
Abstract
Phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoimidazole succinocarboxamide synthetase (PAICS), an essential enzyme involved in de novo purine biosynthesis, is connected with formation of various tumors. However, the specific biological roles and related mechanisms of PAICS in gastric cancer (GC) remain unclear. In the present study, we identified for the first time that PAICS was significantly upregulated in GC and high expression of PAICS was correlated with poor prognosis of patients with GC. In addition, knockdown of PAICS significantly induced cell apoptosis, and inhibited GC cell growth both in vitro and in vivo. Mechanistic studies first found that PAICS was engaged in DNA damage response, and knockdown of PAICS in GC cell lines induced DNA damage and impaired DNA damage repair efficiency. Further explorations revealed that PAICS interacted with histone deacetylase HDAC1 and HDAC2, and PAICS deficiency decreased the expression of DAD51 and inhibited its recruitment to DNA damage sites by impairing HDAC1/2 deacetylase activity, eventually preventing DNA damage repair. Consistently, PAICS deficiency enhanced the sensitivity of GC cells to DNA damage agent, cisplatin (CDDP), both in vitro and in vivo. Altogether, our findings demonstrate that PAICS plays an oncogenic role in GC, which act as a novel diagnosis and prognostic biomarker for patients with GC.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is the fifth most common malignancy and third leading cause of cancer-related death worldwide with the annual death rate in China accounting for more than 40% of the world1. Except for early surgical resection, chemotherapy is the main treatment for GC, which effectively prolongs survival and improves related symptoms of patients in stage IV2,3. However, the prognosis of most patients with advanced GC remains poor mostly because of chemotherapy resistance, which is typically caused by the highly heterogeneous and numerous genetic alterations of this malignancy4. Therefore, numerous studies should been conducted to explore the precise molecular mechanisms underlying the initiation and progression of GC to develop more effective interventions.
Phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoimidazole succinocarboxamide synthetase (PAICS) is a putative bifunctional enzyme involved in the de novo purine biosynthesis pathway with both 5-aminoimidazole ribonucleotide carboxylase and 4-(N-succinylcarbox-amide)-5-aminoimidazole ribonucleotide synthetase activities5. Previous study has demonstrated that cancer cells rely on the PAICS-dependent metabolic pathway for adenosine monophosphate and guanosine monophosphate synthesis, and inactivation of the de novo pathway inhibits cancer cell proliferation both in vitro and in vivo6. PAICS has been shown to be highly expressed in various types of cancer such as breast cancer7, prostate cancer8, bladder cancer9, lung cancer6,10, and acute lymphoblastic leukemia11. Moreover, overexpression of PAICS is predictive of the poor survival of patients with tumors6,8,9,10,12. A study of the oncogenic mechanism of PAICS has revealed that PAICS can induce epithelial-mesenchymal transition in bladder cancer by positively regulating SNAI1 and reducing E-cadherin expression9. In addition, the loss of MYC occupancy was observed on the PAICS promoter in presence of JQ1 in prostate cancer8. However, the specific biological roles and related mechanisms of PAICS in GC remain unclear.
The DNA damage response (DDR) greatly impacts genomic stability through DNA repair pathways, which ultimately determine cell fate13. Normal cells use precisely regulated DDR processes to protect their DNA from exogenous and endogenous DNA-damaging factors14,15. DNA double-strand breaks (DSBs) are the most serious type of DNA damage, as they result in the loss of chromosomal regions. The non-homologous DNA end joining (NHEJ) pathway and homologous recombination repair (HR) pathway are predominantly responsible for the restoration of DSBs16. Upon DDR, DNA damage sites are identified by the MRE11-DAD51-NBS1 (MRN) complex, which then recruits ataxia telangiectasia mutated kinase to these sites17,18,19. This kinase is activated immediately by phosphorylation, which then phosphorylates downstream effectors such as H2AX20. Mediator of DNA damage checkpoint protein 1 then binds to the phosphorylated H2AX (γH2AX) as a platform for DNA repair18,21,22. NHEJ repair mainly occurs in G1-phase through the recruitment of effectors such as 53BP1, Ku70/Ku80 complexes, which can promote the downstream proteins such as XRCC to repair the damaged DNA23. HR repair mainly occurs in S-G2M phase and involves the recruitment of effectors such as BRCA1/2 and DAD51, which can use sister chromatid as template for precise repair24,25,26. When DNA repair processes are dysfunctional, genetic predisposition to cancer tends to arise27.
In the present study, we investigated the role of PAICS in GC and explored related mechanisms of action. We found that PAICS expression was exceptionally upregulated in GC tissues, and high expression of PAICS was correlated with poor prognosis of patients with GC. In addition, PAICS inhibited GC cell apoptosis and promoted GC cell proliferation both in vitro and in vivo. More importantly, PAICS was engaged in DDR by directly interacting with HDAC1/2 and affected DNA damage repair by targeting DAD51. Significantly, knockdown of PAICS enhanced the sensitivity of GC cells to cisplatin (CDDP) both in vitro and in vivo, which provides new insights into GC therapy.
Results
PAICS is upregulated in GC tissues and related to the prognosis of patients with GC
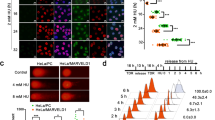
We first analyzed the expression of PAICS using the GEPIA database. The results indicated that PAICS was significantly overexpressed in various cancer tissues, including GC (Fig. 1a). We then confirmed the upregulated expression of PAICS in GC using the ULCAN database which contains 449 GC samples (p < 1 × 10−12) (Fig. 1b). In addition, we examined the immunohistochemistry staining results from the Human Protein Atlas project and found that PAICS was strongly expressed in GC tissues but weakly expressed in normal tissues (Fig. 1c). The Kaplan–Meier survival curves showed that GC patients with high PAICS expression had a poorer prognosis and worse overall survival rates compared to those with low PAICS expression (p = 5.7 × 10−7) (Fig. 1d). However, subsequent analysis revealed that PAICS expression was not correlated with cancer stage, subtype, or TNM stage (Fig. 1e–g).

a Boxplots represent PAICS expression level in breast invasive carcinoma (BRCA), prostate adenocarcinoma (PRAD), stomach adenocarcinoma (STAD), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC) and paired non-tumor tissues. b Relative expression of PAICS mRNA in normal and primary GC samples from TCGA. c Immunohistochemical analysis of PAICS in normal gastric tissue and GC tissue provided by the Human Protein Atlas project. Scale bar 50 μm. d Kaplan–Meier survival curves of overall survival in 499 GC patients based on PAICS expression level. e Relative expression of PAICS mRNA transcript in normal and stage 1, 2, 3 and 4 GC patients. f Relative expression of PAICS mRNA transcript in different histological subtypes. g Relative expression of PAICS mRNA transcript in normal and stage N0, N1, N2, and N3 GC patients.
Knockdown of PAICS inhibits GC cell proliferation and promotes cell apoptosis in vitro
We further investigated the role of PAICS in GC cell proliferation and apoptosis in vitro. Considering the high PAICS expression in GC, PAICS expression in the GC (AGS and SGC-7901) cell lines was therefore downregulated using short hairpin RNA-encoding lentiviruses (shCON, shPAICS#1, or shPAICS#2). Results of qRT-PCR and western blotting confirmed the consistent knockdown of PAICS in both the AGS and SGC-7901 cell lines (Fig. 2a). Data from CCK8 assay indicated that cell proliferation was suppressed with knockdown of PAICS (Fig. 2b). Consistently, knockdown of PAICS significantly decreased cell colony formation rate (Fig. 2c). In addition, cell apoptosis analysis by flow cytometry demonstrated an increased percentage of apoptosis in PAICS-knockdown cells (Fig. 2d). This finding was further supported by increases in cleaved caspases 3/8/9 proteins and a decrease in antiapoptotic protein Bcl-2 in the shPAICS groups compared with the shCON group (Supplementary Fig. S1a). Meanwhile, knockdown of PAICS significantly increased the activity of caspase-3 (Supplementary Fig. S1b). We also performed cell-cycle analysis and observed a significant decrease in the G1-phase and an increase in the S-phase, which suggested that PAICS knockdown disrupted cell-cycle progression with S-phase arrest (Supplementary Fig. S1c). Western blots showed that PAICS knockdown significantly decreased levels of S-phase-related proteins including CDC25A, cyclin A2 and CDK2 (Supplementary Fig. S1d). We further overexpressed PAICS in the stable knockdown cell line SGC-7901 (Supplementary Fig. S2a) and performed rescue assays to confirm the effects of PAICS on cell proliferation. Data from CCK8 assay showed that PAICS overexpression rescued the growth-inhibitory effect of GC cells induced by PAICS knockdown (Supplementary Fig. S2b). Moreover, we knockeddown expression of PAICS in the normal gastric epithelial cell line GES-1 and performed CCK8 assay to detect cell proliferation. Our results showed that knockdown of PAICS in normal gastric cells had little effect on cell proliferation (Supplementary Fig. S3a).

a Western blot and qRT-PCR showed PAICS-knockdown efficiency in AGS and SGC-7901 cells. Cell proliferation (b), colony formation (c), and apoptosis (d) of GC cell lines (AGS and SGC-7901) with or without PAICS knockdown. All data are shown as mean ± SD from three independent experiments. ***p < 0.001, **p < 0.01, *p < 0.05.
Knockdown of PAICS inhibits gastric carcinogenesis in vivo
The effect of PAICS on tumor growth in vivo was further evaluated by establishing a subcutaneous xenograft tumor model in nude mice using PAICS-knockdown or the control AGS cells. As expected, tumor growth was dramatically slower in the PAICS-knockdown group (shPAICS) compared with the control group (shCON) (Fig. 3a, b). In addition, similar results were observed in the mean tumor weight (Fig. 3c). Consistently, we also observed a significant decrease in the positive rate of Ki67 in the PAICS-knockdown group by immunohistochemistry analysis (Fig. 3d, e). In addition, we performed flow cytometry to measure the Ki67 level in PAICS-knockdown (shPAICS#1, shPAICS#2) and the corresponding control (shCON) cells and found that knockdown of PAICS decreased the percentage of Ki67 positive cells (Supplementary Fig. S3b).

a Subcutaneous tumor model of shCON- or shPAICS-AGS cells. b, c Tumor volume and weight were measured at the indicated weeks after mice were transplanted. d, e IHC analysis of Ki67 expression levels in tumor tissues obtained from the xenograft mice. The histogram indicates the Ki67 positive cells from panel. Data represent the mean ± SD from three independent experiments, ***p < 0.001, **p < 0.01, *p < 0.05.
PAICS is engaged in DDR in GC cells
To explore the functions of PAICS, CancerSEA was first used for single-cell analysis. The results indicated that PAICS was primarily involved in regulating the DNA damage and repair (Fig. 4a–d). To confirm this results, we first performed western blotting to investigate the expression levels of the DNA damage-related protein γH2AX in PAICS-knockdown or the control GC cell lines and found that knockdown of PAICS prominently increased γH2AX expression (Fig. 4e). In addition, the neutral comet assay showed that knockdown of PAICS significantly increased the tail DNA in GC cell lines, suggesting that PAICS deletion sustained the DNA damage (Fig. 4f). Concurrently, immunofluorescence analysis confirmed a marked increase in γH2AX foci formation with the knockdown of PAICS (Fig. 4g, h). We also performed rescue assays to confirm the effects of PAICS on DNA damage. Results of the neutral comet assay showed that PAICS knockdown induced DNA damage, which was indicated by a significant increase in tail DNA of GC cells; PAICS overexpression rescued the phenotype of GC cells induced by PAICS knockdown (Supplementary Fig. S2c).

a Single-cell analysis indicates that PAICS is primarily involved in regulation of the DNA damage and repair pathway. b–d Data from Kim KT (No. of cells = 126) demonstrate that PAICS mRNA expression is positively correlated with regulation of the DNA damage (c) and repair (d). e Western blot analysis of expression of γH2AX protein in shCON- or shPAICS-GC cells. f Representative images and quantification of comet assay of shCON- or shPAICS-GC cells. Scale bar 500 μm. g, h Immunofluorescence analysis of the formation of γH2AX foci in shCON- or shPAICS-GC cells. Scale bar 50 μm. Data represent the mean ± SD from three independent experiments, ***p < 0.001, **p < 0.01, *p < 0.05.
PAICS is involved in DDR by interacting with HDAC1 and HDAC2
To further explore the detailed mechanism of PAICS involved in DDR, co-immunoprecipitation and mass spectrometry assays were performed to identity the potential binding proteins of PAICS. The results suggested that PAICS interacted with HDAC1 (Fig. 5a and Table S1). Considering that HDAC1 often forms a complex with HDAC2 to participate in DDR, we therefore evaluated the interaction of PAICS with both HDAC1 and HDAC2 using co-immunoprecipitation assay. We transfected HEK293T cells with GFP-vector or GFP-tagged PAICS in the presence or absence of CDDP treatment and observed that PAICS interacted with both HDAC1 and HDAC2 (Fig. 5b). Concurrently, the interaction was confirmed by co-transfecting Flag-tagged HDAC1 with GFP-tagged PAICS in HEK293T cells (Fig. 5c). We also confirmed the interaction of PAICS and HDAC1/2 in SGC-7901 cells via endogenous immunocoprecipitation (Fig. 5d). In addition, we observed a much stronger interaction of PAICS with HDAC1/2 in response to CDDP treatment (Fig. 5e, f), suggesting that the HDAC-PAICS complex was involved in the DDR.

a, b HEK293T cells transfected with GFP-vector or GFP-PAICS were stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2-h recovery). Cell lysates were immunoprecipitated using anti-GFP. Immunoprecipitants or the whole-cell lysates (Input) separated by SDS-PAGE gel were stained with coomassie brilliant blue for mass spectrometric analysis (a); immunoprecipitants or the Input were analyzed by western blot with anti-HDAC1 and anti-HDAC2 (b). c HEK293T cells co-transfected with Flag-vector/GFP-vector or Flag-HDAC1/GFP-PAICS were stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2-h recovery). Cell lysates were immunoprecipitated using anti-GFP and immunoprecipitants or the Input were analyzed by western blot with anti-GFP and anti-Flag. d SGC-7901 cells lysates were immunoprecipitated using anti-HDAC1 and immunoprecipitants or the Input were analyzed by western blot with anti-PAICS, anti-HDAC1 or anti-HDAC2. e, f SGC-7901 cells stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2-h recovery) were collected. Cell lysates were immunoprecipitated using anti-HDAC1 or anti-HDAC2 and immunoprecipitants or the Input were analyzed by western blot with anti-PAICS, anti-HDAC1 or anti-HDAC2.
PAICS deficiency decreases DAD51 expression and inhibited its recruitment to DNA damage sites
We further detected the mRNA levels of PAICS in SGC-7901 cells treated with CDDP at different time points to explore the detailed regulatory mechanism of HDAC-PAICS in the DDR. The results showed increased PAICS mRNA levels in response to the DDR induced by CDDP as well as a decrease in the level of this gene with recovery (Fig. 6a). We also evaluated the PAICS protein levels. Interestingly, we found no obvious changes in PAICS protein expression either in CDDP-induced DDR or its recovery (Fig. 6b). Moreover, no obvious changes in HDAC1 or HDAC2 protein levels were observed (Fig. 6b). We next determined whether PAICS could regulate HDAC1/2 protein expression in the presence or absence of CDDP treatment and found no obvious effects on the expression of HDAC1 or HDAC2 protein (Fig. 6c). Upon DNA damage, HDAC1/2 is rapidly recruited to DNA damage sites to participate in DNA damage repair by promoting histone H3 lysine 56 (H3K56) deacetylation28,29. To further explore whether PAICS could affect the deacetylase activity of HDAC1/2, we then determined the acetylation level of H3K56 in the presence or absence of CDDP treatment and found a decrease in acetylated-H3K56 in CDDP-induced DDR, which was blocked by PAICS knockdown (Fig. 6d). This finding suggested that knockdown of PAICS affected the deacetylase activity of HDAC1/2. To confirm the effect of PAICS on HDAC1/2 deacetylation activity, we performed HDAC activity assay and found that knockdown of PAICS significantly decreased HDAC deacetylation activity in the presence of CDDP treatment (Supplementary Fig. S4). Notably, our results showed that knockdown of PAICS had no obvious effect on HDAC deacetylation activity in the absence of CDDP treatment (Supplementary Fig. S4). Previous studies have demonstrated that inhibiting the deacetylase activity of HDAC1/2 can reduce the expression and recruitment of repair-related proteins such as DAD51 to DNA damage sites, thus affecting the efficiency of DNA damage repair30,31,32,33. We then evaluated the expression of DAD51 and found that knockdown of PAICS significantly decreased the levels of DAD51 protein and mRNA in the presence of CDDP treatment (Fig. 6d, e). Concurrently, results of immunofluorescence assay showed that PAICS deficiency reduced the recruitment of DAD51 to DNA damage sites in the presence of CDDP-induced DDR (Fig. 6f). In addition, we performed the HR and NHEJ assays and found that knockdown of PAICS inhibited both HR and NHEJ repair pathways (Fig. 6g).

a qRT-PCR analysis of PAICS mRNA expression in SGC-7901 cells treated with CDDP (10 µg/mL for 3, 6, or 12 h followed by 2- or 12-h recovery). b Western blot analysis of PAICS, HDAC1/2 proteins levels in SGC-7901 cells treated with CDDP (10 µg/mL for 3, 6, or 12 h followed by 2- or 12-h recovery). c Western blot analysis of HDAC1 or HDAC2 protein levels in shCON- or shPAICS-SGC-7901 cells stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2 h recovery). d Western blot analysis of acetylated-H3K56 (Ace H3), DAD51 proteins levels in shCON- or shPAICS-SGC-7901 cells stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2 h recovery). e qRT-PCR analysis of DAD51 mRNA expression in shCON- or shPAICS-SGC-7901 cells stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2-h recovery). f Immunofluorescence analysis of the formation of DAD51 foci in shCON- or shPAICS-SGC-7901 cells stimulated with CDDP (10 µg/mL for 12 h followed by a 2-h recovery). Scale bar 50 μm. g Flow cytometry detection of HR and NHEJ repair efficiency in 293 T cells transiently transfected with siPAICS or siCON for 48 h. Data represent the mean ± SD from three independent experiments, ***p < 0.001, **p < 0.01, *p < 0.05.
PAICS deficiency enhances the sensitivity of GC cells to CDDP both in vitro and in vivo
Impaired DNA damage repair induced by HDAC inhibition can sensitize tumor cells to DNA-damaging agents34,35,36. We therefore investigated whether knockdown of PAICS could enhance the sensitivity of GC cells to CDDP, a well-known DNA-damaging agent for GC. Results of immunofluorescence showed that knockdown of PAICS in both AGS and SGC-7901 cell lines significantly increased the formation of γH2AX foci induced by CDDP treatment (Fig. 7a, b), suggesting that PAICS deletion enhanced the sensitivity of GC cells to CDDP. Flow cytometry evaluation confirmed that PAICS knockdown markedly sensitized GC cell lines to apoptosis induction by CDDP (Fig. 7c, d). We also observed that knockdown of PAICS significantly enhanced the inhibition of cell viability in SGC-7901 cells induced by CDDP treatment (Fig. 7e). Moreover, knockdown of PAICS significantly enhanced the growth inhibition of GC cells induced by CDDP treatment in the subcutaneous xenograft tumor model in vivo (Fig. 7f–h).

a, b Immunofluorescence analysis of the formation of γH2AX foci in shCON- or shPAICS-GC cells stimulated with or without CDDP (10 µg/mL for 12 h followed by a 2-h recovery). Scale bar 50 μm. c, d Flow cytometry detection of cell apoptosis in shCON- or shPAICS-GC cells (AGS and SGC-7901) treated with or without CDDP (10 µg/mL for 12 h followed by a 2-h recovery). e CCK-8 detection of cell viability in shCON- or shPAICS-SGC-7901 cells treated with increasing doses of CDDP for 48 h. f Subcutaneous tumor model of shCON- or shPAICS-SGC-7901 treated with CDDP or saline solution (0.9% NaCl). g, h Tumor volume and weight were measured at the indicated days after mice were transplanted. All data are shown as mean ± SD from three independent experiments. ***p < 0.001, **p < 0.01, *p < 0.05.
Discussion
As a key regulatory enzyme involved in the de novo purine biosynthesis pathway, PAICS plays an important role in maintaining normal metabolism5,6. Recent studies have demonstrated elevated expression of PAICS in various malignancies6,8,9,10,12, suggesting that PAICS affects the initiation and progression of tumors. Here, we identified for the first time that PAICS was upregulated in GC. Moreover, knockdown of PAICS in GC cells significantly inhibited cell growth in vitro and in vivo. Analysis of the detailed mechanism revealed that PAICS was involved in the DDR by interacting with HDAC1/2. In addition, the interaction between PAICS and HDAC1/2 was increased significantly in the presence of CDDP-induced DNA damage. Knockdown of PAICS impaired the deacetylase activity of HDAC1/2, reducing the expression and recruitment to DNA damage sites of DAD51, eventually interfering with DNA damage repair. More importantly, the deficiency of PAICS increased the sensitivity of GC cells to CDDP both in vitro and in vivo (Fig. 8).

Proposed mechanism for PAICS-induced gastric carcinogenesis and cisplatin sensitivity.
Our results showed that both HR and NHEJ efficiency were inhibited with PAICS knockdown. We further explored the detailed mechanism and found that knockdown of PAICS reduced the expression of DAD51 and inhibited the recruitment of DAD51 to DNA damage sites. DAD51 is well-known as an important DNA damage repair protein in HR repair37,38,39. The deficiency of DAD51 expression or its recruitment to DNA damage sites can greatly reduce the repair efficiency of HR37,38,39. Our results suggested that PAICS regulated HR by targeting DAD51. It has been reported that the HDAC1/2, as an essential deacetylase of histone, plays an important role in promoting DNA damage and repair40,41. Previous studies reported that the inhibition of HDAC1/2 activity affected the recruitment of NHEJ downstream genes, such as Ku70/80 and XRCC4 to the DNA damage sites, thus impairing the repair efficiency of NHEJ41. We found that knockdown of PAICS partially inhibited the deacetylase activity of HDAC1/2, suggested that PAICS regulated NHEJ by impairing HDAC1/2 activity. However, further studies are needed to explore the in-depth molecular mechanisms of PAICS in regulating NHEJ pathways.
In this study, we observed no obvious changes in HDAC1/2 protein levels either in CDDP-induced DDR or its recovery. Further analysis found that knockdown of PAICS significantly decreased HDAC deacetylation activity upon CDDP treatment. Notably, our results showed that knockdown of PAICS had no obvious effect on HDAC deacetylation activity in the absence of CDDP treatment. Moreover, the interaction of PAICS and HDAC1/2 was weak in GC cells without CDDP stimulation, which was significantly enhanced in CDDP-induced DNA damage. Therefore, we speculate that PAICS might impair HDAC1/2 deacetylation activity by interacting with the functional domains of HDAC1/2, which is regulated with a “switch-like” mechanism. Under normal conditions, PAICS interacts HDAC1/2 weakly and PAICS deficiency does not impaired HDAC1/2 deacetylation activity; upon DNA damage, the interaction of PAICS and HDAC1/2 is enhanced, which induces “switch on” and enhances HDAC deacetylation activity. In this case, knockdown of PAICS causes reduced HDAC deacetylation activity and series of events. The detailed mechanism of PAICS in regulating HDAC1/2 deacetylation activity remains unclear and needs further exploration.
Cisplatin is widely used as a first-line chemotherapy drug for patients with GC. However, the clinical application of this agent is largely limited because of inevitable drug toxicity and resistance42. HDAC inhibitors (HDACis) singly or in combination with other chemotherapy drugs have been demonstrated to be promising therapeutic strategies for cancer43,44,45. Despite of numerous HDACis being evaluated in clinical trials, SAHA and romidepsin have been approved by the FDA for treating cutaneous T-cell lymphoma46,47. HDACis have also been reported to enhance the cytotoxicity of cisplatin on tumor cells34. Consistently, we observed that knockdown of PAICS impaired HDAC1/2 deacetylase activity, thus increasing the sensitivity of GC cells to cisplatin. Cisplatin forms a cross-linked complex with DNA, which can induce DNA damage that leads to cancer cell death and apoptosis48. Increased histone acetylation during DNA damage repair causes the structure of DNA to change from tight to loose, providing favorable conditions for the recruitment of DNA damage repair proteins to DNA damage sites29. Under PAICS-deficient conditions, DNA damage could not be repaired immediately together when HDAC1/2 deacetylase activity was also impaired, maintaining the chromatin in an open state. This loose structure of chromatin promotes the binding of cisplatin to DNA, which induces DNA damage and increases cell death. Therefore, epigenetic chromatin modification may be an important factor that increases cisplatin sensitivity caused by PAICS deficiency.
Taken together, our findings support that PAICS, a novel oncogene of GC, is involved in the DDR by interacting with HDAC1/2. In addition, PAICS deficiency enhances the sensitivity of GC cells to CDDP both in vitro and in vivo, suggesting that PAICS is a new therapeutic target for patients with GC.
Materials and methods
Cell culture and CDDP administration
HEK293T cells and human GC cell lines (AGS and SGC-7901) were purchased from the American Type Culture Collection (Manassas, VA, USA). The normal gastric epithelial cell line GES-1 was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cell lines were certificated by STR analysis (Shanghai Biotechnology Co., Ltd., Shanghai, China). AGS, SGC-7901 and GES-1 cells were cultured in RPMI 1640 medium (Gibco, CA, USA) and HEK293T cells were cultured in Dulbecco’s modified Eagle’ medium (Gibco). Both media were supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco). All cells were maintained in a humidified incubator at 37 °C with 5% CO2. Cisplatin (CDDP) was purchased from Sigma (St. Louis, MO, USA) and dissolved in saline solution at a concentration of 1 mg/ml. For induction of DNA damage, cells were treated with 10 µg/mL CDDP for 12 h followed by a 2-h recovery.
Plasmids and transfection
Plasmids and transfection is described in detail in Supplementary Methods.
Gene expression analysis using publicly available datasets
Gene expression level of PAICS in GC and adjacent normal samples was obtained from GEPIA (http://gepia.cancer-pku.cn/detail.php), UALCAN (http://ualcan.path.uab.edu), Kaplan–Meier plotter (http://kmplot.com/analysis/) and Human Protein Atlas (https://www.proteinatlas.org) web portal, which provide histopathological images and boxplots depicting PAICS expression based on TCGA datasets. CancerSEA (http://biocc.hrbmu.edu.cn/CancerSEA/) that depicts single-cell functional status maps was used to analyse the roles of PAICS in DNA damage and repair.
Cell proliferation, colony formation, apoptosis, caspase-3 activity aassays, cell-cycle analysis, and flow cytometric analysis of Ki67
The assays were performed as detailed in Supplementary Methods.
Tumor xenograft model
The male BALB/c nude mice (5 weeks) were purchased from the Beijing Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). To determine the role of PAICS in vivo, AGS cells (5 × 106 cells/mouse) with stable knockdown of PAICS and the corresponding control were injected subcutaneously into the mice (7 mice/group). Tumor size was measured every seven days and calculated using the following formula: volume = (length × width2) × 0.5. The mice were sacrificed after 4–5 weeks and tumors were removed, weighed, fixed for immunohistochemistry detection of Ki67 (#9449, Cell Signaling Technology, CST, Danvers, MA, USA). For drug sensitivity assay, mice were injected subcutaneously with AGS cells (5 × 106 cells/mouse). When the tumors were measurable, the mice (4 mice/group) were randomly and blindly divided into the control group receiving saline solution (100 µL) and CDDP-treated group receiving CDDP (5 mg/kg dissolved in 100 µL saline solution) by intraperitoneal injection three times a week for 2–3 weeks. All animal studies were approved by the Animal Research Ethics Committee of Shanghai Tenth People’s Hospital (SHHDSRMYY-2019-4642).
RNA isolation, qRT-PCR, western blot analysis, and histone deacetylase activity assay
The assays were performed as detailed in Supplementary Methods.
HR and NHEJ assays
HEK293T cells were co-transfected with siPAICS or siCON (NC), together with DR-GFP (the negative control), GFP (the positive control) or DR-GFP + Isce-I (HR) or NHEJ, separately. After 48 h of transfection, cells were collected and subjected to examine the percentage of GFP (GFP%) using the FACSC antoTM II flow cytometer (BD). The HR or NHEJ repair efficiency was calculated using the following formula: (GFP% of HR/NHEJ − GFP% of negative control)/GFP% of positive control.
Immunofluorescence assay
Cells cultured on confocal dishes were fixed with 4% paraformaldehyde (Sigma) for 15 min at room temperature, and permeabilized with 0.5% Triton X-100 for 20 min at 37 °C. After blocking with 5% BSA for 1 h at room temperature, cells were incubated with the primary antibodies against PAICS (Abclone, 1:100), γH2AX (CST, 1:200) and DAD51 (Abcam, 1:100) at 4 °C overnight. The next day, cells were incubated with corresponding secondary antibodies against Cy™3 AffiniPure Donkey Anti-Mouse IgG (H+L) or Alexa Fluor 647 AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson) at 37 °C for 30 min. 4′,6-diamidino-2-phenylindole (DAPI, Sigma) was then applied to stain the nuclei. Finally, the images of immunofluorescence were obtained with the confocal laser-scanning microscope (Carl Zeiss, Jena, Germany).
Neutral comet assay
AGS and SGC-7901 cells with stable knockdown of PAICS and the corresponding control were collected and mixed with 0.5% low-melting point agarose, followed by being added to the slides covered with 1% normal-melting agarose. After solidifying at 4 °C for 20 min, the slides were immersed in the iced neutral lysis buffer at 4 °C for 4 h. Then, the electrophoresis was performed with untwisted DNA in the pre-cooled neutral electrophoresis buffer. Finally, the slides were stained with SYBR Green I and photographed by the inverted fluorescence microscope (Carl Zeiss). DNA damage was visualized as the percentage of tail DNA and tail moment.
Immunoprecipitation
Cells were collected and lysed with the NETN buffer (20 mM Tris-HCl pH 8.0, 0.5% Nonidet P-40, 200 mM NaCl, 1 mM EDTA) containing protease and phosphatase inhibitors (Sigma) for 30 min on ice. Then, equivalent proteins (1000 μg) were incubated with specific antibodies together with 30 μL protein A/G plus-agarose (Santa Cruz, Texas, USA) rocking at 4 °C overnight. The next day, the immunocomplexes were washed five times with cold NETN buffer, boiled in sample buffer and finally subjected to western blot analysis.
Statistical analysis
All data are shown as mean ± standard deviation (SD) from three independent experiments. Statistical analysis was performed using SPSS v22.0 (IBM, Armonk, NY, USA). Differences were analyzed by Student’s t-test for two groups or one-way ANOVA for multiple groups. All experiments were performed three times and the p values < 0.05 were considered statistically significant.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.68, 394–424 (2018).
Smyth, E. C. et al. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol.27, v38–v49 (2016).
Haj Mohammad, N. et al. Optimal first-line chemotherapeutic treatment in patients with locally advanced or metastatic esophagogastric carcinoma: triplet versus doublet chemotherapy: a systematic literature review and meta-analysis. Cancer Metastasis Rev.34, 429–441 (2015).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature499, 214–218 (2013).
Li, S. X. et al. Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J. Mol. Biol.366, 1603–1614 (2007).
Zhou, S. et al. Roles of highly expressed PAICS in lung adenocarcinoma. Gene692, 1–8 (2019).
Meng, M., Chen, Y., Jia, J., Li, L. & Yang, S. Knockdown of PAICS inhibits malignant proliferation of human breast cancer cell lines. Biol. Res.51, 24 (2018).
Chakravarthi, B. et al. Expression and role of PAICS, a de novo purine biosynthetic gene in prostate cancer. Prostate78, 693–694 (2018).
Chakravarthi, B. et al. A role for de novo purine metabolic enzyme PAICS in bladder cancer progression. Neoplasia20, 894–904 (2018).
Goswami, M. T. et al. Role and regulation of coordinately expressed de novo purine biosynthetic enzymes PPAT and PAICS in lung cancer. Oncotarget6, 23445–23461 (2015).
Zaza, G. et al. Acute lymphoblastic leukemia with TEL-AML1 fusion has lower expression of genes involved in purine metabolism and lower de novo purine synthesis. Blood104, 1435–1441 (2004).
Huang, Q., Liu, F. & Shen, J. Bioinformatic validation identifies candidate key genes in diffuse large-B cell lymphoma. Per Med.16, 313–323 (2019).
Spampinato, C. P. Protecting DNA from errors and damage: an overview of DNA repair mechanisms in plants compared to mammals. Cell Mol. Life Sci.74, 1693–1709 (2017).
Sugasawa, K. Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair44, 110–117 (2016).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature461, 1071–1078 (2009).
Chatterjee, N. & Walker, G. C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen58, 235–263 (2017).
Wilson, M. D. & Durocher, D. Reading chromatin signatures after DNA double-strand breaks. Philos. Trans. R Soc. Lond. B Biol. Sci.372, 20160280 (2017).
Stucki, M. et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell123, 1213–1226 (2005).
Shiloh, Y. & Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol.14, 197–210 (2013).
Matsuoka, S. et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science316, 1160–1166 (2007).
Stucki, M. & Jackson, S. P. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair5, 534–543 (2006).
Gottlieb, T. M. & Jackson, S. P. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell72, 131–142 (1993).
Menon, V. & Povirk, L. F. End-processing nucleases and phosphodiesterases: An elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair43, 57–68 (2016).
Wright, W. D., Shah, S. S. & Heyer, W. D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem.293, 10524–10535 (2018).
Truong, L. N. et al. Homologous recombination is a primary pathway to repair DNA double-strand breaks generated during DNA rereplication. J. Biol. Chem.289, 28910–28923 (2014).
McGlynn, P. & Lloyd, R. G. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol.3, 859–870 (2002).
Roos, W. P., Thomas, A. D. & Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer16, 20–33 (2016).
Tjeertes, J. V., Miller, K. M. & Jackson, S. P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J.28, 1878–1889 (2009).
Van, H. T. & Santos, M. A. Histone modifications and the DNA double-strand break response. Cell Cycle17, 2399–2410 (2018).
Adimoolam, S. et al. HDAC inhibitor PCI-24781 decreases DAD51 expression and inhibits homologous recombination. Proc. Natl Acad. Sci. USA104, 19482–19487 (2007).
Krumm, A. et al. Enhanced histone deacetylase activity in malignant melanoma provokes DAD51 and FANCD2-triggered drug resistance. Cancer Res.76, 3067–3077 (2016).
Lai, T. H. et al. HDAC inhibition induces microRNA-182, which targets DAD51 and impairs HR repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin. Cancer Res.22, 3537–3549 (2016).
Zhao, J. et al. Histone deacetylases 1 and 2 cooperate in regulating BRCA1, CHK1, and DAD51 expression in acute myeloid leukemia cells. Oncotarget8, 6319–6329 (2017).
Diyabalanage, H. V., Granda, M. L. & Hooker, J. M. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett.329, 1–8 (2013).
Liu, X. et al. HDAC1 silencing in ovarian cancer enhances the chemotherapy response. Cell Physiol. Biochem48, 1505–1518 (2018).
Pchejetski, D. et al. Histone deacetylases as new therapy targets for platinum-resistant epithelial ovarian cancer. J. Cancer Res Clin. Oncol.142, 1659–1671 (2016).
Godin, S. K., Sullivan, M. R. & Bernstein, K. A. Novel insights into DAD51 activity and regulation during homologous recombination and DNA replication. Biochem Cell Biol.94, 407–418 (2016).
Holthausen, J. T., Wyman, C. & Kanaar, R. Regulation of DNA strand exchange in homologous recombination. DNA Repair9, 1264–1272 (2010).
Krejci, L., Altmannova, V., Spirek, M. & Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res.40, 5795–5818 (2012).
Murr, R. et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol.8, 91–99 (2006).
Miller, K. M. et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol.17, 1144–1151 (2010).
Venerito, M., Vasapolli, R., Rokkas, T. & Malfertheiner, P. Gastric cancer: epidemiology, prevention, and therapy. Helicobacter23(Suppl 1), e12518 (2018).
Laschanzky, R. S. et al. Selective inhibition of histone deacetylases 1/2/6 in combination with gemcitabine: a promising combination for pancreatic cancer therapy. Cancers11, 1327 (2019).
Schizas, D. et al. Concept of histone deacetylases in cancer: Reflections on esophageal carcinogenesis and treatment. World J. Gastroenterol.24, 4635–4642 (2018).
Zhang, H., Shang, Y. P., Chen, H. Y. & Li, J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol. Res.47, 149–159 (2017).
Furumai, R. et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res.62, 4916–4921 (2002).
Grant, C. et al. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther.10, 997–1008 (2010).
Nikolova, T., Kiweler, N. & Kramer, O. H. Interstrand crosslink repair as a target for HDAC inhibition. Trends Pharm. Sci.38, 822–836 (2017).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81871727, 81572330, 81772941).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Amelio
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, N., Xu, C., Deng, L. et al. PAICS contributes to gastric carcinogenesis and participates in DNA damage response by interacting with histone deacetylase 1/2. Cell Death Dis 11, 507 (2020). https://doi.org/10.1038/s41419-020-2708-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-2708-5
This article is cited by
-
Targeting purine metabolism in ovarian cancer
Journal of Ovarian Research (2022)
-
Tumor suppressive role of microRNA-4731-5p in breast cancer through reduction of PAICS-induced FAK phosphorylation
Cell Death Discovery (2022)
-
Meta-analysis of mucosal microbiota reveals universal microbial signatures and dysbiosis in gastric carcinogenesis
Oncogene (2022)
