
Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most common malignant tumors with poor survival. It is urgent to search for new efficient drugs with good stability and safety for clinical therapy. This study aims to identify potential anticancer drugs from a compound library consisting of 429 natural products. Echinatin, a compound isolated from the Chinese herb Glycyrrhiza uralensis Fisch, was found to markedly induce apoptosis and inhibit proliferation and colony-formation ability in ESCC. Confocal fluorescence microscopy data showed that echinatin significantly induced autophagy in ESCC cells, and autophagy inhibitor bafilomycinA1 attenuated the suppressive effects of echinatin on cell viability and apoptosis. Mechanistically, RNA sequencing coupled with bioinformatics analysis and a series of functional assays revealed that echinatin induced apoptosis and autophagy through inactivation of AKT/mTOR signaling pathway, whereas constitutive activation of AKT significantly abrogated these effects. Furthermore, we demonstrated that echinatin had a significant antitumor effect in the tumor xenograft model and markedly suppressed cell migration and invasion abilities of ESCC cells in a dose-dependent manner. Our findings provide the first evidence that echinatin could be a novel therapeutic strategy for treating ESCC.
Similar content being viewed by others


Introduction
Esophageal cancer is one of the most common malignant tumors, the mortality of which ranks 6th around the world with poor overall survival1. Esophageal cancer has two major histologic subtypes, esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma2, and ESCC is the main histologic subtype with ~90% of cases in Asia-Pacific region including China. Failure of chemotherapy and radiotherapy leads to tumor recurrence and poor prognosis mainly due to limited efficiency and side effects, for example, conventional chemotherapeutics target both cancer cells and normal cells resulting in toxic effects on the body3,4. Currently, esophagectomy remains the chief strategy for treatment of esophageal cancer5, therefore, it has become a top priority to find more stable, effective and safe anticancer drugs.
Natural product is an important resource of new drug candidates, especially for anticancer drugs6,7,8. ~80–83% of the approved anticancer drugs were naturally occurring agents or mimicked natural products9,10. As bioactive components from natural products, vincristine, irinotecan, etoposide, and paclitaxel have been historically used in clinical cancer treatment11,12,13,14. By screening novel chemotherapeutics from natural products, we identified echinatin as an effective anticancer drug candidate from a natural product-based library consisting of 429 compounds. Echinatin is an active component of licorice, which is derived from the roots and rhizomes of Glycyrrhiza species (Leguminosae) and related species15. Licorice extract has many biological effects, including antioxidant, anti-inflammatory, and anticancer, partly through regulation of Nrf2, NO, and NFĸB pathways16,17. However, up to now, the biological function of echinatin in cancer treatment has not been reported, and the molecular mechanism remains unknown to a large extent.
Autophagy plays a dual role in tumorigenesis, although some studies reported that autophagy exerts pro-survival effect, most agents-induced autophagy mainly leads to cell death in cancer18,19. Autophagy can be regulated by many signaling pathways including PI3K/AKT/mTOR pathways, which is crucial in tumor initiation and progression. Our previous studies found that activation of PI3K/AKT signaling plays a vital role in ESCC growth and metastasis20,21. Therefore, novel therapeutic strategies that target autophagy and AKT/mTOR signaling are urgently needed. In this study, we examined the effects of echinatin on growth, chemosensitivity, and metastatic potential of ESCC cells in vitro and in vivo. Moreover, RNA sequencing (RNA-seq), ingenuity pathway analysis (IPA), and a series of functional assays were performed to investigate whether echinatin exerts the anticancer effects through regulation of autophagy and AKT/mTOR pathways.
Results
Echinatin induces apoptosis and decreases cell proliferation in ESCC
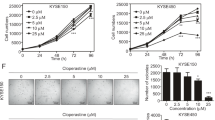
We have identified effective anticancer agents from a food-source natural product library consisting of 429 compounds previously22. In this study, echinatin, an active component of licorice derived from the roots and rhizomes of Glycyrrhiza species (Leguminosae)15 (Fig. 1a), was identified as a candidate agent in inhibiting ESCC cell proliferation. To evaluate the effect of echinatin on ESCC cell proliferation, KYSE30 and KYSE270 cells were treated with increasing concentrations of echinatin for up to 5 days. As shown in Fig. 1b, echinatin inhibited cell proliferation substantially in both dose-and time-dependent manner. The colony-formation assay indicated that echinatin markedly declined the number of colony formation (Fig. 1c). Collectively, the above results demonstrated that echinatin might exhibit tumor-suppressive bioactivity in ESCC. To determine the effect of echinatin on apoptosis in ESCC cells, the cells were treated with echinatin at the indicated concentrations (up to 40 μM) for 24 and 48 h, and the results showed that echinatin-induced apoptosis in a dose-dependent way (Figs. 1d and S1A), indicating that echinatin elicited apoptosis in ESCC cells. The induction of apoptosis by echinatin was further evidenced by the increase of apoptotic markers, including cleaved caspase-3 and cleaved poly ADP ribose polymerase (PARP), upon echinatin treatment (Figs. 1e and S1B). Taken together, these data suggest that echinatin can trigger apoptosis to repress proliferation of ESCC cells.

a Chemical structure of echinatin. b KYSE30 and KYSE270 cells were treated with echinatin at different concentrations (for up to 40 μM), and cell viability determined by CCK-8 assay. c The colony formation of KYSE30 and KYSE270 cells was inhibited upon exposure to echinatin. d The detection of apoptosis in ESCC cells treated with different concentrations of echinatin for 48 h by an Annexin V-FITC/PI double staining assay. e Western blot analysis was performed to detect the expression of cleaved caspase-3, caspase-3, and cleaved PARP in the ESCC cells treated with indicated concentrations of echinatin for 48 h. Bars, SD; **P < 0. 01, ***P < 0.001.
Echinatin induces autophagy in ESCC cells
We next examined whether echinatin could induce autophagy in ESCC cells. The confocal analysis showed that exposure of KYSE30 and KYSE270 cells to echinatin resulted in cellular LC3 puncta accumulation (Fig. 2a, b). In addition, the western blot results confirmed the increased LC3 expression in echinatin-treated cells in a dose-dependent manner (Fig. 2c). Furthermore, as shown in Fig. 2d, e, pretreatment with bafilomycinA1 (BafA1), an inhibitor of autophagy, could restore the cell viability and colony-formation ability in echinatin-treated KYSE30 and KYSE270 cells. The echinatin-induced apoptosis was reversed in presence of BafA1 treatment (Fig. 2f), and the echinatin-induced cleaved caspase-3 and cleaved PARP expressions were also significantly decreased after blockade of autophagy with BafA1 (Fig. 2g). These results indicate that echinatin induces ESCC cells apoptosis probably via the activation of autophagy.

The expression of LC3 in the KYSE30 and KYSE270 cells exposed to echinatin was compared by immunofluorescence (a) and Western blot (c), b The number of LC3 puncta per cell and the percentage of the cells with LC3 puncta were counted under florescence microscope. KYSE30 and KYSE270 cells were treated with different concentrations of echinatin, with or without pretreated with BafA1 (0.5 nM, 12 h), and then the viability and colony-formatoin ability determined by CCK-8 assay (d) and colony-formation assay (e), respectively. Comparison of apoptosis and expression of apoptotic markers in ESCC cell treated with different concentrations of echinatin with or without BafA1 pretreatment (0.5 nM, 12 h) by an Annexin V-FITC/PI double staining assay (f) and western blot (g). Bar = 10 μm. Bars, SD; *P < 0.05, **P < 0. 01, ***P < 0.001.
The AKT/mTOR signaling pathway mediates the effect of echinatin on autophagy and apoptosis in ESCC cells
To investigate the molecular mechanisms of how echinatin induces apoptosis and autophagy in ESCC cells, the RNA-seq was performed to profile the differentially expressed genes in the KYSE30 cells treated with echinatin (20 μM) for 48 h. A total of 688 genes were identified to be significantly regulated by echinatin (fold change ≥4), including 105 upregulations and 583 downregulations (Supplementary Table S1). IPA analysis was used to characterize the canonical pathways. As shown in Fig. 3a, a cluster of echinatin-regulated genes constructed a signaling network that strongly pointed to the AKT pathway. As expected, echinatin was found to significantly reduce expression levels of p-AKT and p-mTOR (Fig. 3b), indicating that echinatin could inhibit mTOR/AKT pathway, which has been reported to inhibit autophagy in various types of cancer23. In order to investigate whether echinatin induces autophagy and apoptosis via AKT/mTOR pathway, a vector expressing AKT (T308D/S473D), the constitutively active form of AKT24, was transfected into KYSE30 and KYSE270 cells and the anticancer effects of echinatin were determined (Fig. 3c). The results showed that the ectopic expression of AKT (T308D/S473D) significantly increased the viability of echinatin-treated KYSE30 and KYSE270 cells (Fig. 3d). In addition, the echinatin-induced apoptosis was abrogated by AKT (T308D/S473D) overexpression (Fig. 3e), and the western blot assay further confirmed that echinatin-induced cleavage of caspase-3 and PARP were significantly decreased in AKT (T308D/S473D)-overexpressing ESCC cells (Fig. 3f). Moreover, increased LC3 protein expression upon echinatin treatment was also reversed by AKT (T308D/S473D) overexpression (Fig. 3f). Our results suggest that echinatin induces autophagic apoptosis in ESCC cells by inhibiting the AKT/mTOR pathway.

a Ingenuity pathway analysis (IPA) suggested a dysregulation of AKT pathway in echinatin-treated KYSE30 cells. b KYSE30 and KYSE270 cells were exposed to different concentrations of echinatin for 48 h, and western blot analysis was performed to detect the expression levels of AKT, p-AKT, mTOR, and p-mTOR. c–f The KYSE30 and KYSE270 cells transfected with AKT (T308D/S473D)-expressing plasmid or vector control were treated with echinatin at the indicated concentrations for 48 h, and then compared for p-mTOR expression (c), cell viability (d), apoptosis (e), and expression levels of AKT, p-AKT, mTOR, p-mTOR, LC3, cleaved caspase-3 and cleaved PARP (f). Bars, SD; *P < 0.05, **P < 0. 01.
Echinatin enhances the sensitivity of ESCC cells to fluorouracil (5-FU)
The poor prognosis of ESCC was related to chemotherapy resistance. To study the biological function of echinatin in cancer chemoresistance, we examined the sensitivity of ESCC cells to 5-FU with or without echinatin by CCK-8, colony-formation and western blot assays. A combination of low-dose echinatin and low-dose 5-FU significantly suppressed cell growth (Fig. 4a) and colony formation (Fig. 4b) compared to low-dose echinatin or 5-FU alone. The expression of cleaved caspase-3 and cleaved PARP were increased in the ESCC cells treated with a combination of echinatin and 5-FU (Fig. 4c). Collectively, these data demonstrated that echinatin can increase the sensitivity of ESCC cells to 5-FU by inducing apoptosis.

a, b The viability and colony-formation ability of the KYSE30 and KYSE270 cells treated with 5-FU (1.25 or 0.3 μM), echinatin (20 μM) alone, or the combination of 5-FU and echinatin for up to 5d was determined by CCK-8 assay (a) and colony-formation assay (b), respectively. c Echinatin increased apoptosis in the 5-FU-treated ESCC cells, indicated by higher expression levels of cleaved caspase-3 and cleaved PARP. Bars, SD; *P < 0.05, **P < 0. 01, ***P < 0.001.
Echinatin inhibits migration and invasion in ESCC cells
We next determined the effect of echinatin on the migration and invasion properties of ESCC cells, and the results showed that echinatin significantly inhibited the capability of KYSE30 and KYSE270 cells to migrate and invade (Fig. 5a, b). The expression of EMT markers, vimentin, β-catenin and E-cadherin, were also detected by western blot. The increased E-cadherin and the decreased β-catenin and vimentin expression indicated that echinatin inhibited the migration and invasion in ESCC cells by reversing EMT. In order to investigate whether echinatin inhibits migration and invasion via AKT pathway, the plasmid expression constitutively active form of AKT, AKT (T308D/S473D), was transfected into KYSE30 and KYSE270 cells, the results showed that activation of AKT signaling significantly abrogated the inhibitory effects of echinatin on ESCC cell migration and invasion (Fig. 5d, e).

The migration (a) and invasion (b) abilities of KYSE30 and KYSE270 cells treated with echinatin at different concentrations for 24 h were determined by chamber migration and invasion assays. c Western blot analysis of expressions of vimentin, β-catenin and E-cadherin in the KYSE30 and KYSE270 cells treated with echinatin at the indicated concentrations for 24 h. d, e The KYSE30 and KYSE270 cells transfected with AKT (T308D/S473D)-expressing plasmid or vector control were treated with echinatin at the indicated concentrations for 24 h, and then compared for migration ability (d), invasion ability (e). Bars, SD; *P < 0.05, **P < 0. 01, ***P < 0.001.
Echinatin suppresses growth of ESCC tumor xenografts in vivo
The nude mice bearing KYSE270-derived tumor xenografts were orally administrated with echinatin (20 mg/kg and 50 mg/kg), and its effect on tumor growth was monitored. The tumor burden was found to be markedly suppressed with decreases of 57% and 48% in the group receiving 20 and 50 mg/kg of echinatin, respectively (Fig. 6a). Western blot data showed that echinatin inhibited the AKT/mTOR pathway, indicated by decreased expression levels of p-AKT and p-mTOR (Fig. 6b). There was no significant difference between the treatment and control groups in terms of body weight (Fig. 6c). Moreover, echinatin treatment did not exert any overt change in terms of the morphology of the vital organs, including lungs, liver, and kidneys (Fig. 6d). Furthermore, no significant difference in serum alanine transaminase (ALT) or aspartate transaminase (AST) levels of nude mice was observed (Fig. 6e), suggesting that echinatin had no toxic effect on animals.

The nude mice bearing KYSE270-derived tumor xenografts were orally administrated with echinatin (20 mg/kg or 50 mg/kg) every 2 days (n = 6 per group), whereas the control group received the vehicle only. a Tumor curves showed that echinatin significantly suppressed the growth of tumor xenografts. b Expression levels of AKT, p-AKT, mTOR, p-mTOR and LC3 in the tumors from mice treated with echinatin or vehicle were detected by Western blot. c Body weight of nude mice during the experimental period. d Hematoxylin and eosin (H&E) staining of lung, liver, and kidney specimens collected from mice of the treatment and control groups. e Comparison of serum ALT and AST level between echinatin-treated and control groups. Bars, SD; ***P < 0.001.
Discussion
Autophagy is a tightly regulated catabolic process of cellular self-digestion, in which cytoplasm, organelles, and proteins were separated into autophagosomes and then fused with lysosomes for degeneration to maintain cell renewal and homeostasis during starvation, stress, microbial invasion, and inflammation25. The dysregulation of autophagy is closely related to a number of diseases, including cancer, cardiovascular disease, and autoimmunity26,27. Many of the stimulators that lead to apoptosis can trigger autophagy, in which autophagy usually appeares before cell apoptosis. Autophagy activation beyond a certain threshold may lead to the breakdown of its functions, resulting in autophagic cell death or other types of cell death. The genetic experiments-related study also reported that autophagy may have an important role in controlling apoptosis28. Therefore, it has been well appreciated that autophagy can induce cell death to inhibit cancer progression, thus may be a target for cancer therapy29,30. In the present study, we found that echinatin increased the level of LC3, a marker protein of autophagy, indicating that echinatin can activate autophagy. In addition, when ESCC cells were pretreated with BafA1, an inhibitor of autophagy, the effects of echinatin on apoptosis, cell viability and colony-formation ability were markedly attenuated, indicating that echinatin acts as an activator of autophagy to induce apoptosis in ESCC cells.
Emerging evidences suggest that the AKT/mTOR pathway can activate a series of processes involved in tumor initiation and progression, including cell proliferation, differentiation, survival, chemoresistance and metastasis, and therefore may be a potential target for anticancer therapy16,31,32. To elucidate the action mechanism of echinatin in cancer cells, RNA-seq was performed to screen the differentially expressed genes in echinatin-treated ESCC cells, and IPA analysis suggested that AKT pathway, which has been documented to block autophagy through regulation of mTOR signaling33, may play an important role in the bioactivity of echinatin. The rescue experiment by using the AKT (T308D/S473D) plasmid expressing constitutively activation of AKT and the subsequent functional assays indicated that reactivation of AKT/mTOR signaling significantly abrogated the effects of echinatin on apoptosis, proliferation, autophagy, migration, and invasion abilities of cancer cells (Figs. 3, 5), suggesting that echinatin exerts anticancer effects through AKT/mTOR-autophagy pathway. The link between inflammation and cancer has been clearly demonstrated by epidemiological and experimental data34,35, for example, inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis are associated with an increased risk of colon adenocarcinoma36. Some anti-inflammatory therapies showed great efficacy in cancer prevention and treatment37, which increases the rationale of study in anticancer bioactivity of anti-inflammatory drugs. The fact that echinatin was originally described as licorice extract that displayed antioxidant and anti-inflammatory activities, corroborates our finding on is anticancer effect. Furthermore, whether anticancer bioactivity of echinatin is related to its antioxidant and anti-inflammatory properties warrants further investigation.
Natural product such as traditional Chinese medicines has been used in the treatment of cancer for a long time. In view of its multi-component and multi-target characteristics in pharmacology, natural product is a promising resource for screening anticancer drugs. Vincristine, irinotecan, etoposide, and paclitaxel are classical examples of plant-derived compounds, and more new-generation drugs have been developed and used in clinic11. Echinatin is an active component of licorice, which possesses multiple biological activities. Although the chemistry of licorice has been extensively studied, the active components that contribute to its biological functions are far from clear. Licochalcone A, another flavonoid isolated from licorice, has been reported to inhibit cell viability in non-small cell lung cancer and breast cancer21,38. Although echinatin and licochalcone A have similar structures39, the anticancer activity of echinatin has not been reported up to now12. In this study, our results showed that echinatin decreased cell proliferation and colony formation in a dose-dependent manner, and more importantly, inhibited the growth of ESCC tumor xenografts in vivo, without observed damage to the normal esophageal epithelial cells or the vital organs of animals. Cancer, which still has no cure, has an increasing mortality rate year by year. Cancer metastasis and the resistance to chemotherapy often predict a poor prognosis40. Our data showed that echinatin not only significantly reduced the migration and invasion of ESCC cells, accompanied by EMT reversal, but also enhanced the antitumor effect of 5-FU, therefore, supporting the significance of echinatin in postoperative adjuvant therapy. Whether echinatin inhibits ESCC metastasis and sensitizes ESCC cells to 5-FU in vivo or not warrants further investigation.
Taken together, we uncovered that echinatin is a potential anticancer natural product with an inhibitory effect on ESCC tumorigenesis and metastatic potential, and sensitizes ESCC cells to 5-FU treatment. Mechanistically, echinatin induces cell autophagy and apoptosis via the AKT/mTOR signaling pathway (Fig. 7). It is the first time to demonstrate the anticancer bioactivity of echinatin and the underlying mechanism. These findings provide solid evidence on the potential of echinatin as therapeutic options for the treatment of ESCC.

Activation of AKT/mTOR signaling inhibits autophagy to regulate the balance between proliferation and apoptosis in ESCC cells, while echinatin induces cell autophagy and apoptosis via inhibiting AKT/mTOR pathway, leading to the suppression of cell growth and tumorigensis.
Materials and methods
Cell culture and treatment
Human ESCC cell lines, KYSE30 and KYSE270 were obtained from DSMZ (Braunschweig, Germany). The cell lines were authenticated by short tandem repeat profiling, and tested for mycoplasma contamination. These cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and penicillin and streptomycin at 37 °C in 5% CO2.
Plasmid and transfection
The plasmid expressing constitutively active form of AKT, the AKT (T308D/S473D), was a gift from Dr. Robert Weinberg41. ESCC cells were transfected with AKT (T308D/S473D) or vector control using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Cell viability assay
Cells were cultured in 96-well plate for 24 h, and then treated with echinatin (TargetMol, Boston, MA, USA) at different concentrations. The cell viability was determined by Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies Inc., Rockville, MD, USA) according to the manufacturer’s guidelines, and the absorbance was quantified by an automated microplate spectrophotometer (BioTek Instruments Winooski, VT, USA) at 450 nm22.
Colony-formation assay
Colony-formation assay was performed as described previously4. Cells were seeded in six-cell plate and cultured with echinatin. After 14 days, the cells were washed gently with PBS three times, fixed with 75% ethanol for 15 min, and stained with crystal violet. The numbers of colonies were counted for analysis.
Annexin V-FITC/PI staining assay
Cell apoptosis was determined using an Annexin V-FITC/PI Apoptosis Detection kit (KeyGen, Nanjing, Jiangsu, China)4. In brief, cells were suspended in binding buffer and stained with annexin V-FITC and propidium iodide (PI) for 15 min at room temperature in darkness. Apoptosis was assessed and analyzed using a C6 flow cytometer (BD Biosciences, San Diego, CA, USA).
Immunofluorescence staining
The cells were fixed with 4% paraformaldehyde for 10 min, and blocked with 10% goat serum for 2 h after being permeabilized with 0.1% TrintonX-100 for 10 min. After washed with 1% TBST three times, cells were incubated with the primary antibody against LC3A/B at 4 °C overnight, and then incubated with the appropriate fluorescent secondary antibody at room temperature for 2 h. The cells were counter-stained with 40,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, Waltham, MA, USA), and the images were visualized by laser scanning confocal microscopy (Carl Zeiss AG, Jena, Thuringia, Germany)26.
RNA-seq and IPA
The RNA-seq was performed in the Beijing Genomics Institute Tech (Shenzhen, Guangzhou, China). Gene profiles in two conditions were analyzed by DESeq R package (1.10.1). The genes with a P value less than 0.05 determined by DESeq were represented as differentially expressed. The IPA software (Ingenuity Systems, Redwood City, CA, USA) was used for pathway analysis16.
The detection of cell migration and invasion assay in vitro
Cell migration ability was detected by using uncoated Transwell chambers (8 μm pore size; BD Biosciences, Bedford, MA, USA). Cells in serum-free medium were seeded in the upper chamber, and the complete growth medium as the chemoattractant was added to the lower chamber. After 24 h, the migrated cells were fixed with methanol and then stained with crystal violet. The images of each well were obtained in three different fields. The matrigel-coated chambers were used in invasion assay with a similar protocol in accordance with cell migration assay42.
Western blot analysis
Preparation of cell lysates and details of western blot were described previously43. The antibodies used included E-cadherin (1:5000; Proteintech, 20874-1-AP), β-Catenin (1:1000, Cell Signaling Technology, CST9562), vimentin (1:1000, Cell Signaling Technology, CST5741), LC3A/B (1:1000, Cell Signaling Technology, CST4108), caspase-3 (1:1000, Cell Signaling Technology, CST9662), cleaved caspase-3 (1:1000, Cell Signaling Technology, CST 9661), PARP (1:1000, Cell Signaling Technology, CST9532), cleaved PARP (1:1000, Cell Signaling Technology, CST 5625), phospho-AKT (p-AKT) (1:2000, Cell Signaling Technology, CST4060), phospho-mTOR (p-mTOR) (1:1000, Cell Signaling Technology, CST5536), mTOR (1:1000, Cell Signaling Technology, CST2972) and AKT (1:2000, Cell Signaling Technology, CST2920). The reaction was visualized using Clarity Western ECL substrate (Bio-Rad, Hercules, CA, USA) and detected by exposure to autoradiographic film.
Tumor xenograft experiment
Tumor xenograft experiments were performed as described previously44,45. The male nude mice aged 6–8 weeks were maintained in standard conditions and cared for according to the institutional guidelines for animal care. KYSE270 cells in PBS and matrigel with equal volume were subcutaneously injected into mice to establish tumor xenografts. When the diameter of the tumor reached about 5 mm, the mice were randomly divided into three groups (n = 6 mice/group) and treated with echinatin (20 mg/kg and 50 mg/kg) by oral gavage, as well as vehicle as control, respectively, every 2 days. Tumor volumes were measured every 2 days in a blinded fashion, calculated by the formula of V = (length × width2)/2. At the end of the experiment, the tumors, livers, lungs, and kidneys were collected for western blot and histological analysis. All the animal experiments were approved by the Ethics Committee for Animal Experiments of Jinan University.
Statistical analysis
All in vitro experiments were performed in three independent experiments. Data were expressed as the mean ± SD and compared by GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). Comparison between groups were analyzed by a Student’s t test method. P values < 0.05 were deemed significant.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clin.68, 394–424 (2018).
Xie, X. et al. Gossypetin is a novel MKK3 and MKK6 inhibitor that suppresses esophageal cancer growth in vitro and in vivo. Cancer Lett.442, 126–136 (2019).
Zheng, W.-B. et al. Propafenone suppresses esophageal cancer proliferation through inducing mitochondrial dysfunction. Am. J. Cancer Res.7, 2245 (2017).
Wang, Y. et al. Liensinine perchlorate inhibits colorectal cancer tumorigenesis by inducing mitochondrial dysfunction and apoptosis. Food Funct.9, 5536–5546 (2018).
Takeuchi, H. et al. A risk model for esophagectomy using data of 5354 patients included in a Japanese nationwide web-based database. Ann. Surg.260, 259–266 (2014).
Butler, M. S. The role of natural product chemistry in drug discovery. J. Nat. Prod.67, 2141–2153 (2004).
Newman, D. J., Cragg, G. M. & Snader, K. M. Natural products as sources of new drugs over the period 1981− 2002. J. Nat. Prod.66, 1022–1037 (2003).
Min, H. Y. et al. The natural compound gracillin exerts potent antitumor activity by targeting mitochondrial complex II. Cell Death Dis.10, 810, https://doi.org/10.1038/s41419-019-2041-z (2019).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod.75, 311–335 (2012).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod.79, 629–661 (2016).
Nobili, S. et al. Natural compounds for cancer treatment and prevention. Pharmacol. Res.59, 365–378 (2009).
Li-Weber, M. Targeting apoptosis pathways in cancer by Chinese medicine. Cancer Lett.332, 304–312 (2013).
Walsh, V. & Goodman, J. Cancer chemotherapy, biodiversity, public and private property: the case of the anti-cancer drug Taxol. Soc. Sci. Med.49, 1215–1225 (1999).
Mugera, G. M. Useful drugs and cancer causing chemicals in Kenya medical and toxic plants. 12th Inaugural Lecture (University of Nairobi, 1977).
Ji, S. et al. Bioactive constituents of Glycyrrhiza uralensis (Licorice): discovery of the effective components of a traditional herbal medicine. J. Nat. Prod.79, 281–292 (2016).
Asl, M. N. & Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res.22, 709–724 (2008).
Mae, T. et al. A licorice ethanolic extract with peroxisome proliferator-activated receptor-γ ligand-binding activity affects diabetes in KK-Ay mice, abdominal obesity in diet-induced obese C57BL mice and hypertension in spontaneously hypertensive rats. J. Nutr.133, 3369–3377 (2003).
Degenhardt, K. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell10, 51–64 (2006).
Turcotte, S. et al. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell14, 90–102 (2008).
Li, B. et al. Suppression of esophageal tumor growth and chemoresistance by directly targeting the PI3K/AKT pathway. Oncotarget5, 11576 (2014).
Li, B. et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget8, 38755 (2017).
Xu, W. W. et al. Synephrine hydrochloride suppresses esophageal cancer tumor growth and metastatic potential through inhibition of galectin-3-AKT/ERK signaling. J. Agric. food Chem.66, 9248–9258 (2018).
Mathew, R., Karantza-Wadsworth, V. & White, E. Role of autophagy in cancer. Nat. Rev. Cancer7, 961 (2007).
Haendeler, J., Hoffmann, J., Rahman, S., Zeiher, A. M. & Dimmeler, S. Regulation of telomerase activity and anti‐apoptotic function by protein–protein interaction and phosphorylation. FEBS Lett.536, 180–186 (2003).
Gao, L. et al. Paeonol induces cytoprotective autophagy via blocking the Akt/mTOR pathway in ovarian cancer cells. Cell Death Dis.10, 609, https://doi.org/10.1038/s41419-019-1849-x (2019).
Xu, W. W. et al. Cancer cell-secreted IGF2 instigates fibroblasts and bone marrow-derived vascular progenitor cells to promote cancer progression. Nat. Commun.8, 14399 (2017).
Jeon, S. J. et al. TIPRL potentiates survival of lung cancer by inducing autophagy through the eIF2alpha-ATF4 pathway. Cell Death Dis.10, 959, https://doi.org/10.1038/s41419-019-2190-0 (2019).
Lozy, F. & Karantza, V. Autophagy and cancer cell metabolism. Seminars Cell Develop. Biol.23, 395–401 (2012).
Green, D. R. & Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell157, 65–75 (2014).
Fulda, S. & Kögel, D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene34, 5105 (2015).
Hsieh, A., Truitt, M. & Ruggero, D. Oncogenic AKTivation of translation as a therapeutic target. Br. J. Cancer105, 329 (2011).
Mann, K. M., Ying, H., Juan, J., Jenkins, N. A. & Copeland, N. G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther.168, 29–42 (2016).
Moosavi, M. A. et al. Phytochemicals as potent modulators of autophagy for cancer therapy. Cancer Lett.424, 46–69 (2018).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell140, 883–899 (2010).
Grivennikov, S. I. & Karin, M. Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev.20, 65–71 (2010).
Gillen, C., Walmsley, R., Prior, P., Andrews, H. & Allan, R. Ulcerative colitis and Crohn’s disease: a comparison of the colorectal cancer risk in extensive colitis. Gut35, 1590–1592 (1994).
Gonda, T. A., Tu, S. & Wang, T. C. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle8, 2005–2013 (2009).
Huang, W.-C., Su, H.-H., Fang, L.-W., Wu, S.-J. & Liou, C.-J. Licochalcone A inhibits cellular motility by suppressing E-cadherin and MAPK signaling in breast cancer. Cells8, 218 (2019).
Liang, M., Li, X., Ouyang, X., Xie, H. & Chen, D. Antioxidant mechanisms of echinatin and licochalcone A. Molecules24, 3 (2019).
Wang, S., Huang, S. & Sun, Y. L. Epithelial-mesenchymal transition in pancreatic cancer: a review. BioMed Res. Int.2017, 2646148 (2017).
Meyerson, M. et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell90, 785–795 (1997).
Li, B. et al. MicroRNA-377 suppresses initiation and progression of esophageal cancer by inhibiting CD133 and VEGF. Oncogene36, 3986–4000 (2017).
Huang, X.-H. et al. Benzethonium chloride suppresses lung cancer tumorigenesis through inducing p38-mediated cyclin D1 degradation. Am. J. Cancer Res.9, 2397 (2019).
Yang, J. et al. Adefovir dipivoxil sensitizes colon cancer cells to vemurafenib by disrupting the KCTD12-CDK1 interaction. Cancer Lett.451, 79–91 (2019).
Xu, W. W. et al. IGF2 induces CD133 expression in esophageal cancer cells to promote cancer stemness. Cancer Lett.425, 88–100 (2018).
Acknowledgements
We thank Dr. Robert Weinberg (Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, USA) for providing the AKT (T308D/S473D) plasmid. This work was supported by the National Natural Science Foundation of China (81803551, 81773085, 81672953, 81973339, 31770888), and the National Key Research and Development Program of China (2017YFA0505100).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G.M. Fimia
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, P., Liu, QW., Xie, Y. et al. Echinatin suppresses esophageal cancer tumor growth and invasion through inducing AKT/mTOR-dependent autophagy and apoptosis. Cell Death Dis 11, 524 (2020). https://doi.org/10.1038/s41419-020-2730-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-2730-7
This article is cited by
-
Application of Natural Medicinal Plants Active Ingredients in Oral Squamous Cell Carcinoma
Chinese Journal of Integrative Medicine (2024)
-
Autophagy-amplifying nanoparticles evoke immunogenic cell death combined with anti-PD-1/PD-L1 for residual tumors immunotherapy after RFA
Journal of Nanobiotechnology (2023)
-
Diosmetin suppresses the progression of ESCC by CDK2/Rb/E2F2/RRM2 pathway and synergies with cisplatin
Oncogene (2023)
-
Blockade of NMT1 enzymatic activity inhibits N-myristoylation of VILIP3 protein and suppresses liver cancer progression
Signal Transduction and Targeted Therapy (2023)
-
N4-acetylcytidine modification of lncRNA CTC-490G23.2 promotes cancer metastasis through interacting with PTBP1 to increase CD44 alternative splicing
Oncogene (2023)
