverage genome sequence data. Bioinformatics. 2011;27:1481–8.
Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016;26:108–18.
Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, et al. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018;46:D1144–D1149.
Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–45.
Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2.
de Pagter MS, Kloosterman WP. The diverse effects of complex chromosome rearrangements and chromothripsis in cancer development. Recent Results Cancer Res. 2015;200:165–93.
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40.
Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15:371–81.
Davare MA, Tognon CE. Detecting and targetting oncogenic fusion proteins in the genomic era. Biol Cell. 2015;107:111–29.
Jividen K, Li H. Chimeric RNAs generated by intergenic splicing in normal and cancer cells. Genes Chromosomes Cancer. 2014;53:963–71.
Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012;2:598–607.
Liu S, Tsai WH, Ding Y, Chen R, Fang Z, Huo Z, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016;44:e47.
Efanov AA, Brenner AV, Bogdanova TI, Kelly LM, Liu P, Little MP, et al. Investigation of the relationship between radiation dose and gene mutations and fusions in post-chernobyl thyroid cancer. J Natl Cancer Inst. 2018;110:371–8.
Klugbauer S, Lengfelder E, Demidchik EP, Rabes HM. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene. 1995;11:2459–67.
Smida J, Salassidis K, Hieber L, Zitzelsberger H, Kellerer AM, Demidchik EP, et al. Distinct frequency of ret rearrangements in papillary thyroid carcinomas of children and adults from Belarus. Int J Cancer. 1999;80:32–38.
Su X, Li Z, He C, Chen W, Fu X, Yang A. Radiation exposure, young age, and female gender are associated with high prevalence of RET/PTC1 and RET/PTC3 in papillary thyroid cancer: a meta-analysis. Oncotarget. 2016;7:16716–30.
Leeman-Neill RJ, Kelly LM, Liu P, Brenner AV, Little MP, Bogdanova TI, et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer. 2014;120:799–807.
Dacic S, Luvison A, Evdokimova V, Kelly L, Siegfried JM, Villaruz LC, et al. RET rearrangements in lung adenocarcinoma and radiation. J Thorac Oncol. 2014;9:118–20.
Chen Y, Li G, Lei Y, Yang K, Niu H, Zhao J, et al. Lung cancer family history and exposure to occupational/domestic coal combustion contribute to variations in clinicopathologic features and gene fusion patterns in non-small cell lung cancer. Thorac Cancer. 2019;10:695–707.
Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–90.
Navarrete-Meneses MP, Salas-Labadia C, Sanabrais-Jimenez M, Santana-Hernandez J, Serrano-Cuevas A, Juarez-Velazquez R, et al. Exposure to the insecticides permethrin and malathion induces leukemia and lymphoma-associated gene aberrations in vitro. Toxicol Vitr. 2017;44:17–26.
Egashira S, Jinnin M, Ajino M, Shimozono N, Okamoto S, Tasaki Y, et al. Chronic sun exposure-related fusion oncogenes EGFR-PPARGC1A in cutaneous squamous cell carcinoma. Sci Rep. 2017;7:12654.
Holly JMP, Broadhurst J, Mansor R, Bahl A, Perks CM. Hyperglycemia promotes TMPRSS2-ERG gene fusion in prostate cancer cells via upregulating insulin-like growth factor-binding protein-2. Front Endocrinol. 2017;8:305.
Eguchi-Ishimae M, Eguchi M, Ishii E, Miyazaki S, Ueda K, Kamada N, et al. Breakage and fusion of the TEL (ETV6) gene in immature B lymphocytes induced by apoptogenic signals. Blood. 2001;97:737–43.
Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, Varambally S, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230.
Swanson TA, Krueger SA, Galoforo S, Thibodeau BJ, Martinez AA, Wilson GD, et al. TMPRSS2/ERG fusion gene expression alters chemo- and radio-responsiveness in cell culture models of androgen independent prostate cancer. Prostate. 2011;71:1548–58.
Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83.
Peterson LF, Boyapati A, Ahn EY, Biggs JR, Okumura AJ, Lo MC, et al. Acute myeloid leukemia with the 8q22;21q22 translocation: secondary mutational events and alternative t(8;21) transcripts. Blood. 2007;110:799–805.
Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33.
de Alava E, Gerald WL. Molecular biology of the Ewing’s sarcoma/primitive neuroectodermal tumor family. J Clin Oncol. 2000;18:204–13.
Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85.
Brien GL, Stegmaier K, Armstrong SA. Targeting chromatin complexes in fusion protein-driven malignancies. Nat Rev Cancer. 2019;19:255–69.
Yu YP, Tsung A, Liu S, Nalesnick M, Geller D, Michalopoulos G, et al. Detection of fusion transcripts in the serum samples of patients with hepatocellular carcinoma. Oncotarget. 2019;10:3352–60.
Chen ZH, Yu YP, Tao J, Liu S, Tseng G, Nalesnik M, et al. MAN2A1-FER fusion gene is expressed by human liver and other tumor types and has oncogenic activity in mice. Gastroenterology. 2017;153:1120–32 e1115.
Kron KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, Shiah YJ, et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat Genet. 2017;49:1336–45.
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6.
Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14.
Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Investig. 2013;123:855–65.
De Keersmaecker K, Rocnik JL, Bernad R, Lee BH, Leeman D, Gielen O, et al. Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Mol Cell. 2008;31:134–42.
Vanden Bempt M, Demeyer S, Broux M, De Bie J, Bornschein S, Mentens N, et al. Cooperative enhancer activation by TLX1 and STAT5 drives development of NUP214-ABL1/TLX1-positive T cell acute lymphoblastic leukemia. Cancer Cell. 2018;34:271–85 e277.
Martinez-Soria N, McKenzie L, Draper J, Ptasinska A, Issa H, Potluri S, et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell. 2018;34:626–42 e628.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Wilson MK, Pujade-Lauraine E, Aoki D, Mirza MR, Lorusso D, Oza AM, et al. Fifth ovarian cancer consensus conference of the gynecologic cancer intergroup: recurrent disease. Ann Oncol. 2017;28:727–32.
Agostini A, Brunetti M, Davidson B, Goran Trope C, Heim S, Panagopoulos I, et al. Identification of novel cyclin gene fusion transcripts in endometrioid ovarian carcinomas. Int J Cancer. 2018;143:1379–87.
Zhuo L, Gong J, Yang R, Sheng Y, Zhou L, Kong X, et al. Inhibition of proliferation and differentiation and promotion of apoptosis by cyclin L2 in mouse embryonic carcinoma P19 cells. Biochem Biophys Res Commun. 2009;390:451–7.
Li HL, Wang TS, Li XY, Li N, Huang DZ, Chen Q, et al. Overexpression of cyclin L2 induces apoptosis and cell-cycle arrest in human lung cancer cells. Chin Med J. 2007;120:905–9.
Christie EL, Pattnaik S, Beach J, Copeland A, Rashoo N, Fereday S, et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat Commun. 2019;10:1295.
Gottesman MM, Pastan IH. The role of multidrug resistance efflux pumps in cancer: revisiting a JNCI publication exploring expression of the MDR1 (P-glycoprotein) gene. J Natl Cancer Inst. 2015;107:13.
Shi S, Gu S, Han T, Zhang W, Huang L, Li Z, et al. Inhibition of MAN2A1 enhances the immune response to anti-PD-L1 in human tumors. Clin Cancer Res. 2020;26:5990–6002.
Greer P. Closing in on the biological functions of Fps/Fes and Fer. Nat Rev Mol Cell Biol. 2002;3:278–89.
Kannan K, Coarfa C, Rajapakshe K, Hawkins SM, Matzuk MM, Milosavljevic A, et al. CDKN2D-WDFY2 is a cancer-specific fusion gene recurrent in high-grade serous ovarian carcinoma. PLoS Genet. 2014;10:e1004216.
Walz HA, Shi X, Chouinard M, Bue CA, Navaroli DM, Hayakawa A, et al. Isoform-specific regulation of Akt signaling by the endosomal protein WDFY2. J Biol Chem. 2010;285:14101–8.
Wang J, Chen X, Tong S, Zhou H, Sun J, Gou Y, et al. Overexpression of WDFY2 inhibits prostate cancer cell growth and migration via inactivation of Akt pathway. Tumour Biol. 2017;39:1010428317704821.
De Grandis M, Cambot M, Wautier MP, Cassinat B, Chomienne C, Colin Y, et al. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood. 2013;121:658–65.
Bartolini A, Cardaci S, Lamba S, Oddo D, Marchio C, Cassoni P, et al. BCAM and LAMA5 mediate the recognition between tumor cells and the endothelium in the metastatic spreading of KRAS-Mutant Colorectal Cancer. Clin. Cancer Res. 2016;22:4923–33.
Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–36.
Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–80.
Kannan K, Coarfa C, Chao PW, Luo L, Wang Y, Brinegar AE, et al. Recurrent BCAM-AKT2 fusion gene leads to a constitutively activated AKT2 fusion kinase in high-grade serous ovarian carcinoma. Proc Natl Acad Sci USA. 2015;112:E1272–1277.
Kim SH, Da Cruz Paula A, Basili T, Dopeso H, Bi R, Pareja F, et al. Identification of recurrent FHL2-GLI2 oncogenic fusion in sclerosing stromal tumors of the ovary. Nat Commun. 2020;11:44.
Cai T, Sun D, Duan Y, Qiu Y, Dai C, Yang J, et al. FHL2 promotes tubular epithelial-to-mesenchymal transition through modulating beta-catenin signalling. J Cell Mol Med. 2018;22:1684–95.
Sun L, Yu S, Xu H, Zheng Y, Lin J, Wu M, et al. FHL2 interacts with EGFR to promote glioblastoma growth. Oncogene. 2018;37:1386–98.
Grzelak CA, Sigglekow ND, McCaughan GW. GLI2 as a marker of hedgehog-responsive cells. Hepatology. 2015;61:1770.
Raleigh DR, Choksi PK, Krup AL, Mayer W, Santos N, Reiter JF. Hedgehog signaling drives medulloblastoma growth via CDK6. J Clin Investig. 2018;128:120–4.
Smebye ML, Agostini A, Johannessen B, Thorsen J, Davidson B, Trope CG, et al. Involvement of DPP9 in gene fusions in serous ovarian carcinoma. BMC Cancer. 2017;17:642.
Krzyzanowski PM, Sircoulomb F, Yousif F, Normand J, La Rose J, K EF, et al. Regional perturbation of gene transcription is associated with intrachromosomal rearrangements and gene fusion transcripts in high grade ovarian cancer. Sci Rep. 2019;9:3590.
Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
Tamura R, Yoshihara K, Yamawaki K, Suda K, Ishiguro T, Adachi S, et al. Novel kinase fusion transcripts found in endometrial cancer. Sci Rep. 2015;5:18657.
Li N, Zheng J, Li H, Deng J, Hu M, Wu H, et al. Identification of chimeric TSNAX-DISC1 resulting from intergenic splicing in endometrial carcinoma through high-throughput RNA sequencing. Carcinogenesis. 2014;35:2687–97.
Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer. 2002;2:59–65.
Zou Y, Turashvili G, Soslow RA, Park KJ, Croce S, McCluggage WG, et al. High-grade transformation of low-grade endometrial stromal sarcomas lacking YWHAE and BCOR genetic abnormalities. Mod Pathol. 2020;33:1861–70.
Tamura R, Yoshihara K, Saito T, Ishimura R, Martinez-Ledesma JE, Xin H, et al. Novel therapeutic strategy for cervical cancer harboring FGFR3-TACC3 fusions. Oncogenesis. 2018;7:4.
Daly C, Castanaro C, Zhang W, Zhang Q, Wei Y, Ni M, et al. FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene. 2017;36:471–81.
Lin ZR, Wang MY, He SY, Cai ZM, Huang WR. TACC3 transcriptionally upregulates E2F1 to promote cell growth and confer sensitivity to cisplatin in bladder cancer. Cell Death Dis. 2018;9:72.
Zhou DS, Wang HB, Zhou ZG, Zhang YJ, Zhong Q, Xu L, et al. TACC3 promotes stemness and is a potential therapeutic target in hepatocellular carcinoma. Oncotarget. 2015;6:24163–77.
Frattini V, Pagnotta SM, Tala, Fan JJ, Russo MV, Lee SB, et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. 2018;553:222–7.
Acharya S, Hensley ML, Montag AC, Fleming GF. Rare uterine cancers. Lancet Oncol. 2005;6:961–71.
Dickson BC, Lum A, Swanson D, Bernardini MQ, Colgan TJ, Shaw PA, et al. Novel EPC1 gene fusions in endometrial stromal sarcoma. Genes Chromosomes Cancer. 2018;57:598–603.
Micci F, Panagopoulos I, Bjerkehagen B, Heim S. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res. 2006;66:107–12.
Huang X, Spencer GJ, Lynch JT, Ciceri F, Somerville TD, Somervaille TC. Enhancers of Polycomb EPC1 and EPC2 sustain the oncogenic potential of MLL leukemia stem cells. Leukemia. 2014;28:1081–91.
Wang Y, Alla V, Goody D, Gupta SK, Spitschak A, Wolkenhauer O, et al. Epigenetic factor EPC1 is a master regulator of DNA damage response by interacting with E2F1 to silence death and activate metastasis-related gene signatures. Nucleic Acids Res. 2016;44:117–33.
Wu X, Liu M, Zhu H, Wang J, Dai W, Li J, et al. Ubiquitin-specific protease 3 promotes cell migration and invasion by interacting with and deubiquitinating SUZ12 in gastric cancer. J Exp Clin Cancer Res. 2019;38:277.
Astolfi A, Fiore M, Melchionda F, Indio V, Bertuccio SN, Pession A. BCOR involvement in cancer. Epigenomics. 2019;11:835–55.
Liu R, Gao J, Yang Y, Qiu R, Zheng Y, Huang W, et al. PHD finger protein 1 (PHF1) is a novel reader for histone H4R3 symmetric dimethylation and coordinates with PRMT5-WDR77/CRL4B complex to promote tumorigenesis. Nucleic Acids Res. 2018;46:6608–26.
Panagopoulos I, Micci F, Thorsen J, Gorunova L, Eibak AM, Bjerkehagen B, et al. Novel fusion of MYST/Esa1-associated factor 6 and PHF1 in endometrial stromal sarcoma. PLoS ONE. 2012;7:e39354.
Micci F, Brunetti M, Dal Cin P, Nucci MR, Gorunova L, Heim S, et al. Fusion of the genes BRD8 and PHF1 in endometrial stromal sarcoma. Genes Chromosomes Cancer. 2017;56:841–5.
Brunetti M, Gorunova L, Davidson B, Heim S, Panagopoulos I, Micci F. Identification of an EPC2-PHF1 fusion transcript in low-grade endometrial stromal sarcoma. Oncotarget. 2018;9:19203–8.
Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature. 2017;549:287–91.
Laugesen A, Hojfeldt JW, Helin K. Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb Perspect Med. 2016;6:19.
Croce S, Hostein I, Ribeiro A, Garbay D, Velasco V, Stoeckle E, et al. YWHAE rearrangement identified by FISH and RT-PCR in endometrial stromal sarcomas: genetic and pathological correlations. Mod Pathol. 2013;26:1390–1400.
Lee CH, Ou WB, Marino-Enriquez A, Zhu M, Mayeda M, Wang Y, et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci USA. 2012;109:929–34.
Barbash O, Lee EK, Diehl JA. Phosphorylation-dependent regulation of SCF(Fbx4) dimerization and activity involves a novel component, 14-3-3varepsilon. Oncogene. 2011;30:1995–2002.
Kim J, Parrish AB, Kurokawa M, Matsuura K, Freel CD, Andersen JL, et al. Rsk-mediated phosphorylation and 14-3-3varepsilon binding of Apaf-1 suppresses cytochrome c-induced apoptosis. EMBO J. 2012;31:1279–92.
Hrzenjak A. JAZF1/SUZ12 gene fusion in endometrial stromal sarcomas. Orphanet J Rare Dis. 2016;11:15.
Nakajima T, Fujino S, Nakanishi G, Kim YS, Jetten AM. TIP27: a novel repressor of the nuclear orphan receptor TAK1/TR4. Nucleic Acids Res. 2004;32:4194–204.
Sung Y, Park S, Park SJ, Jeong J, Choi M, Lee J, et al. Jazf1 promotes prostate cancer progression by activating JNK/Slug. Oncotarget. 2018;9:755–65.
Huang L, Cai Y, Luo Y, Xiong D, Hou Z, Lv J, et al. JAZF1 suppresses papillary thyroid carcinoma cell proliferation and facilitates apoptosis via regulating TAK1/NF-kappaB pathways. Oncol Targets Ther. 2019;12:10501–14.
Muller J, Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev. 2009;19:150–8.
Ma X, Wang J, Wang J, Ma CX, Gao X, Patriub V, et al. The JAZF1-SUZ12 fusion protein disrupts PRC2 complexes and impairs chromatin repression during human endometrial stromal tumorogenesis. Oncotarget. 2017;8:4062–78.
Dewaele B, Przybyl J, Quattrone A, Finalet Ferreiro J, Vanspauwen V, Geerdens E, et al. Identification of a novel, recurrent MBTD1-CXorf67 fusion in low-grade endometrial stromal sarcoma. Int J Cancer. 2014;134:1112–22.
Mandato VD, Valli R, Mastrofilippo V, Bisagni A, Aguzzoli L, La Sala GB. Uterine inflammatory myofibroblastic tumor: more common than expected: case report and review. Medicine. 2017;96:e8974.
Mohammad N, Haimes JD, Mishkin S, Kudlow BA, Leong MY, Chew SH, et al. ALK is a specific diagnostic marker for inflammatory myofibroblastic tumor of the uterus. Am J Surg Pathol. 2018;42:1353–9.
Haimes JD, Stewart CJR, Kudlow BA, Culver BP, Meng B, Koay E, et al. Uterine inflammatory myofibroblastic tumors frequently harbor ALK fusions with IGFBP5 and THBS1. Am J Surg Pathol. 2017;41:773–80.
Ducray SP, Natarajan K, Garland GD, Turner SD, Egger G. The transcriptional roles of ALK fusion proteins in tumorigenesis. Cancers. 2019;11:21.
Blake EA, Sheridan TB, Wang KL, Takiuchi T, Kodama M, Sawada K, et al. Clinical characteristics and outcomes of uterine tumors resembling ovarian sex-cord tumors (UTROSCT): a systematic review of literature. Eur J Obstet Gynecol Reprod Biol. 2014;181:163–70.
Lee CH, Kao YC, Lee WR, Hsiao YW, Lu TP, Chu CY, et al. Clinicopathologic characterization of GREB1-rearranged uterine sarcomas with variable sex-cord differentiation. Am J Surg Pathol. 2019;43:928–42.
Cheng M, Michalski S, Kommagani R. Role for growth regulation by estrogen in breast cancer 1 (GREB1) in hormone-dependent cancers. Int J Mol Sci. 2018;19:22.
Wu Y, Zhang Z, Cenciarini ME, Proietti CJ, Amasino M, Hong T, et al. Tamoxifen resistance in breast cancer is regulated by the EZH2-Eralpha-GREB1 transcriptional axis. Cancer Res. 2018;78:671–84.
Blanchard Z, Vahrenkamp JM, Berrett KC, Arnesen S, Gertz J. Estrogen-independent molecular actions of mutant estrogen receptor 1 in endometrial cancer. Genome Res. 2019;29:1429–41.
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51.
Hokland P, Ommen HB. Towards individualized follow-up in adult acute myeloid leukemia in remission. Blood. 2011;117:2577–84.
Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Evidence of recurrent gene fusions in common epithelial tumors. Trends Mol Med. 2006;12:529–36.
Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013;13:772–87.
Acknowledgements
We would like to thank Elixigen (www.elixigen.com) for English language editing.
Funding
This study was supported by National Natural Scientific Foundation of China (NO. 82072854, 81772776).
Ethics
No ethics approvals were required for this paper.
Author information
Authors and Affiliations
Contributions
Guarantor of the article: Y.Z. designed and revised the review. B.L. wrote and revised the manuscript. R.J., B.X., and W.W. revised the contents of the manuscript. B.L. constructed and revised the figures. All authors approved the final manuscript and agreed to be responsible for this review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Stephanou
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, B., Jiang, R., Xie, B. et al. Fusion genes in gynecologic tumors: the occurrence, molecular mechanism and prospect for therapy. Cell Death Dis 12, 783 (2021). https://doi.org/10.1038/s41419-021-04065-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04065-0
