
Abstract
Ferroptosis, a newly defined mode of regulated cell death caused by unbalanced lipid redox metabolism, is implicated in various tissue injuries and tumorigenesis. However, the role of ferroptosis in stem cells has not yet been investigated. Glutathione peroxidase 4 (GPX4) is a critical suppressor of lipid peroxidation and ferroptosis. Here, we study the function of GPX4 and ferroptosis in hematopoietic stem and progenitor cells (HSPCs) in mice with Gpx4 deficiency in the hematopoietic system. We find that Gpx4 deletion solely in the hematopoietic system has no significant effect on the number and function of HSPCs in mice. Notably, hematopoietic stem cells (HSCs) and hematopoietic progenitor cells lacking Gpx4 accumulated lipid peroxidation and underwent ferroptosis in vitro. α-Tocopherol, the main component of vitamin E, was shown to rescue the Gpx4-deficient HSPCs from ferroptosis in vitro. When Gpx4 knockout mice were fed a vitamin E-depleted diet, a reduced number of HSPCs and impaired function of HSCs were found. Furthermore, increased levels of lipid peroxidation and cell death indicated that HSPCs undergo ferroptosis. Collectively, we demonstrate that GPX4 and vitamin E cooperatively maintain lipid redox balance and prevent ferroptosis in HSPCs.
Similar content being viewed by others


Introduction
Continuous replenishment of the hematopoietic system depends on hematopoietic stem cell (HSC) self-renewal and differentiation into progenitors and eventually mature blood cells [1]. Most HSCs remain quiescent under homeostatic conditions but are stimulated to proliferate and differentiate upon encountering intrinsic or extrinsic stresses [2]. Reactive oxygen species (ROS), mainly generated endogenously during mitochondrial oxidative phosphorylation, are involved in the determination of HSC fate. Low levels of ROS are essential to maintain the quiescent status of HSCs, while high levels of ROS abolish quiescence, limit lifespan, and induce cell death [2, 3].
Scavenger networks have been developed to protect cells from excess ROS. Among them, glutathione peroxidase 4 (GPX4), a kind of selenoenzyme, specifically reduces lipid peroxidation by utilizing glutathione (GSH). Indeed, as the major hydroperoxide scavenger, GPX4 is a vital suppressor of ferroptosis [4, 5], a newly defined type of programmed cell death caused by iron-dependent lipid peroxidation accumulation [6, 7]. Failure of the lipid peroxide reducing system caused by GSH depletion or Gpx4 deficiency results in ferroptosis [8], which has been implicated in a plethora of diseases involving multiple tissues and cells. Mice with loss of Gpx4 did not survive during embryonic development [9]. Conditional induction of Gpx4 knockout in adult mice impaired the integrity of various organs, such as the brain and kidney [4, 10]. Cell type-specific Gpx4 depletion led to cell death and dysfunction in the retina, brain, liver, and reproductive tissue [11,12,13,14,15]. Moreover, GPX4 was revealed to be essential in hematopoietic cells including T cells, erythroid precursors, and myeloid cells [16,17,18]. However, the function of GPX4 and the influence of ferroptosis on hematopoietic stem and progenitor cells (HSPCs) remain unclear.
Vitamin E is a well-known lipophilic antioxidant that naturally exists in foods such as plant seeds, vegetables, and eggs. α-Tocopherol (α-Toc), the major component of vitamin E, reduces cell lipid peroxides and prevents ferroptosis [4, 12]. In contrast, hydrophilic antioxidants such as NAC are ineffective against ferroptosis induced by Gpx4 deficiency. Moreover, dietary supplementation with vitamin E alleviates phenotypes resulting from Gpx4 deficiency in endothelial cells, hepatocytes, T cells, myeloid cells, and reticulocytes [15, 16, 18,19,20].
Here we found that Gpx4 depletion in the hematopoietic system alone had no significant effect on the number and function of HSPCs in mice. Intriguingly, HSPCs lacking Gpx4 accumulated lipid peroxidation and dramatically underwent ferroptosis in vitro. Further investigation indicated that Vitamin E/α-Toc is the crucial factor leading to the discrepant fate of HSPCs. Gpx4 knockout mice fed a vitamin E-depleted diet showed a reduced number of HSPCs and impaired function of the hematopoietic system. High levels of total ROS and lipid peroxidation indicated that HSPCs underwent ferroptosis. In summary, our results showed that GPX4 cooperates with vitamin E, maintaining the balance of lipid redox and preventing ferroptosis in HSPCs.
Results
GPX4 deficiency results in HSPCs ferroptosis in vitro
HSCs rapidly proliferate and differentiate to generate single-cell colonies when they are cultured in vitro. For determination of whether GPX4 is essential for HSC function, individual long-term HSCs (LT-HSCs) from wild-type mice were sorted and treated with RSL3, a widely used GPX4 inhibitor. Strikingly, RSL3 completely blocked the colony formation of LT-HSCs (Fig. 1a). To exclude the off-target effects of RSL3, we generated Gpx4flox/flox Vav-Cre mice in which Gpx4 was specifically knocked out in the hematopoietic system and Gpx4flox/flox Mx-Cre mice in which Gpx4 was depleted in the hematopoietic system by pIpC treatment [21]. Then, the LT-HSCs isolated from the Gpx4flox/flox Vav-Cre mice and the pIpC-treated Gpx4flox/flox Mx-Cre mice were evaluated by a single-cell colony-forming assay. Similar to the RSL3-treated LT-HSCs, the Gpx4-depleted (Gpx4−/− LT-HSCs lost the ability to produce single-cell colonies (Fig. 1b, c). Next, we assessed the susceptibility of different types of HSPCs to RSL3. We observed that LT-HSCs and short-term HSCs (ST-HSCs) were more resistant to RSL3 than multipotent progenitors (MPPs) and granulocyte-macrophage progenitors (GMPs) (Supplementary Fig. 1a). Consistently, the expression levels of GPX4 in the LT-HSCs and ST-HSCs were higher than those in the MPPs and GMPs (Supplementary Fig. 1b). These results suggest that GPX4 is indispensable for HSC function in vitro.

a LT-HSC single cells were sorted and cultured in a medium with or without RSL3 for 14 days. Colony numbers were counted (n = 4 mice). Scale bars = 0.5 mm. LT-HSCs derived from the Gpx4flox/flox mice and Gpx4flox/flox Vav-Cre mice (b) or the pIpC-treated Gpx4flox/flox Mx-Cre mice (c) were tested with single-cell colony-forming assay as in (a) (n = 2 Vav-Cre mice or 4 Mx-Cre mice). d The viability of LSK cells isolated from the wild-type mice after 48 h of culture with the indicated drugs (n = 3 mice). e LSK cells isolated from the wild-type mice were cultured with the indicated drugs for 24 h, and lipid ROS were measured with C11-BODIPY and detected by flow cytometry (n = 3 mice). f The viability of LSK cells isolated from the Gpx4flox/flox mice or the Gpx4flox/flox Vav-Cre mice after 48 h of culture with the indicated drugs (n = 3 mice). g LSK cells isolated from the Gpx4flox/flox mice or the Gpx4flox/flox Vav-Cre mice were cultured with the indicated drugs for 24 h, and lipid ROS were measured with C11-BODIPY and detected by flow cytometry (n = 3 mice). Data are the mean ± SD. (ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001 vs. the RSL3 group in (d, e), vs. the Gpx4−/− group in (f, g).
Given that GPX4 is a vital suppressor of ferroptosis, we wondered whether HSPCs undergo ferroptosis in the absence of GPX4. Hence, lineage−sca1+c-kit+ (LSK) cells isolated from the wild-type (Fig. 1d, e) or Gpx4-depleted mice (Fig. 1f, g) were cultured in vitro and treated with different drugs. Both the RSL3-treated LSK cells and the Gpx4-depleted LSK cells accumulated lipid peroxides and underwent cell death within two days (Fig. 1d−g). Moreover, neither Nec-1s nor Z-VAD affected cell viability, excluding the involvement of necroptosis and apoptosis. In contrast, the lipophilic antioxidant ferrostatin 1 (Fer-1) and the iron chelator deferiprone (DFO) reduced lipid peroxide levels and suppressed cell death (Fig. 1d−g). We noticed that although lipid peroxidation was diminished, nearly half of the RSL3-treated LSK cells failed to be rescued by DFO (Fig. 1d). Considering the essential biological function of iron in cells, we speculated that DFO may be toxic to HSPCs as they are highly dependent on iron. Indeed, after treatment with DFO alone, both LSK cells and GMPs died in vitro (Supplementary Fig. 1c, d). These results demonstrate that GPX4 deficiency leads to LSK cell ferroptosis in vitro. Furthermore, a similar scenario was observed in GMPs: both RSL3-treated and Gpx4-depleted GMPs accumulated lipid peroxides and succumbed to ferroptosis (Supplementary Fig. 1e–h). In summary, the above results suggest that HSPCs undergo ferroptosis in vitro when GPX4 is inhibited or depleted.
Gpx4 deletion does not affect homeostasis of the hematopoietic system
It has been reported that GPX4 is indispensable in embryonic development and the integrity of multiple tissues [9,10,11, 13]. To further explore the function of GPX4 in HSPCs in vivo, we studied the phenotype of HSPCs in mice in which Gpx4 was knocked out in the hematopoietic system. Gpx4 depletion in the hematopoietic system of the Gpx4flox/flox Vav-Cre mice was confirmed by Western blots (Fig. 2a). Surprisingly, no significant difference was observed between the Gpx4flox/flox Vav-Cre mice (Gpx4−/−) and the Gpx4flox/flox mice (Gpx4f/f) in the numbers of total bone marrow (BM) cells (Fig. 2b) and HSPCs (Fig. 2c, d) including LT-HSCs, ST-HSCs, MPPs, LSK cells, GMPs, and common lymphoid progenitors (CLPs). Moreover, Gpx4 ablation had no significant impact on either the ROS levels of LT-HSCs and lineage−c-kit+ (LK) cells or the lipid ROS levels of BM cells (Fig. 2e, f). Thus, the HSPC phenotype in the Gpx4flox/flox Vav-Cre mice at homeostasis was not affected by Gpx4 depletion. Given that Gpx4 deletion is detrimental in numerous tissues and Gpx4 was deleted at the embryonic stage in the Gpx4flox/flox Vav-Cre mice, it is possible that some compensatory mechanism conferred HSPC resistance to the Gpx4 deficiency. To rule out this possibility, we constructed Gpx4flox/flox Mx-Cre mice, in which Gpx4 deletion in the hematopoietic system was induced by pIpC treatment (Supplementary Fig. 2a). Similar to that in the Gpx4flox/flox Vav-Cre mice, the HSPC phenotype was not impaired in the Gpx4flox/flox Mx-Cre mice treated with pIpC (Supplementary Fig. 2b–g). These results demonstrate that Gpx4 deficiency does not affect homeostasis of the hematopoietic system in mice.

a Gpx4 deletion in the blood cells of the Gpx4flox/flox Vav-Cre was verified by Western blots. b The BM cell numbers were counted. c, d The numbers of HSPCs were measured by flow cytometry. Left panels: representative flow cytometric plots. Right panels: statistical data. e The relative MFI of ROS in LK cells, LSK cells, and LT-HSCs (CD34−Flt3−LSK cells). f The relative MFI of lipid peroxidation in BM cells was detected by C11-BODIPY. N = at least five mice in each group. Data are the mean ± SD. (ns not significant, **P < 0.01).
Gpx4 deficiency does not affect HSC function in vivo
During homeostasis, most HSCs remain quiescent, with low metabolic activity and low ROS levels [2]. We wondered whether GPX4 is essential for HSCs under stress in vivo. The Gpx4-deleted mice were treated with 5-fluorouracil (5-FU) to remove cycling HSCs, which are susceptible to 5-FU. The remaining quiescent HSCs then proliferated and differentiated to restore the hematopoietic system. Nine days post 5-FU treatment, we found no significant difference in the number of HSPCs in the Gpx4-deleted mice compared with the wild-type mice (Fig. 3a). Neither the levels of ROS nor the proliferation rates of LSK cells and LT-HSCs were affected by Gpx4 depletion (Fig. 3b, c). Similar results were found in mice 22 days post 5-FU treatment (Supplementary Fig. 3a, b). These results suggest that the hematopoietic system recovered normally in the Gpx4-deleted mice.

a The number of HSPCs, b the relative MFI of ROS in LSK cells and LT-HSCs, and c the cell cycle of LSK cells and LT-HSCs from the pIpC-treated Gpx4flox/flox Mx-Cre mice 7 days after 5-FU treatment were measured. b The experimental schematic for serial competitive transplantation with LT-HSCs. e The chimerism of peripheral blood cells, f the chimerism of HSPCs at the 4th month after primary transplantation, g the chimerism of HSPCs at the 6th months after secondary transplantation in the recipient mice receiving LT-HSCs from the Gpx4flox/flox Vav-Cre mice. h The chimerism of peripheral blood cells, i the chimerism of HSPCs at the 7th month after primary transplantation, j the chimerism of HSPCs at the 4th month after secondary transplantation receiving LT-HSCs from the Gpx4flox/flox Mx-Cre mice. N = at least three recipient mice in each group in (e−j). Data are the mean ± SD.
To further investigate the function of HSCs in vivo, we conducted competitive HSC transplantation to evaluate the long-term rebuilding capacity of HSCs in the Gpx4-depleted mice. LT-HSCs obtained from the Gpx4f/f mice or Gpx4−/− mice (CD45.2) together with BM cells obtained from competitor mice (CD45.1) were transplanted into recipient mice (CD45.1.2), whose hematopoietic system was destroyed by a lethal dose of X-ray irradiation (Fig. 3d). Once transplanted, donor-derived HSCs home to bone marrow and replenish the hematopoietic system in recipient mice. However, no obvious difference in the chimerism rates of peripheral blood cells within four months (Fig. 3e) was observed even though the T cell chimerism rates were lower in the mice that received Gpx4-depleted HSCs (Supplementary Fig. 3c). Furthermore, the chimerism rates of various HSPCs especially CLPs in the primary recipient at the fourth month were not affected by Gpx4 depletion (Fig. 3f), indicating that the difference in T cell chimerism rates was not due to the difference in HSC differentiation. Indeed, it has been reported that Gpx4-deficient T cells have intrinsic defects in homeostatic balance maintenance and cell expansion upon stress [16]. After secondary transplantation, there was still no significant difference in the chimerism rates of the HSPCs between the recipients of the Gpx4-deficient and Gpx4-sufficient LT-HSCs (Fig. 3g). Likewise, transplantation with LT-HSCs derived from the pIpC-treated Gpx4flox/flox Mx-Cre mice resulted in no significant change in the chimerism rates of the Gpx4-deficient HSPCs during either primary or secondary transplantation (Fig. 3h−j). Thus, our results show that Gpx4 deficiency does not affect HSC self-renewal and differentiation in vivo, which is quite distinct from the scenario in vitro.
α-Toc rescues HSPCs from ferroptosis ex vivo
What leads to the entirely different destiny of the Gpx4-deficient HSPCs in vivo and ex vivo? The levels of oxygen are much higher in the culture medium than in the bone marrow niche, which may inflict more ROS stress on HSPCs [22]. We then tested whether ferroptosis is mediated by higher oxygen levels in HSPCs in vitro. The results revealed that RSL3-induced ferroptosis in both LSK cells and GMPs, was only partially relieved when they were cultured at low oxygen levels. In addition, the oxygen levels did not affect the viability of either the Gpx4-deleted LSK cells or the Gpx4-deleted GMPs (Supplementary Fig. 4a, b). Consistently, the lipid ROS levels of HSPCs were not reduced under low levels of oxygen (Supplementary Fig. 4c, d). These results suggest that oxygen levels are not the main reason for ferroptosis in the Gpx4-deficient HSPCs.
It has been reported that dietary vitamin E alleviates phenotypes resulting from lipid peroxidation in T cells and reticulocytes [16, 17]. α-Toc, the prominent component of vitamin E, is a lipophilic antioxidant and has been proven to reduce lipid peroxidation and block ferroptosis in vitro [4, 12] (Fig. 4a). We suspected that α-Toc can inhibit HSPC ferroptosis in vitro. Certainly, we observed that α-Toc, instead of N-acetyl-L-cysteine (NAC), a hydrophilic antioxidant, significantly reduced lipid ROS and inhibited ferroptosis in both LSK cells and GMPs (Fig. 4b−e). Furthermore, α-Toc endowed Gpx4-deficient LT-HSCs with the ability to generate colonies (Fig. 4f). Thus, α-Toc protects HSPCs from ferroptosis ex vivo.

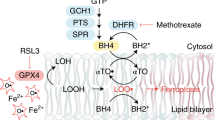
a Schematic illustration of the ferroptosis pathway and the targets of different drugs. Ferroptosis was triggered by iron-dependent accumulation of lipid peroxidation. GPX4 makes use of GSH to reduce lipid peroxidation and inhibit ferroptosis. Ferroptosis can be induced by the GPX4 inhibitor RSL3 and inhibited by the iron chelator DFO or lipophilic antioxidants such as Fer-1 and α-Toc. N-Acetyl-L-cysteine (NAC) is the precursor of cysteine, which promotes GSH synthesis. LSK cells (b, c) or GMPs (d, e) from the Gpx4flox/flox Vav-Cre mice were cultured with NAC or α-Toc. The viability and lipid ROS levels of these cells were measured (n = 3 mice). f LT-HSCs derived from the Gpx4flox/flox Vav-Cre mice were tested with single-cell colony-forming assay in a medium containing NAC or α-Toc (n = 3 mice). # indicates no colony was formed. Data are the mean ± SD. (ns not significant, ***P < 0.001).
Deficiency of both GPX4 and vitamin E induces HSPC ferroptosis and impairs homeostasis of the hematopoietic system in vivo
Vitamin E is absent in the cell culturing medium but is abundantly supplied from experimental animal food. Given that α-Toc rescues HSPCs in vitro, we hypothesized that dietary vitamin E is critical to protect the Gpx4-deficient HSPCs from ferroptosis in vivo. In previous experiments, mice were fed natural ingredient diets containing ≥120 IU/kg vitamin E. To verify our hypothesis, we then fed mice a fixed formulation diet (containing 75 IU/kg vitamin E) or a vitamin E-depleted diet for 3 weeks. Strikingly, we found that combined depletion of Gpx4 and vitamin E led to the loss of body weight, splenomegaly, and reduced BM cell number (Fig. 5a−c). Moreover, in peripheral blood, the proportion of both T cells and B cells decreased significantly while the proportion of M cells increased (Fig. 5d). More importantly, the numbers of LT-HSCs, CMPs, GMPs, and CLPs decreased significantly upon the removal of Gpx4 and vitamin E together (Fig. 5e). However, Gpx4 deletion alone did not impair the HSPC phenotype, consistent with previous results (Fig. 5a−e). Similarly, the depletion of vitamin E alone did not affect the HSPC phenotype (Fig. 5a−e). These data suggest that vitamin E and GPX4 cooperate to maintain hematopoietic system homeostasis.

a The body weight, b spleen weight, and c total BM cell counts of the Gpx4flox/flox Vav-Cre mice were measured after 3 weeks of a VE-depleted diet. d Left panel: representative flow cytometric plots of the T cell, B cell, and myeloid cell groups in peripheral blood. Right panel: statistical data. e The number of HSPCs in the Gpx4flox/flox Vav-Cre mice after 3 weeks of a VE-depleted diet. N = at least four mice in each group. Data are the mean ± SD. (*P < 0.05, **P < 0.01, ***P < 0.001).
To determine, how combined depletion of Gpx4 and vitamin E impaired hematopoietic system homeostasis, we first detected the cell cycle of HSPCs. In the absence of Gpx4 and vitamin E, no significant change in the percentage of LT-HSCs and LSK cells in G0 phase was found (Fig. 6a), indicating that the proliferation of these cells was not blocked. In contrast, significant death of HSPCs was found in the mice deficient in both Gpx4 and vitamin E (Fig. 6b), which was responsible for the decreased number of these HSPCs. Next, we observed enhanced lipid ROS levels in c-kit+ cells (Fig. 6c). Moreover, the ROS levels dramatically increased in LK cells, LSK cells, and LT-HSCs of the Gpx4-deficient mice fed vitamin E depleted chow (Fig. 6d). Taken together, our results reveal that GPX4 and vitamin E cooperatively protect HSPCs from ferroptosis in vivo.

a The cell cycle of LK cells, LSK cells, and LT-HSCs in the Gpx4flox/flox Vav-Cre mice after 3 weeks of a VE-depleted diet. b The HSPCs in the Gpx4flox/flox Vav-Cre mice after 3 weeks of a VE-depleted diet were determined to be DAPI negative. c Left panel: representative flow cytometric plots of lipid ROS in c-kit+ cells of the Gpx4flox/flox Vav-Cre mice after 3 weeks of a VE-depleted diet. Right panel: statistical data of the relative MFI of lipid ROS. d Left panel: representative flow cytometric plots of ROS in LK cells, LSK cells, and LT-HSCs of the Gpx4flox/flox Vav-Cre mice after 3 weeks of a VE-depleted diet. Right panel: statistical data of the relative MFI of ROS. N = at least three mice in each group. Data are the mean ± SD. (ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001).
Discussion
Exponential progress has been made in the field of ferroptosis in recent years in identifying ferroptosis regulators, unveiling their pathological function, and developing drugs or therapies [23]. The relevance of ferroptosis to diseases related to multiple tissues has been well documented [24]. However, previous reports have mainly focused on differentiated cells, leaving the role of ferroptosis in stem cells poorly understood. Here, we focused on HSPCs, which are the most widely studied adult stem cells. We showed that GPX4 deficiency in HSPCs caused by inhibitors or genetic deletion leads to lipid peroxidation and ferroptosis in vitro. In contrast, dietary vitamin E is sufficient to protect HSPCs from ferroptosis and prevent hematopoietic system impairment in the Gpx4-depleted mice. Our work identified ferroptosis in HSPCs for the first time and demonstrates the cooperative effects of Gpx4 and vitamin E on the maintenance of HSPC function and the homeostasis of the hematopoietic system.
As a critical lipid peroxidation scavenger in cells, GPX4 is a key regulator of ferroptosis and is essential to the functional integrity of various tissues [25]. Previous studies reported that Gpx4 is involved in erythropoiesis and that Gpx4 deficiency in the hematopoietic system results in RIP3-dependent necroptosis of erythroid precursors [17]. Another report showed that myeloid-specific deficiency of Gpx4 drove pyroptosis during polymicrobial sepsis [18]. In contrast, our work mainly focuses on HSPCs, the top of the hematopoietic pyramid, and we found that Gpx4-deficient HSPCs tend to undergo ferroptosis. These diverse outcomes of Gpx4 deficiency in different kinds of hematopoietic cells indicate diverse regulation of lipid peroxidation and cell death types.
Long-term ex vivo expansion of HSCs has long been a critical challenge for HSC transplantation, which is a curative therapy for numerous blood disorders [26, 27]. Here we found GPX4 is essential for HSC survival ex vivo, implicating the importance of lipid peroxidation scavenging. Therefore, whether lipophilic antioxidants such as a-Toc may improve the efficiency of HSC expansion ex vivo is worth future study.
Vitamin E is well known as a hydrophobic antioxidant and an essential nutrient. Vitamin E deficiency is associated with neuron abnormalities, ataxia, and hemolytic anemia [28]. Dietary supplementation with vitamin E ameliorates membrane fragility of red blood cells and thus prevents hemolysis [29]. More importantly, it has been previously reported that vitamin E can compensate for GPX4 deficiency in some tissues including endothelial cells, hepatocytes, T cells, myeloid cells, and reticulocytes [15, 16, 18,19,20]. Similarly, our results highlight the importance of dietary vitamin E in protecting HSPCs from lipid peroxidation and ferroptosis in addition to the GPX4/GSH system. Given that vitamin E is an effective ferroptosis inhibitor supplied from normal animal chow, caution needs to be taken to evaluate the influence of vitamin E on the phenotypes related to lipid peroxidation and ferroptosis.
The potential physiological application of ferroptosis has been highlighted in recent years. Ferroptosis inhibitors including vitamin E, have the potential to alleviate diseases resulting from ferroptosis such as ischemic perfusion injury, neurodegeneration, and acute renal injury. Recently, ferroptosis was reported to be involved in radiation-induced cell death [30,31,32]. This finding may partially explain the radioprotective function of vitamin E and selenium in the hematopoietic system [33, 34]. Nevertheless, it has also been reported that vitamin E may promote tumor metastasis [35] and increase the risk of cancer [36]. Indeed, ferroptosis is considered cancer-suppressive. Many types of cancer especially therapy-resistant cancer cells are sensitive to GPX4 deficiency[37, 38]. The development of cancer therapies based on the induction of ferroptosis alone or combined with other therapeutic approaches has shown promise [39]. Thus, on one side, there may be a potential dosing window to kill malignant hematopoietic cells by targeting GPX4, leaving healthy HSPCs intact. On the other side, it needs to be elucidated whether tumor cells or malignant hematopoietic cells can hijack vitamin E against ferroptosis-inducing therapy.
Materials and methods
Mice
C57BL/6 WT mice were purchased from Beijing HFK BioScience Company (Beijing, China). Gpx4flox/flox mice (CD45.2) were a kind gift from Professor Fudi Wang. Gpx4flox/flox mice were crossed with Vav-Cre mice and Mx-Cre mice to generate the Gpx4flox/flox Vav-Cre mice and Gpx4flox/flox Mx-Cre mice, respectively. For Gpx4 deletion, Gpx4flox/flox Mx-Cre mice were intraperitoneally injected with 20 mg/kg pIpC (Sigma) every other day for two weeks. CD45.1/45.2 mice and CD45.1 mice on a C57BL/6 background were used as competitor and recipient mice, respectively, in the competitive transplantation assay. Mice were fed natural ingredient diets containing ≥120 IU/kg vitamin E. A fixed formulation diet with or without 75 IU/kg vitamin E (Beijing HFK BioScience Company, Beijing, China) was fed to the mice involved in the vitamin E depletion experiments. For 5-FU treatment, mice were intraperitoneally injected with 150 mg/kg 5-FU (Sigma). The hematopoietic phenotype was analyzed 9 and 22 d after treatment. Two to four-month age mice of both sexes were utilized, and all experimental protocols were approved by the Animal Care and Ethics Committee of Jinan University.
Flow cytometry and cell sorting
For flow cytometric analysis, cells were stained and labeled with fluorophore-conjugated antibodies purchased from BD Bioscience, Invitrogen, and Biolegend. All the antibody clones and fluorescent labels for FACS and flow cytometry are listed in Supplementary Table 1. After staining, red blood cells were eliminated by lysis buffer (BD Biosciences) and analyzed on a BD Fortessa system. For FACS, c-kit+ cells were separated from BM cells with anti-APC microbeads (Miltenyi) and then stained with antibodies for surface markers. HSPCs were then sorted by BD Arial III. The data were analyzed using FlowJo software.
Cell culture
For in vitro experiments, HSPCs were cultured in DMEM/F12 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 20% BIT9500 (StemCell), 2 mM L-glutamine (Gibco), penicillin/streptomycin, 5 × 10–5 M β-ME, 10 ng/ml stem cell factor (PeproTech), 10 ng/ml thrombopoietin (PeproTech), and 15 ng/ml interleukin-3 (PeproTech). For the single-cell colony-forming assay, single LT-HSCs were sorted in 96-well plates and cultured for 14 days in the medium described above. The numbers and sizes of colonies were assessed under microscopy.
HSC transplantation
For the HSC primary transplantation assay, 400 LT-HSCs from donor mice were injected via the orbital route into irradiated (8 Gy X-ray) recipient mice together with 5 × 105 BM cells from competitor mice. For the second transplantation, 1 × 106 BM cells from primary recipient mice were transplanted into irradiated (8 Gy X-ray) secondary recipient mice.
Measurement of ROS
After surface marker staining, BM cells were stained with 20 μM DCFDA following the manual of the DCFDA/H2DCFDA-Cellular ROS Assay Kit (Abcam) at 37 °C in the dark for 30 min. Cells were then washed and analyzed by flow cytometry.
Measurement of lipid ROS
The cells to be tested were incubated with 5 μM C11-BODIPY (Life Technology) at 37 °C for 30 min, washed, and detected by flow cytometry. The levels of lipid ROS were measured in the FITC channel.
Analysis of cell cycle
For cell cycle analysis, cells were fixed by incubating with 120 μl of BD Cytofix/Cytoperm buffer at RT for 30 min in the dark. The cells were then washed with BD Perm/Wash Buffer and permeabilized with 0.4% Triton for 2–3 min. After washing, the cells were stained with Ki67 monoclonal antibody for 40 min at RT in the dark. Finally, the cells were stained with 1 mg/ml DAPI before flow cytometry.
Q-PCR
Total RNA was purified from HSPCs using the RNAqueous®-Micro Kit (Life Technology) and then reverse-transcribed using PrimeScript™ RT Master Mix (TaKaRa). Real-time PCR was performed on a QuantStudio 6 Flex system (Applied Biosystems). The amount of target mRNA was normalized to that of β-actin mRNA. The gene expression quantities were determined according to the relative Ct.
Western blot
For Western blot, BM cells were lysed in RIPA buffer. Then, the proteins were run on standard 12% SDS-PAGE gels, transferred onto PVDF membranes (Bio-Rad), and blocked with TBST containing 5% milk for 1 h at RT. The membranes were then incubated with primary antibodies overnight at 4 °C, washed with TBST, incubated with secondary antibodies for 1 h at RT, and detected on an Amersham Imager 600 System (GE). Primary antibodies against GPX4 (Abcam, ab125066) and GAPDH (CST, 5174) and secondary antibodies against rabbit IgG (CST, 7074) and mouse IgG (CST, 7076) were used. The data were analyzed using ImageJ.
Statistics
The sample sizes were described in the figure legend and were determined as at least three biologically independent animals according to previous studies performed by our group. No exclusions of data were made that would significantly impact the results or conclusions. Animals with the same genotype and gender and similar age were randomly assigned to experimental groups. Investigators were not blinded during the group allocation during the experiment. Analysis of flow cytometric data was performed using FlowJo version 10. GraphPad Prism 8 was used for statistical analysis. The results are shown as the mean ± SD. For data that were fitted with a Gaussian distribution, unpaired Student’s two-tailed t-test was used to determine the statistical significance. For data that were not fitted with a Gaussian distribution, the Mann−Whitney test was used. (ns not significant, * P < 0.05, **P < 0.01, ***P < 0.001.)
References
Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008;132:631–44.
Chandel NS, Jasper H, Ho TT, Passegue E. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol. 2016;18:823–32.
Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development 2014;141:4206–18.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317–31.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017;171:273–85.
Stockwell BR, Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. 2020;27:365–75.
Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502.
Yoo SE, Chen L, Na R, Liu Y, Rios C, Van Remmen H, et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820–7.
Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17.
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
Ueta T, Inoue T, Furukawa T, Tamaki Y, Nakagawa Y, Imai H, et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J Biol Chem. 2012;287:7675–82.
Imai H, Hakkaku N, Iwamoto R, Suzuki J, Suzuki T, Tajima Y, et al. Depletion of selenoprotein GPx4 in spermatocytes causes male infertility in mice. J Biol Chem. 2009;284:32522–32.
Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–68.
Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016;127:139–48.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97–108 e4.
Wortmann M, Schneider M, Pircher J, Hellfritsch J, Aichler M, Vegi N, et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circulation Res. 2013;113:408–17.
Altamura S, Vegi NM, Hoppe PS, Schroeder T, Aichler M, Walch A, et al. Glutathione peroxidase 4 and vitamin E control reticulocyte maturation, stress erythropoiesis and iron homeostasis. Haematologica 2020;105:937–50.
Joseph C, Quach JM, Walkley CR, Lane SW, Lo Celso C, Purton LE. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell. 2013;13:520–33.
Kobayashi H, Morikawa T, Okinaga A, Hamano F, Hashidate-Yoshida T, Watanuki S, et al. Environmental optimization enables maintenance of quiescent hematopoietic stem cells ex vivo. Cell Rep. 2019;28:145–58 e9.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology, and role in disease. Nat Rev Mol Cell Biol. 2021;22:266−282.
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49.
Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52.
Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019;571:117–21.
Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354:1813–26.
Zingg JM. Vitamin E: a role in signal transduction. Annu Rev Nutr. 2015;35:135–73.
Wang F, Wang T, Lai J, Li M, Zou C. Vitamin E inhibits hemolysis induced by hemin as a membrane stabilizer. Biochem Pharmacol. 2006;71:799–805.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15:469–84.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–62.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9:1673–85.
Weiss JF, Srinivasan V, Kumar KS, Landauer MR. Radioprotection by metals: selenium. Adv Space Res: Off J Comm Space Res. 1992;12:223–31.
Srinivasan V, Weiss JF. Radioprotection by vitamin E: injectable vitamin E administered alone or with WR-3689 enhances survival of irradiated mice. Int J Radiat Oncol, Biol, Phys. 1992;23:841–5.
Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 2019;178:330–45 e22.
Yang CS, Suh N, Kong AN. Does vitamin E prevent or promote cancer? Cancer Prev Res. 2012;5:701–5.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017;551:247–50.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017;547:453–7.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280−296.
Acknowledgements
We thank Mr. Hanchuan Mou (Jinan University) and Dr. Xuexian Fang (Hangzhou Normal University) for providing technical assistance.
Funding
This work was supported by the National Key R&D Program of China [2016YFA0100602, 2017YFA0103302], the National Natural Science Foundation of China [32000602, 92049304, 91749203, 82030039], the Guangdong Basic and Applied Basic Research Foundation [2020A1515110479, 2020A1515011522], the China Postdoctoral Science Foundation [2019M663370], the Program for Guangdong Introducing Innovative and Entrepreneurial Teams [2017ZT07S347], Innovative Team Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory [2018GZR110103002], the Open Project of the State Key Laboratory of Trauma, Burn and Combined Injury, Third Military Medical University [SKLKF202002].
Author information
Authors and Affiliations
Contributions
ZJ oversaw the project. QH, FW, and ZJ initiated the study and developed the concept of the paper. QH, YZ, HL, ZO, JL, and WD performed the experiments. HW, JM, and FW provided Gpx4flox/flox mice. QH, YZ, HL, YG, FW, and ZJ analyzed and interpreted the data. QH, YZ, and ZJ wrote the paper, with help from ZO, YG, and FW.
Corresponding authors
Ethics declarations
Ethics
No human participants, human data, or human tissue is involved in this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Agostini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, Q., Zhang, Y., Lou, H. et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis 12, 706 (2021). https://doi.org/10.1038/s41419-021-04008-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04008-9
This article is cited by
-
Mitochondrial regulation of GPX4 inhibition–mediated ferroptosis in acute myeloid leukemia
Leukemia (2024)
-
Ferritin-mediated mitochondrial iron homeostasis is essential for the survival of hematopoietic stem cells and leukemic stem cells
Leukemia (2024)
-
Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies
Signal Transduction and Targeted Therapy (2024)
-
Ferroptosis: potential targets and emerging roles in pancreatic diseases
Archives of Toxicology (2024)
-
Deciphering Ferroptosis: From Molecular Pathways to Machine Learning-Guided Therapeutic Innovation
Molecular Biotechnology (2024)
