
Abstract
Autophagy, a well-observed intracellular lysosomal degradation process, is particularly important to the cell viability in diabetic cardiomyopathy (DCM). Peroxidasin (PXDN) is a heme-containing peroxidase that augments oxidative stress and plays an essential role in cardiovascular diseases, while whether PXDN contributes to the pathogenesis of DCM remains unknown. Here we reported the suppression of cell viability and autophagic flux, as shown by autophagosomes accumulation and increased expression level of LC3-II and p62 in cultured H9C2 and human AC16 cells that treated with 400 μM palmitate acid (PA) for 24 h. Simultaneously, PXDN protein level increased. Moreover, cell death, autophagosomes accumulation as well as increased p62 expression were suppressed by PXDN silence. In addition, knockdown of PXDN reversed PA-induced downregulated forkhead box-1 (FoxO1) and reduced FoxO1 phosphorylation, whereas did not affect AKT phosphorylation. Not consistent with the effects of si-PXDN, double-silence of PXDN and FoxO1 significantly increased cell death, suppressed autophagic flux and declined the level of FoxO1 and PXDN, while the expression of LC3-II was unchanged under PA stimulation. Furthermore, inhibition of FoxO1 in PA-untreated cells induced cell death, inhibited autophagic flux, and inhibited FoxO1 and PXDN expression. Thus, we come to conclusion that PXDN plays a key role in PA-induced cell death by impairing autophagic flux through inhibiting FoxO1, and FoxO1 may also affect the expression of PXDN. These findings may develop better understanding of potential mechanisms regarding autophagy in insulin-resistant cardiomyocytes.
Similar content being viewed by others


Introduction
Diabetic cardiomyopathy (DCM), promoted by insulin resistance, hyperlipidema, and hyperglycaemia1, is described as the existence of abnormal myocardial structure and function independent of recognized cardiac risk factors in individuals with diabetes2. As the progression of DCM, diastolic dysfunction often develops into systolic dysfunction, heart failure, and eventually cardiac death3, which accounts for significant morbidity and mortality in developed countries4. Multiple mechanisms are involved in the pathogenesis of DCM, among which recent researches have highlighted the importance of abnormal autophagy and subsequent cardiomyocyte death5.
Autophagy refers to a cellular protective mechanism that removes damaged proteins and organelles via lysosomal degradation pathways6. Impaired autophagy is associated with cardiomyocyte death and plays a key role in DCM7,8. In OVE26 diabetes mice, decreased AMPK activity and subsequent reduction of autophagy were correlated with cardiomyocyte death and cardiac dysfunction9. Drugs such as resveratrol, trehalose, curcumin, and liraglutide are demonstrated to reduce cell death by promoting autophagy in insulin-resistant cardiomyocytes and diabetic animals via various pathways10,11,12,13, Among which forkhead box-1 (FoxO1), a transcriptional factor, has been recognized as a key regulator. It can promote autophagy by stimulating autophagy-related genes and accelerate lysosomal proteolysis via rab7, MURF1, etc.14,15.
Peroxidasin (PXDN), also named vascular peroxidase 1 (VPO1), is a member of the heme-containing peroxidase family which is highly expressed in cardiovascular tissue and plays an important role in cardiovascular diseases16,17,18. Liu et al.19 found that PXDN was significantly increased in the aorta of type 2 diabetes rats. Previous studies also revealed that PXDN is capable to induce cell death via promoting apoptosis20 and programmed necrosis21 in endothelial cells. However, whether PXDN is involved in cardiomyocyte death in the diabetic heart has yet not been investigated.
In this study, we firstly investigated the role of PXDN in insulin-resistant H9C2 and AC16 cells, and our results suggest that PXDN inhibition may improve autophagic flux to reduce cell death in insulin-resistant cardiomyocytes. Moreover, PXDN was found to suppress the expression and dephosphorylation of FoxO1 under PA treatment while downregulaton of FoxO1 in turn inhibited PXDN expression. These findings may develop better understanding of potential mechanisms regarding autophagy in insulin-resistant cardiomyocytes.
Results
PXDN expression increases in the progress of cell death induced by PA
To explore whether the expression of PXDN was affected in the progress of fatty acid-induced cell death, H9C2 was treated with 0, 200 μM, or 400 μM of PA for 0, 12, and 24 h, respectively. We found that PXDN increased significantly after treated with PA (400 μM) for 24 h (Fig. 1A and B). Increased cell death was also observed via cell viability analyses after treatment with PA (400 μM) for 24 h (Fig. 1C and D) in H9C2. Thus, 400 μM palmitate for 24 h was used for subsequent experiments. In addition, insulin resistance was induced in cultured H9C2 cells, as shown by a less glucose consumption at 12 and 24 h after 200 or 400 μM PA treatment compared with the control group (Fig. 1E).

A H9C2 cells were treated with different concentrations of PA (0, 200, and 400 μM) for 24 h. PXDN protein level was measured by western blot and quantitative analysis was shown on the right side (n = 3). B H9C2 cells were treated with 400 μM PA for different duration (0, 12, and 24 h). PXDN protein level was measured and quantitative analysis was shown on the right side (n = 3). C H9C2 cells were treated with different concentrations of PA for 24 h; cell death rate was measured by LIVE/DEAD viability kit. Live cells were stained by vital dyes calcein acetoxymethyl ester (Calcein-AM) and exhibited green fluorescence, while dead cells were stained by ethidium homodimer-1 (EthD-1) and exhibited red fluorescence. The percentage of cell death was shown on the right side (scale bar = 50 μM, n = 4). D Cells were treated with 400 μM PA for different duration; the cell death rate was measured by LIVE/DEAD viability kit. The percentage of cell death was shown on the right side (scale bar = 50 μM, n = 4). E Cells were treated with different concentrations of PA for different duration, and glucose consumption was determined (n = 3). Data were presented as mean ± SEM. One-way ANOVA test was used. *P < 0.05, **P < 0.01, ***P < 0.001.
PXDN silence improves impaired autophagic flux and reduces subsequent cell death induced by PA
Next, we explored whether PXDN was necessary for cell death stimulated by PA. siRNA was used to achieve PXDN inhibition in H9C2 cells before treated with PA. It was shown in Fig. 2D that PXDN is significantly knockdown by si-PXDN in H9C2 cells whether in control or PA-treated groups. Meanwhile, si-PXDN significantly reduced cell death under PA treatment (Fig. 2A). Transmission electron microscopy (TEM) was used to investigate whether autophagic flux was involved in PXDN-mediated cell death subsequently. A remarkable increase of both autophagosomes and autolysosomes was observed in PA group, while si-PXDN significantly decreased the number of autophagosomes (Fig. 2C). To further confirm whether PXDN impaired autophagosome turnover, DC661 was used to block autophagosome turnover by inhibiting lysosomal acidification. Immunoblotting showed that PA significantly increased the protein expression level of LC3II and p62. si-PXDN didn’t change LC3II and p62 levels in normal conditions, but significantly decreased the accumulation of p62 without affecting LC3II expression after PA. Treatment with DC661 increased the protein expression level of LC3II and p62 in the vehicle and si-PXDN group, but failed to further enhance LC3II and p62 expression level in the PA group. In addition, DC661 reversed the effect of si-PXDN on p62 after PA (Fig. 2D). Similar to H9C2 cells, the silence of PXDN significantly reduced cell death and decreased the accumulation of p62 under PA treatment in AC16 human cells (Fig. 2B and Fig. S1A).

A H9C2 cells were treated with 50 μM si-negative control (NC) or si-PXDN in serum-free medium for 24 h, and treated with 400 μM palmitate acid (PA) or bovine serum albumin (BSA) for another 24 h. Cell survival was evaluated using fluorescence staining with calcein acetoxymethyl ester (Calcein-AM) for live cells in green and ethidium homodimer-1 (EthD-1) for dead cells in red. The percentage of cell death was shown in the right side (scale bar = 50 μM, n = 4). B AC16 cells were treated with si-NC or si-PXDN in serum-free medium for 24 h, and treated with PA or BSA for another 24 h. Cell survival was evaluated using fluorescence staining. The percentage of cell death was shown in the right side (scale bar = 50 μM, n = 4). C Autophagosomes and autolysosomes in H9C2 with si-NC or si-PXDN in the absence or presence of PA were detected by transmission electron microscopy. Black Arrow referred to autophagosomes and white arrow referred to autolysosomes. The number of autophagosomes and autolysosomes per cell was shown in the right side (scale bar = 20 μm in top and 6 μm in bottom, respectively. n = 3). D H9C2 cells were treated with 50 μM si-NC or si-PXDN in serum-free medium for 24 h, and treated with 400 μM PA or BSA with or without 1 μM DC661 for another 24 h. PXDN, LC3II, and p62 levels were detected by Western blot and quantitative analyses were shown below (n = 4–8). Data were presented as mean ± SEM. One-way ANOVA test was used. *P < 0.05, **P < 0.01, ***P < 0.001.
These results revealed that PA treatment impaired autophagosome turnover and contributed to cell death in cardiomyocytes, while si-PXDN promoted autophagic flux and reduced cell death.
PXDN silence enhances the expression and dephosphorylation of FoxO1
To explore the potential pathways in PXDN regulating autophagic flux, we investigated several recognized regulators of autophagic flux including p-AKT/AKT and FoxO1 pathways. As shown in Fig. 3A, the decline of p-AKT/AKT after PA was not affected by si-PXDN treatment. However, si-PXDN restored the PA-induced decrease of FoxO1 protein level and reduced FoxO1 phosphorylation, indicating FoxO1 as a potential mediator between PXDN and autophagic flux. In addition, si-PXDN alleviated PA-induced insulin resistance in the form of increased glucose consumption (Fig. 3B).

A H9C2 cells were treated with 50 μM si-negative control (NC) or si-PXDN in serum-free medium for 24 h, and treated with 400 μM palmitate acid (PA) or bovine serum albumin (BSA) for another 24 h. p-AKT, AKT, p-FoxO1 and FoxO1 levels were detected by western blot and quantitative analyses were shown below (n = 4). B H9C2 cells were treated with si-NC or si-PXDN in the absence or presence of PA; glucose consumption was determined (n = 3). Data were presented as mean ± SEM. One-way ANOVA test was used. NS referred to no significance. *P < 0.05, **P < 0.01, ***P < 0.001.
PXDN reduces autophagic flux to induce cell death via FoxO1 inhibition
We further investigated the relationship between PXDN and FoxO1 in regulating autophagic flux and cell death in PA-treated cardiomyocytes. As shown in Figs. 4A and B, si-PXDN reduced cell death under PA treatment, while double silence of PXDN and FoxO1 blocked this effect in H9C2 and AC16 cells. Autophagic flux was measured subsequently. It was shown that, compared with downregulation of PXDN alone, double silence of PXDN and FoxO1 showed a severe autophagosome aggregation, while autolysosomes remained unchanged (Fig. 4C). LC3II protein level was basically unchanged between different groups under the stimulation of PA. si-PXDN significantly decreased the accumulation of p62, and this effect can be blocked by additional silence of FoxO1. DC661 didn’t further enhance p62 level except for si-PXDN group (Fig. 4D and Fig. S1B). These results suggested that FoxO1 may act as a downstream factor of PXDN in PA-medicated autophagosome accumulation and cell death.

A H9C2 cells were treated with 50 μM si-negative control (NC), si-PXDN, si-FoxO1, or si-PXDN plus si-FoxO1 in serum-free medium for 24 h, and treated with 400 μM palmitate acid (PA) for another 24 h. Cell survival was evaluated using fluorescence staining with calcein acetoxymethyl ester (Calcein-AM) for live cells in green and ethidium homodimer-1 (EthD-1) for dead cells in red. The percentage of cell death was shown on the right side (scale bar = 50 μM, n = 4). B AC16 cells were treated with NC, si-PXDN, si-FoxO1, or si-PXDN plus si-FoxO1 in serum-free medium for 24 h, and treated with 400 μM PA for another 24 h. Cell survival was evaluated using fluorescence staining. The percentage of cell death was shown on the right side (scale bar = 50 μM, n = 4). C Autophagosomes and autolysosomes in PA-treated H9C2 with si-NC, si-PXDN, si-FoxO1, or si-PXDN plus si-FoxO1 were detected by transmission electron microscopy. Black Arrow referred to autophagosomes and white arrow referred to autolysosomes. The number of autophagosomes and autolysosomes per cell was shown on the right side (scale bar = 20 μm in top and 6μm in bottom, respectively. n = 3). D H9C2 cells were treated with 50 μM si-NC, si-PXDN, si-FoxO1, or si-PXDN plus si-FoxO1 in serum-free medium for 24 h, and treated with 400 μM PA with or without 1 μM DC661 for another 24 h. PXDN, FoxO1, LC3II, and p62 levels were detected by western blot. Quantitative analyses were shown below (n = 4–8). E Western blot of PXDN、p-FoxO1, and FoxO1 in PA-treated H9C2 with si-NC, si-PXDN, si-FoxO1, or si-PXDN plus si-FoxO1 (n = 4). Quantitative analyses were shown below (n = 4). Data were presented as mean ± SEM. One-way ANOVA test was used. *P < 0.05, **P < 0.01, ***P < 0.001.
As shown in Fig. 4E, additional FoxO1 silence could counteract the increase of FoxO1 induced by si-PXDN, while did not influence the phosphorylation of FoxO1. In contrast, FoxO1 phosphorylation decreased in the presence of si-PXDN. Interestingly, PXDN protein level decreased after treated with either si-PXDN or si-FoxO1, and was further downregulated while double silence. These results indicated that PXDN may suppress the expression and activity of FoxO1 under PA treatment while the silence of FoxO1 may also inhibit PXDN expression.
FoxO1 inhibition induces cell death, reduces autophagic flux, and suppresses PXDN expression
To determine the effect of FoxO1 on cell death, autophagic flux, and PXDN under normal conditions, si-FoxO1 was used. It was shown in Fig. 5A–C that si-FoxO1, similar to PA stimulation, induced cell death and autophagosome accumulation, as well as up-regulated LC3II, p62 protein level, and downregulated FoxO1. However, unlike PA-induced PXDN up-regulation, si-FoxO1 significantly decreased the expression of PXDN. Co-immunoprecipitation (IP) was conducted subsequently and PXDN was not found in the IP products of FoxO1 antibody in both basal and PA conditions (Fig. S2). According to the JASPAR database, FoxO1 was predicted to bind with the promoter of PXDN gene (Table. S2). These results indicated that FoxO1 was indeed a key factor in autophagic flux regulation and could regulate the expression of PXDN.

A H9C2 cells were treated with 50 μM si-negative control (NC), 50 μM si-FoxO1, 400 μM palmitate acid (PA), or 400 μM bovine serum albumin (BSA) for 24 h. Cell survival was evaluated using fluorescence staining with calcein acetoxymethyl ester (Calcein-AM) for live cells in green and ethidium homodimer-1 (EthD-1) for dead cells in red. The percentage of cell death was shown on the right side (scale bar = 50 μM, n = 4). B Autophagosomes and autolysosomes in PA-treated H9C2 with 50 μM NC, 50 μM si-FoxO1, 400 μM PA, or 400 μM BSA were detected by transmission electron microscopy. Black Arrow referred to autophagosomes and white arrow referred to autolysosomes. The number of autophagosomes and autolysosomes per cell was shown on the right side (scale bar = 20 μm in top and 6μm in bottom, respectively. n = 3). C H9C2 cells were treated with 50 μM si-NC, 50 μM si-FoxO1, 400 μM PA, or 400 μM BSA for 24 h (with or without 1 μM DC661). FoxO1, LC3II, and p62 levels were detected by western blot and quantitative analyses were shown below (n = 4–8). D H9C2 cells were treated with 50 μM NC, 50 μM si-FoxO1, 400 μM PA, or 400 μM BSA for 24 h. PXDN, p-FoxO1, and FoxO1 levels were detected by western blot and quantitative analyses were shown below (n = 4–8). Data were presented as mean ± SEM. One-way ANOVA test was used. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
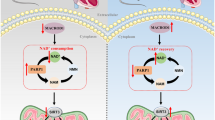
The main findings of this study were as follows: (1) PXDN increased in the progress of cell death in PA-treated cardiomyocytes; (2) PXDN silence improved autophagic flux and inhibited subsequent cell death induced by PA; (3) The effect of PXDN on autophagic flux was mediated via the expression and phosphorylation of FoxO1; (4) FoxO1 may affect the expression of PXDN (Fig. 6). Our findings firstly demonstrate that PXDN plays a key role in PA-induced cell death via regulating FoxO1 and autophagic flux, while FoxO1 may also affect the expression of PXDN, suggesting that PXDN may play an important role in insulin-resistant cardiomyocytes.

The increased protein level of PXDN stimulated by PA impaired autophagic flux to induce cell death in insulin-resistant cardiomyocytes via down-regulating the expression of FoxO1 and promoting FoxO1 phosphorylation.
Autophagy is an important cell-protective mechanism in various condition22,23, and dysfunction of the autophagic signals is observed in DCM in the previous studies8,24. Here in this study, TEM was applied to evaluate the progress of autophagic flux visually, while LC3II (the active isoform of LC3-II, indicating the initiation of autophagy) and p62 (LC3-binding protein, indicating the degradation of autophagosomes) were used as markers to evaluate the degree of autophagic flux. Compared with the control group, increased cell death and impaired autophagic flux were observed in cardiomyocytes stimulated with PA, which was consistent with the work of Bharat et al.25.
PXDN is reported to play essential roles in several cardiovascular diseases via catalyzing hydrogen peroxide to form hypochlorous acid and augment oxidative stress18,26. Previous studies reported that PXDN significantly increased in the aorta of streptozocin-induced diabetes rats19, but the relationship between PXDN and DCM remains unclear. In addition, Increased PXDN under ox-LDL treatment may lead to endothelial cell death via promoting apoptosis by activating p38-mitogen-activated protein kinases/caspase 3 pathway and inducing programmed necrosis by activating β-catenin signaling pathway20,21. However, whether PXDN participates in cardiomyocyte death has not been studied yet. In this study, we found PXDN increased in the progress of autophagy flux restrain and cell death induced by PA, and the effect of PA was abolished by PXDN-siRNA transfection, suggesting that PXDN may play an important role in PA-induced cell death.
Transcriptional factor FoxO1 is recently found to involve in the pathology of DCM through regulating autophagy14,27. Post-transcriptional regulation is the main regulatory method of FoxO1 activity, and phosphorylation of FoxO1 at specific sites such as Tyr25, Ser256, Ser249, and Ser319 are reported to inactivate FoxO1 by inhibiting the entry of FoxO1 into the nucleus. Ser256, which was detected in this study, is the most widely studied site involving in the inactivation of FoxO128. In addition, it was reported that the decrease of p-FoxO1 (Ser256) was associated with enhanced autophagic signal in insulin-resistant heart12. Our study found that FoxO1 expression was decreased by PA stimulation, while si-PXDN reversed this effect and further activated FoxO1 by inhibiting the phosphorylation of FoxO1 at ser256, indicating that PXDN may be able to modulate the expression and activation of FoxO1. Previous studies have shown that phosphorylation of FoxO1 at Ser256 leads to the nuclear exclusion of FoxO1, which triggers its poly-ubiquitination and degradation, among which the phosphorylation of AKT plays an essential role28. However, the ratio of p-AKT/AKT was not affected by si-PXDN treatment in our study, indicating that PXDN may affect FoxO1 phosphorylation through another regulatory pathway. In addition, few studies investigated the upstream factors that regulated the expression of total FOXO1 protein level, thus how PXDN affects FoxO1 remains unknown. We believe that genome and proteomic sequencing may be helpful to find possible pathways regulating FoxO1 expression and phosphorylation. Furthermore, the silence of FoxO1 was found to inhibit the expression of PXDN, suggesting that FoxO1 may regulate PXDN in turn to maintain homeostasis. In other words, the decreased expression of PXDN induced by FoxO1 suppression may partially reduce cell damage. Co-IP was conducted to investigate whether FoxO1 could directly bind to PXDN protein. Unfortunately, stable interaction was not found between FoxO1 and PXDN. Since FoxO1, as a transcriptional factor, may regulate the expression of PXDN by binding to the promoter of the gene rather than direct bind to the protein, JASPAR database was applied to predict the potential binding site of FoxO1 and PXDN and results showed that FoxO1 may bind the promoter of PXDN, indicating that FoxO1 may be able to directly target PXDN gene.
Certainly, this study bore some limitations. Firstly, though we have investigated the role of PXDN in vitro, animal models of DCM are demanded to further confirm our results. In addition, the regulatory mechanisms between PXDN and FoxO1 need to be further studied.
In conclusion, this study highlights the pivotal role of PXDN in insulin-resistant cardiomyocytes. Our findings suggest that inhibition of PXDN improves autophagic flux to reduce cell death through suppressing the expression and activity of FoxO1, while FoxO1 may also affect PXDN expression. These findings may develop our understanding of potential mechanisms regarding autophagy in insulin-resistant cardiomyocytes.
Methods
Materials
High-glucose Dulbecco’s Modified Eagle’s Medium (DMEM), trypsin-EDTA solution, fetal bovine serum, penicillin, and streptomycin were purchased from Gibco (Thermofisher Scientific, USA). Rabbit anti-PXDN antibody was obtained from Millipore (USA). Sodium palmitate (p9767), rabbit anti-AKT antibody (sab4500797), rabbit anti-p62 antibody (P0067), rabbit anti-LC3 antibody (L8918) were purchased from Sigma-Aldrich (USA). Antibody against phospho-AKT (S473) (ab18206) was purchased from Abcam (UK), while rabbit anti-FoxO1 antibody (CST2880S) and primary rabbit antibody for phospho-FoxO1 (Ser256) (CST9461S) were obtained from Cell Signaling Technology (USA). DC661 (dimeric chloroquine) was purchased from selleck (USA). Glucose assay reagent (F006) was purchase from Nanjing jiancheng bioengineering Institute (Nanjing, China). Reagents for western blot, IgG, and protein A + G Agarose were purchase from Beyotime Institute of Biotechnology (Jiangsu, China). LIVE/DEAD viability kit was obtained from Invitrogen (Thermofisher Scientific, USA). And small interfering RNAs (siRNAs) for PXDN and FoxO1 and negative controls (sequences were listed in Table S1), as well as transfection kit was purchase from RiboBio Co Ltd (Guangzhou, China).
Cell culture
H9C2, obtained from Cell Bank of China Science Academy (Shanghai, China) was cultured in high-glucose DMEM medium with 10% fetal bovine serum and 0.1% penicillin/streptomycin. AC16 human cell line was obtained from Cellcook Biotech (Guangzhou, China) and cultured in DMEM-F12 medium with 12.5% fetal bovine serum and 0.1% penicillin/streptomycin. The report of AC16 cell line authentication was attached as a supplement material. The incubator was maintained at 37 °C with 5% CO2 and 95% air. Subculture was carried out when cells reach 70–80% of confluence. In addition, cells were cultured without serum for 24 h prior to experiments. 1 μM DC661 (dimeric chloroquine) was used to inhibit lysosomal acidification29.
siRNA transfections
siRNA interference was performed as described previously30,31. Cardiomyocytes were seeded in six-well plates to reach a 50% confluence and treated in serum-free medium for 24 h with (1) PXDN siRNAs (50 nmol/L each), (2) FoxO1 siRNAs (50 nmol/L each), (3) negative control siRNA (50 nmol/L), (4) both PXDN siRNAs (50 nmol/L each) and FoxO1 siRNAs (50 nmol/L each), respectively. Transfection was achieved using a complement transfection kit according to the manufacturer’s protocol.
Fatty acid treatment
Fatty acid was applied to induce insulin resistance32. 0.028 g of palmitic acid (PA) was dissolved in 2 ml 0.1 N NaOH at a temperature of 75 °C until clear, while 1.8 g of BSA was dissolved in 8 ml 0.9% NaCl at 55 °C. The stock solution of 10 mM PA/18% BSA was prepared by adding the PA solution to the BSA solution, then filtered and stored at −40 °C according to the previous study33. 18% BSA solution was prepared as control meanwhile. The stock solutions were heated for 15 min at 55 °C, and then cooled to room temperature before added to the culture medium.
Glucose consumption
Glucose consumption was measured as previously described34. Ten-microliter medium was removed at 0, 12 or 24 h after treatments. Glucose concentration was measured using a glucose assay kit based on the glucose oxidase method. The amount of glucose consumption (GC) was determined by the glucose concentrations of blank wells subtracting the glucose in cell-plated wells.
Cell viability
LIVE/DEAD viability kit was used to determine cell viability according to the manufacturer’s instructions. Briefly, cells were washed with PBS and then incubated with live green and dead red solutions for 30 minutes at room temperature. Images were acquired using a fluorescence microscope subsequently (Eclipse,Nikon,Japan)35.
Transmission electron microscopy (TEM)
For TEM analyse, cell samples were fixed with 2.5% glutaraldehyde in 0.1 mol/L phosphate buffer overnight at 4 °C, then post fixed in 1% osmium tetroxide and dehydrated in ethanol solutions. HT7700 transmission electron microscope was used to obtain TEM images36.
Western blot analysis
Western blot analysis was performed using standard procedures as described before37. Cells were lysed in RIPA buffer spiked with protease and phosphatase inhibitor cocktail. Protein concentration was assayed by BCA Protein Assay Kit before loaded on 8-12% gel. After electrophoresis, the protein was transferred to polyvinylidene fluoride membrane and probed with specific primary and secondary antibodies. The immune complexes were visualized by SuperSignal West Pico Chemiluminescent Substrate and a gel documentation system (Bio-Rad, USA).
Co-IP
H9C2 cells were lysed in NP-40 buffer and incubated 1 h at 4 °C with IgG and protein A + G Agarose to get rid of non-specific binding. Primary antibody against FoxO1 or IgG was coupled with protein A + G Agarose and incubated overnight at 4 °C. The agarose was collected by centrifugation and washed with PBS for three times. Immunoprecipitated proteins were loaded on 8% SDS gel for western blotting.
Online database analyses
UCSC (https://genome.ucsc.edu/)38 was used to obtain the fasta sequence of PXDN and JASPAR (http://jaspar.genereg.net)39 was used to predict the binding sites of FoxO1 and PXDN gene.
Statistics
Data were presented as means ± SEM. One-way ANOVA with Tukey Kramer test was used when the data were normally distributed, a non-parametric test was used otherwise. SPSS version.23 or Graph Prism 6 were used for multiple statistical comparisons. Differences were considered to be significant at P < 0.05.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Jia, G., Whaley-Connell, A. & Sowers, J. R. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 61, 21–28 (2018).
Jia, G., Hill, M. A. & Sowers, J. R. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 122, 624–638 (2018).
Murfitt, L., Whiteley, G., Iqbal, M. M. & Kitmitto, A. Targeting caveolin-3 for the treatment of diabetic cardiomyopathy. Pharm. Ther. 151, 50–71 (2015).
Jia, G., DeMarco, V. G. & Sowers, J. R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 12, 144–153 (2016).
Varma, U., Koutsifeli, P., Benson, V. L., Mellor, K. M. & Delbridge, L. M. D. Molecular mechanisms of cardiac pathology in diabetes – Experimental insights. Biochim Biophys. Acta Mol. Basis Dis. 1864, 1949–1959 (2018).
Nakai, A. et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 (2007).
Sarparanta, J., Garcia-Macia, M., & Singh, R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr. Diabetes Rev. 13, 352–369 (2017).
Huynh, K., Bernardo, B. C., McMullen, J. R. & Ritchie, R. H. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharm. Ther. 142, 375–415 (2014).
Xie, Z. et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60, 1770–1778 (2011).
Zhang, Y. et al. Liraglutide relieves myocardial damage by promoting autophagy via AMPK-mTOR signaling pathway in zucker diabetic fatty rat. Mol. Cell Endocrinol. 448, 98–107 (2017).
Wang, B. et al. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J. Cell Mol. Med. 18, 1599–1611 (2014).
Wang, Q. & Ren, J. mTOR-Independent autophagy inducer trehalose rescues against insulin resistance-induced myocardial contractile anomalies: Role of p38 MAPK and Foxo1. Pharm. Res. 111, 357–373 (2016).
Yao, Q. et al. Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J. Mol. Cell Cardiol. 124, 26–34 (2018).
Chistiakov, D. A., Orekhov, A. N. & Bobryshev, Y. V. The impact of FOXO-1 to cardiac pathology in diabetes mellitus and diabetes-related metabolic abnormalities. Int J. Cardiol. 245, 236–244 (2017).
Hariharan, N. et al. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107, 1470–1482 (2010).
Li, H., Cao, Z., Zhang, G., Thannickal, V. J. & Cheng, G. Vascular peroxidase 1 catalyzes the formation of hypohalous acids: characterization of its substrate specificity and enzymatic properties. Free Radic. Biol. Med. 53, 1954–1959 (2012).
Ge, L. et al. The role of losartan in preventing vascular remodeling in spontaneously hypertensive rats by inhibition of the H2O2/VPO1/HOCl/MMPs pathway. Biochem. Biophys. Res. Commun. 493, 855–861 (2017).
Peng, H. et al. VPO1 modulates vascular smooth muscle cell phenotypic switch by activating extracellular signal-regulated kinase 1/2 (ERK 1/2) in abdominal aortic aneurysms. J. Am. Heart Assoc. 7, e10069 (2018).
Liu, S. et al. Role of vascular peroxidase 1 in senescence of endothelial cells in diabetes rats. Int. J. Cardiol. 197, 182–191 (2015).
Bai, Y. P. et al. Role of VPO1, a newly identified heme-containing peroxidase, in ox-LDL induced endothelial cell apoptosis. Free Radic. Biol. Med. 51, 1492–1500 (2011).
Zhang, Y. et al. Vascular peroxide 1 promotes ox-LDL-induced programmed necrosis in endothelial cells through a mechanism involving β-catenin signaling. Atherosclerosis 274, 128–138 (2018).
Yamaguchi, O., Taneike, M. & Otsu, K. Cooperation between proteolytic systems in cardiomyocyte recycling. Cardiovasc Res. 96, 46–52 (2012).
Rifki, O. F. & Hill, J. A. Cardiac autophagy: good with the bad. J. Cardiovasc Pharm. 60, 248–252 (2012).
Coliva, G., Duarte, S., Perez-Sala, D. & Fedorova, M. Impact of inhibition of the autophagy-lysosomal pathway on biomolecules carbonylation and proteome regulation in rat cardiac cells. Redox Biol. 23, 101123 (2019).
Jaishy, B. et al. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J. Lipid Res. 56, 546–561 (2015).
Liu, Z. et al. Vascular peroxidase 1 is a novel regulator of cardiac fibrosis after myocardial infarction. Redox Biol. 22, 101151 (2019).
Klotz, L. et al. Redox regulation of FoxO transcription factors. Redox Biol. 6, 51–72 (2015).
Xin, Z. et al. FOXOs in the impaired heart: New therapeutic targets for cardiac diseases. Biochim Biophys. Acta Mol. Basis Dis. 1863, 486–498 (2017).
Rebecca, V. W. et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Disco. 9, 220–229 (2019).
You, B. et al. Vascular peroxidase 1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation, apoptosis resistance and migration. Cardiovasc. Res. 114, 188–199 (2018).
Yamagata, K. et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231 (2008).
Li, S. et al. Excessive autophagy activation and increased apoptosis are associated with palmitic acid-induced cardiomyocyte insulin resistance. J. Diabetes Res. 2017, 2376893 (2017).
Pascual, G. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017).
Chang, W. et al. Berberine improves insulin resistance in cardiomyocytes via activation of 5’-adenosine monophosphate-activated protein kinase. Metabolism 62, 1159–1167 (2013).
Li, J. et al. Circulating fibrocytes stabilize blood vessels during angiogenesis in a paracrine manner. Am. J. Pathol. 184, 556–571 (2014).
Xiao, C. et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of miR-125b. Circ. Res. 123, 564–578 (2018).
Wei, C. D. et al. Globular adiponectin protects H9C2 cells from palmitate-induced apoptosis via Akt and ERK1/2 signaling pathways. Lipids Health Dis. 11, 135 (2012).
Haeussler, M. et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47, D853–D858 (2019).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020).
Funding
The work was supported by grants from National Program on Key Basic Research Project of China (Nos. 2019YFF0216304 to R.Z.S.), Outstanding Youth Foundation Project of Hunan Natural Science Foundation (Nos. 2019JJ20036 to R.Z.S.), Key R & D Program of Hunan Provincial Department of Science and technology (Nos. 2018SK2137 to R.Z.S.), Chinese Cardiovascular Association V.G. foundation (Nos.2017-CCA-VG-005 to R.Z.S.), National Nature Science Foundation of China (Nos. 81873479 to G.G.Z.) and National Nature Science Foundation of China (Nos. 81670267 to G.G.Z.).
Author information
Authors and Affiliations
Contributions
All authors made substantive intellectual contributions to this study to qualify as authors. R.Z.S., G.G.Z., and G.J.C. conceived of the design of the study. C.L., Z.Y.L., Q.X., and R.Z.S. contributed to the design of the study. C.L., Z.Y.L., and H.H.P. collected the data. C.L., J.C., and H.H.Z. analyzed the data. G.J.C. provide experimental technical support for this study. C.L. drafted the manuscript. Z.Y.L., Q.X., H.H.P., J.C., and H.H.Z. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. M. Fimia
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, C., Liu, Z., Xu, Q. et al. PXDN reduces autophagic flux in insulin-resistant cardiomyocytes via modulating FoxO1. Cell Death Dis 12, 418 (2021). https://doi.org/10.1038/s41419-021-03699-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03699-4
