
Abstract
Head and neck squamous cell carcinomas (HNSCCs) harbor a subset of cells that are CD44(+) and present with malignancy and radiotherapy resistance. As a key regulator of self-renewal, Nanog expression not only determines cell fate in pluripotent cells but also mediates tumorigenesis in cancer cells; thus, we examined the role of Nanog in CD44(+) HNSCC. Three HNSCC cell lines, tumor xenografts, and patient tumors were examined. Nanog levels were significantly higher in CD44(+) HNSCC spheroids than in CD44(−) spheroids, and further increased when grown as spheroids to enrich for CSCs. CD44(+) spheroids showed a 3.4–7.5-fold increase in migration and invasion compared with CD44(−) spheroids and were resistant to radiation therapy, which was reversed by inhibiting Nanog. Nanog knockdown also decreased spheroid formation by 66.5–68.8%. Moreover, a phosphokinase array identified upregulated ERK1/2 signaling in CD44(+) HNSCC cells compared with that in CD44(−) cells. ERK1/2 signaling was found to regulate Nanog expression, aiding tumor progression, metastasis, and radiotherapy resistance. In xenograft models, the combination of radiation and Nanog or ERK1/2 inhibition inhibited tumor growth by 75.6% and 79.1%, respectively. In lung metastasis models, CD44(+) cells injected into the tail vein of mice led to significantly more lung metastases and higher Nanog expression level compared with that by ERK1/2-knockdown CD44(+) cells. Finally, in tumor tissues, CD44 and Nanog expression levels were correlated with tumorigenesis in HNSCC patients. Thus, targeting Nanog and the ERK1/2 signaling pathway may prevent or reverse CSC phenotypes and epithelial–mesenchymal transition that drive tumor progression, metastasis, and radiotherapy resistance in HNSCC.
Similar content being viewed by others


Introduction
Cancer of the head and neck (HNC) is the sixth most common cancer worldwide, with over 500,000 new cases diagnosed annually worldwide1. Approximately 80–90% of HNCs are squamous cell carcinomas (SCC). HNC globally resulted in >300,000 deaths every year, which made HNC the ninth most frequent cause of death from cancer2,3. Moreover, there are currently no reliable parameters to predict a long-lasting curative response or treatment outcomes.
Cancer stem-like cells (CSCs), also called tumor-initiating cells, are a small population of tumor cells that have the capacity to self-renew like normal stem cells, initiate tumor and drive tumor growth, invasion and metastasis4. A small number of CSCs were able to form tumors in non-obese diabetic/severe combined immunodeficient mice in vivo5. Interest in CSCs is rapidly increasing during the recent years, and existence of such cells in various of tumors has been verified including prostate6, melanoma7, lung8, liver9, breast10, brain11, pancreas12, ovary13, mesenchymal carcinomas,14 and head and neck15. In 2007, Prince et al.16 first identified a cellular subpopulation in head and neck tumors expressing the surface marker CD44 with stem-like characteristics; these cells were capable of reproducing when implanted into immunosuppressed mice. CD44 also has a critical role in cell metastasis and malignancy, and is involved in multiple steps17,18. In head and neck squamous cell carcinomas (HNSCCs), CD44 has been reported to be involved in metastasis and has also been identified as a CSC marker. However, critical molecular mechanisms and roles that maintain the ‘stemness’ of CSCs in the subset of patients with advanced HNSCC with high CD44 expression are largely unknown.
In this study, we sought to identify the role of CD44 and specific signaling pathways in the maintenance of CSC properties of HNSCCs. We demonstrated that the ERK1/2 signaling pathway has a critical role in the self-renewing and tumorigenic abilities of CSCs in HNSCC and in mouse xenograft models. Notably, our findings suggest that ERK1/2 indirectly upregulates Nanog expression via β-catenin, thereby maintaining the stemness of HNSCC CSCs. The ERK1/2-Nanog signaling pathway may thus be a novel target for eliminating CSCs in HNSCC.
Materials and methods
Cell lines and reagents
The human QLL-1 cell line (a SCC cell line originating from metastatic lymph nodes in oral cancer) were a generous gift from Dr. J. Shah (Memorial Sloan-Kettering Cancer Center, New York, NY). The human SCC-15 and SCC-25 cell lines (SCC cell lines originating from the tongue) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in minimum essential medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 0.4 μg/ml hydrocortisone, penicillin (50 mg/mL), and streptomycin (50 mg/mL); this was termed regular media. Cancer cell lines were actively passaged for <6 months from the time that they were received from the ATCC, and UKCCCR guidelines were followed19.
The ERK inhibitor U0216 (#662005), JNK1/2 inhibitor SP600125 (#420119), and p38 inhibitor SB590885 were purchased from Selleckchem (Houston, TX). Accutase (AT104) was obtained from Innovative Cell Technologies, Inc. (San Diego, CA). EGF (E9644) and bFGF (F5392) were purchased from Sigma-Aldrich (St. Louis, MO). B27 (17504044) and N2 (17502001) were obtained from Life Science, USA.
Spheroid formation
The human QLL-1, SCC-15, and SCC-25 Cell lines were resuspended in spheroid media, which consisted of Dulbecco’s modified eagle medium (DMEM)-F12 containing 20 ng/mL of EGF, bFGF, N2 (1×), and B27 (1×), and then plated on Ultra-Low Attachment culture dishes (Corning Inc., Corning, NY) as previously described20. Spheroids were collected after 5–7 days unless otherwise stated. Proteins were then extracted for analysis or cells were dissociated with Accutase and used for other experiments21.
sh.RNA lentiviral particles
Nanog was silenced via lentiviral transduction of human Nanog shRNA (SC-43958-V; Santa Cruz Biotechnology, Dallas, TX). ERK1/2 and β-catenin were silenced via lentiviral transduction of human ERK1 shRNA, ERK2 shRNA and β-catenin shRNA (SC-44252-V, SC-29307-V, and SC-35335-V; Santa Cruz Biotechnology). Scramble shRNA (sh.Scr) control constructs (SC-108080; Santa Cruz Biotechnology) were also used. Maximal knockdown occurred 72–96 h after transduction that was performed according to manufacturer’s instructions (Santa Cruz Biotechnology).
In vitro assays
Spheroids were dissociated using Accutase (#07920; STEMCELL Technologies Inc.), after which monolayer cells were collected with trypsin. To assay proliferation, 1 × 104 cells were plated onto 96-well flat bottom plates and maintained in regular media overnight. Water-soluble tetrazolium salt-1 (ab155902; abcam) assay was used to assess cell number after 3 days via optical density according to manufacturer’s instructions22. Soft agar colony formation from single cells was performed as previously described20. To measure migration and invasion, cells (2 × 104 cells/well) were suspended in 0.2 mL serum-free DMEM and loaded onto the upper wells of Transwell chambers (8-µm pore size, #3422; Corning Inc.); the lower wells were filled with 0.8 mL DMEM supplemented with serum. For the invasion assay, the upper wells of the chambers were precoated with BD Matrigel matrix (354234, BD Biosciences, Franklin Lakes, NJ) and 10 mg/mL growth factor; migration assays employed non-coated Transwell chambers. After incubation for 48 h at 37 °C, cells on the upper surface of the filter were removed with a cotton swab, after which invading or migratory cells on the lower surface of the filter were fixed and stained with a Diff-Quick kit (Thermo Fisher Scientific, Waltham, MA) and imaged at a magnification of ×20. Invasiveness and migration were quantified as the average number of cells in five microscopic fields per well via phase-contrast microscopy.
Fluorescence-activated and magnetic cell sorting
For fluorescence-activated cell sorting (FACS), cells were dissociated using Accutase and resuspended in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA). The cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD44 (BD555478; BD Biosciences) or isotype control antibody (BD555742; BD Biosciences) and then analyzed on a FACSCalibur platform (BD Biosciences) using Cell Quest software. CD44-positive cells were collected using a magnetic cell sorting system (MiltenyiBiotec, BergischGaldbach, Germany). In brief, cells were dissociated using Accutase, stained with CD44-Micro Beads, and passed through a LS magnetic column that retains CD44-positive cells. CD44-positive cells were then eluted from the column after removal of the magnet and quantified by immunofluorescence (IF) using FITC-conjugated CD44 antibodies.
Western blot analysis
Samples were collected in radioimmunoprecipitation (RIPA) buffer (Sigma-Aldrich) containing Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland), after which protein concentrations were determined by the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). Western blotting was performed using the following antibodies, ERK1 (sc-271270), ERK2 (sc-136288), and c-Myc (sc-40) from Santa Cruz Biotechnology; Sox2 (#2748, #3579), Oct-4 (#2750), Nanog (#4893), Slug (#9585), Snail (#3879), phospho-ERK1/2 (#9101), CD44 (#3578, #3570), E-cadherin (#14472), ERK1/2 (#4696), and cleaved caspase-3 (#9661) from Cell Signaling Technology (Danvers, MA); N-cadherin (BD610920) and E-cadherin (BD610181) from BD Biosciences; Zeb1 (NBP-1-05987) from Novus Biologicals (Centennial, CO); and β-actin from Sigma-Aldrich.
Real-time reverse transcription PCR
Total RNA was extracted from all cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer’s protocol. 500 ng of total RNA from cultured cell lines was converted to cDNA using RT2 First Strand kit (Cat.330401, Qiagen) and mixed with SYBR green master mix (Cat.201443, Qiagen) for qPCR using AIIA7 (Life Technologies). All primers were purchased from Qiagen and the glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Cat.PPH00150E, NM_002 NM_002046) gene was used to normalize the expression levels in subsequent quantitative analyses. To amplify the target genes encoding N-cadherin, Snail, Slug, and Zeb1, the following primers were used: N-cadherin (Cat.PPH00636F, NM_001792.3), Snail (Cat.PPH02459B, NM_005985), Slug (Cat.PPH02475A, NM_003068), and Zeb1 (Cat.PPH01922A, NM_030751.5) were used. The experiment was performed in triplicate.
Mouse experiments
All mouse protocols were approved by the MSKCC Institutional Animal Care and Use Committee. Tissues were dissected from the indicated mice. Paraffin-embedded sections were deparaffinized, and sections were examined by IHC as described below. For subcutaneous flank tumors, 1 × 107 CD4(+)-SCC-15 or SCC-25 cells previously transduced with sh.Nanog, sh.ERK1/2, sh.Scr (control) were resuspended in 100 μL Hank’s balanced salt solution (HBSS) and injected subcutaneously into the right flank of athymic, 7–9-week-old male BALB/c nu/nu mice following anesthesia with isoflurane. Mice were randomly assigned to different treatment groups (five mice per group) when tumors reached 100 mm3 in volume, which was designated as day 0. The mice were then treated with IR (8 Gy) intraperitoneally once a week. Tumors were measured 3× per week for 2 weeks, and tumor volume was calculated as length × (width)2 × 0.52. After the mice were exsanguinated, tumors were excised and cut into thirds. To assess tumor initiation in vivo, 5000 CD44(+) SCC-25 cells previously transduced with sh.ERK1/2, sh.Scr were resuspended in 100 mL HBSS and injected subcutaneously into the right flank of athymic, Rag2/ɣC double knockout mice (Taconic Biosciences, Rensselaer, NY) following anesthesia with isoflurane. Mice were monitored weekly for tumor growth for up to 7 weeks. For induction of lung metastases, mice were injected via the tail vein with 10,000 CD44(+) SCC-25 cells transduced with sh.ERK1/2 or sh.Scr. After killing the mice at different time points, tumor colonies in the lungs were counted. Each tumor was fixed in 10% buffered formalin for 24 h, embedded in paraffin, and processed into 5-µm sections.
Immunocytochemistry
Spheroids were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. Following fixation, cells were incubated with antibodies against Sox2 (#4900; Cell Signaling Technology), N-cadherin, Nanog (#8822; Cell Signaling Technology), Snail (ab53519; Abcam, Cambridge, UK), and/or CD44-FITC in a PBS solution with 1% BSA and 0.1% Triton X-100 at 4 °C overnight. The samples were then incubated with the secondary anti-mouse AlexaFluor 488 (A11005; Life Technologies, Carlsbad, CA) and anti-rabbit AlexaFluor 594 (A11012; Life Technologies) antibodies and counterstained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI). Imaging was performed using an inverted confocal microscope and images were processed using Imaris 7.6.
Immunohistochemistry and immunofluorescence
For IHC, formalin fixed, paraffin-embedded sections were deparaffinized by xylene and rehydrated. Sections were either stained with H&E or immunostained using the VECTASTAIN Elite ABC kit (Vector Laboratories Inc.) following manufacturer’s instructions and standard protocols23. Antibodies included CD44 (#3570, Cell Signaling Technology), CD24 (sc-11406, Santa Cruz Biotechnology), CD133 (MBS462020; MyBioSource), Musashi-1 (ab21628; abcam), Sox2 (#4900; Cell Signaling Technology), Oct-4 (#2750; Cell Signaling Technology), Nanog (#4893; Cell Signaling Technology), c-Myc (sc-40; Santa Cruz Biotechnology), Ki-67 (ab197234; abcam), cleaved caspase-3 (#9661; Cell Signaling Technology), ɣH2AX (Millipore 05-636), and Snail (sc-271977; Santa Cruz Biotechnology). Nuclei were also counterstained with DAPI for IF. Stained cells were visualized using an inverted confocal microscope and images were processed using Imaris 7.6.
Tissue microarray
To investigate the expression of CD44, phospho-ERK1/2, and Nanog in normal and HNSCC tissues, commercially available paraffin-embedded tissue array slides containing 45 HNSCC tissue and 5 corresponding normal tissues (A209 II; ISU ABXIS, Seoul, Republic of Korea) were purchased. Sections were deparaffinized, then incubated with anti-human CD44 (#3570; Cell Signaling Technology), CD24 (sc-11406; Santa Cruz Biotechnology), CD133 (MBS462020; MyBioSource), Musashi-1 (ab21628; abcam), Sox2 (#4900; Cell Signaling Technology), Oct-4 (#2750; Cell Signaling Technology), Nanog (#4893; Cell Signaling Technology), and c-Myc (sc-40; Cell Signaling Technology) in a PBS solution with 1% BSA and 0.1% Triton X-100 at 4°C overnight. The slides were then incubated with the secondary anti-mouse AlexaFluor 488 and anti-rabbit AlexaFluor 594 antibodies and counterstained with DAPI. The samples were then imaged using an inverted confocal microscope and images were processed using Imaris 7.6.
Tumor tissues from five patients with HNSCC from department of Head Neck Surgery were also made using a precision tissue array instrument (Chongqing Cancer Hospital, China). These patients presented with oral squamous cell carcinoma and laryngeal squamous cell carcinoma between July 2017 and December 2017 at the Chongqing Cancer Hospital (China). Ethical approval was obtained from hospital ethics committee before the beginning of the study and all participants signed an inform consent form. A representative core biopsy (2 mm in diameter) was obtained from each tumor and embedded. IF for CD44, Nanog, phospho-ERK1/2, and Snail was performed as described above.
Statistical analysis
All experimental data are reported as mean and the error bars represent the standard deviation (±SD) of triplicate samples. Statistical analysis was performed using Instant 3.10 software (GraphPad). P values were calculated using the Student’s t test. For comparisons among two groups or more, treatment groups were compared to the control group using a one-way analysis of variance with Bonferroni adjustment for multiple comparisons.
Results
Nanog protein is increased in HNSCC spheroids, CD44(+) cells, and patient tissues
Self-renewal is one of the important properties employed by CSCs to maintain their proliferative capacities15. To assess the clinical relevance of CSC markers and self-renewal proteins in human HNSCC tissues, we compared the expression levels of CSC markers (CD24, CD44, CD133, and Musashi-1) and self-renewal proteins (Sox2, Oct-4, Nanog, and c-Myc). All self-renewal proteins and CD44 were overexpressed in tumor tissues compared with normal tissues (Figs. 1a, b, Suppl. Fig. 1A, B). Interestingly, Nanog expression in CD44(+) cells was ~38.6% by immunofluorescence (Fig. 1c). We thus hypothesized that Nanog has a major role in CD44(+)-HNSCC cells as a self-renewal protein and a new “master gene” of tumorigenesis in HNSCC. We first confirmed that all HNSCC cell lines (SCC-15, SCC-25, and QLL-1) when grown as spheroids showed increased expression of CD44, whereas the levels of other putative CSC markers such as CD24 and CD133 were not reliably increased (Fig. 1d, Suppl. Fig. 1C). The percentage of CD44(+) cells in spheroid-forming media varied from 7.2% in QLL-1 cells to 9.3% in SCC-15 cells (Suppl. Fig. 1D, E) by FACS. Furthermore, in spheroid cell and CD44(+) HNSCC cells, Sox2 and Nanog levels were found significantly increased, but not Oct-4 and c-Myc (Fig. 1e, f).

a, b Graphs of the positive area (%) per field for the CSC marker proteins CD24, CD44, CD133, and Musashi-1 and the self-renewal proteins Sox2, Oct-4, Nanog, and c-Myc. c Immunofluorescence (IF) staining of commercially available tissue array slides containing 45 HNSCC and 5 corresponding normal tissues with DAPI (blue), CD44 (green), and Nanog (red). Scale bar = 20 µm. d, e Western blotting of CSC marker proteins and self-renewal proteins in three HNSCC cell lines grown as monolayers or as spheroids. f Western blotting of CD44, Sox2, Oct-4, Nanog, and c-Myc following FACS for CD44(−) and CD44(+) cells. g IF of CD44(+) SCC-15 and QLL-1 cells transduced with sh.NANOG or sh.Scr. Scale bar = 50 µm. h GFP-expressing cell in a single cell assay using CD44(+) SCC-15 and QLL-1 cells following transduction with GFP, or NANOG shRNA (sh.Nanog), or scrambled shRNA (sh.Scr). Error bars represent standard deviation. *p < 0.05.
Because CD44(+) HNSCC cells lines were grown in a single cell assay under spheroid formation conditions, whereas the CD44(−) cells were not (Suppl. Fig. 1F), we examined whether Nanog contributes to spheroid formation. First, we knocked down NANOG in the two CD44(+) cells by transduction with sh. NANOG or sh.Scr (control). Nanog- knockdown in vitro did not affect cell growth (Suppl. Fig. 1G). Compared with sh.Scr in CD44(+) cells, sh.NANOG in CD44(+) cells showed decreased Nanog and CD44 protein expression (Fig. 1g). NANOG-knockdown cells showed a 66.5–68.8% decrease in spheroid formation compared with that of control cells transduced with sh.Scr (Fig. 1h). These results suggest that HNSCCs are sustained by the self-renewal and tumor initiation properties mediated by Nanog expression in CD44(+) cells.
Nanog has a critical role in CD44(+)-HNSCC cells with a mesenchymal phenotype
CD44 has an important role in communications and cell–matrix interactions and is involved in cell motility, proliferation, and survival17,18. CD44(+) cells were more malignant than CD44(−) cells (Suppl. Fig. 2A). Epithelial–mesenchymal transition (EMT) is essential for initiation of metastasis for cancer progression23 and is driven by the transcription factors SNAI1, SLUG, ZEB1/2, or TWIST1/224. CD44(+)-HNSCC cells showed decreased expression of E-cadherin and increased Snail expression as EMT-related protein (Suppl. Fig. 2B). CD44(+) cells also showed increased invasion and migration by 3.4–5.1-fold and 5–7.5-fold, respectively, compared with control (Suppl. Fig. 2C). We next examined the role of Nanog in the migration and invasion of CD44(+)-HNSCC cells. CD44(+)-HNSCC cells with sh.NANOG showed a 4.2-12.4-fold decrease in invasion and 4.4-6.3-fold decrease in migration compared with that of CD44(+)- HNSCC cells with sh.Scr (Figs. 2a, b). NANOG-knockdown in CD44(+) cells also exhibited reduced expression of N-cadherin and Snail. (Fig. 2c, d, Suppl. Fig. 2D). Moreover, we confirmed that CD44(+) cells transduced with Sox2, Oct-4, or c-Myc shRNA did not alter invasion or migration in vitro (Suppl. Fig. 2E). Knockdown was confirmed by western blot (Suppl. Fig. 2F). Furthermore, NANOG-knockdown was found to reduce the colony formation of CD44(+) HNSCC cells by 26.6–92.2% (Fig. 2e). Altogether, these results indicate that the self-renewal protein Nanog promotes EMT and acquisition of CSC properties in HNSCC.

a Invasion and b migration assays of CD44(+) cells transduced with sh.NANOG or sh.Scr. c Western blotting of Nanog, CD44, and EMT-related proteins, N-cadherin, Snail, Slug, and Zeb1 in CD44(+) cells transduced with sh.NANOG or sh.Scr. d Immunostaining of CD44(+) cells transduced with sh.NANOG or sh.Scr for N-cadherin and Snail. e Colony formation of CD44(+) cells following transduction sh.NANOG or sh.Scr. Scale bar = 20 µm. Error bars represent standard deviation. *p < 0.05.
Nanog inhibition in CD44(+)-HNSCC cells relieves radiotherapy resistance
Numerous studies have demonstrated that CSCs are generally resistant to chemotherapy25,26,27. We thus assessed the sensitivity of HNSCC cells to radiation treatment (IR), which is commonly used for the treatment of advanced HNSCC. Monolayers showed a moderate response to IR with a 51.5–61.2% reduction in proliferation, whereas spheroids showed a reduced response to IR with only a 17.1–20.1% reduction (Suppl. Fig. 3A). Next, we stably transduced CD44(+) cells with sh.Scr or sh.NANOG (Fig. 3a). NANOG-knockdown alone reduced proliferation in CD44(+) cells by only 7.5–11.5%, whereas the combination of sh.NANOG and radiotherapy showed more than an additive effect with decreases in cell viability ranging from 57.5 to 71.9% (Fig. 3b).

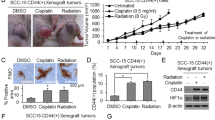
a Western blotting of Nanog in CD44(+) cells transduced with sh.NANOG or sh.Scr. b Proliferation assays of CD44(+) HNSCC cells following treatment with sh.NANOG, sh.Scr, or 8 Gy radiotherapy. c Tumor growth curves of CD44(+) SCC-25 cell xenografts after treatment with sh.NANOG, sh.Scr, or 8 Gy. d Representative tumor images from each treatment group on the 15th day. e Immunofluorescence (IF) analysis of treated tumors for proliferation (Ki-67, green), apoptosis (cleaved caspase-3, red), ɣH2AX (DNA damage, white), and CD44 (green). Scale bar = 20 µm. Error bars represent standard deviation. *p < 0.05.
The effects of Nanog inhibition and radiotherapy on SCC-25 mouse xenografts were examined. The tumor-initiating ability in CD44(+)-SCC-25 cell was substantially reduced in cells with suppressed Nanog expression (Suppl. Fig. 3B). These 1 × 107 CD44(+)-SCC-25 cells were then injected into the flanks of immunodeficient mice. When tumors were ~100 mm3 in volume, the mice were randomized into IR-treated (8 Gy) or untreated groups. Tumors of untreated mice grew to ~ 750–800 mm3 in just 15 days following randomization. On the other hand, tumors of mice injected with sh.NANOG in CD44(+)-SSC-25 cells or treated with 8 Gy grew to an average size of 507.9 mm3 and 421.2 mm3, respectively. The combination of sh.NANOG and 8 Gy dramatically inhibited tumor growth, with tumors growing to an average of 168.4 mm3 (75.6% less). (Fig. 3c). After 15 days, xenografts were harvested and analyzed (Fig. 3d). The combined sh.NANOG and 8 Gy-treated tumors showed a dramatic increase in the apoptosis marker cleaved caspase-3 and ɣ-H2AX as well as a decrease in the CSC marker CD44. (Fig. 3e, Suppl. Fig. 3C). Thus, these findings indicate that CD44(+)-HNSCC cells are relatively resistant to radiotherapy, which can be overcome by inhibiting Nanog.
ERK1/2 signaling pathway promotes EMT and acquisition of CSC phenotypes in CD44(+)-HNSCC cells
Mitogen-activated protein kinases (MAPKs) are a highly conserved family of serine/threonine protein kinases involved in several fundamental cellular processes as well as the survival of cancers and CSCs28,29. To investigate the possible role of specific kinases in the stemness of HNSCC CSCs, we first examined the activation status of MAPKs in CD44(+) and CD44(−) cells. The expression and phosphorylation of 44 proteins were analyzed using SSC-15-CD44(−) and CD44(+) spheroids (Suppl. Fig. 4A). Relative expression levels and protein phosphorylation were quantitated by densitometric analysis and normalized to those of CD44(−) cells (Fig. 4a). We found that the extracellular signal-regulated protein kinases 1/2 (ERK1/2) were highly activated in CD44(+)-HNSCC cells (Fig. 4a). Moreover, the levels of MAPKs including phosphorylation ERK1/2 and JNK1/2 were differentially increased in CD44(+)-HNSCC by western blot (Fig. 4b).

a Phospho-RTK protein array analysis using CD44(+) and CD44(−) cells. b Western blotting of CD44, Nanog, phosphorylated ERK1/2, total ERK2, phosphorylated JNK1/2, total JNK2, phosphorylated p38, total p38, and β-actin in CD44(+) and CD44(−) cells. c Western blotting of total ERK1/2, total ERK1, total ERK2, CD44, and Nanog in CD44(+) cells transduced with sh.ERK1/2 or sh.Scr. d IF of CD44(+) cells transduced with sh.ERK1/2 or sh.Scr for CD44 and Nanog. e Single cell assay and f colony formation assay of CD44(+) SCC-15 and QLL-1 cells following transduction with sh.ERK1/2 or sh.Scr. g Migration and Invasion assay of CD44(+) SCC-15, SCC-25, and QLL-1 cells following transduction with sh.ERK1/2 or sh.Scr. h Western blotting of ERK1/2, N-cadherin, Snail, Slug, Zeb1, and β-actin and immunofluorescent images of N-cadherin and Snail staining in CD44(+) cells transduced with sh.ERK1/2 or sh.Scr. Scale bar = 20 µm. Error bars represent standard deviation. *p < 0.05.
We next examined the contribution of ERK1/2 to stemness and EMT. We established CD44(+)-SCC-15 and -QLL-1 cells were transduced with sh.ERK1 and sh.ERK2 or sh.Scr, and ERK1/2 knockdown was confirmed by western blot (Fig. 4c). ERK1/2 knockdown in vitro did not affect cell growth (Suppl. Fig. 4B), but resulted in a significantly decrease of CD44 and Nanog expression (Figs. 4c, d). Similar results were obtained when the ERK1/2 inhibitor U0126 was used instead of ERK1/2 signaling pathway inhibition (Suppl. Fig. 4C). ERK1/2 inhibition by shRNA or U0126 reduced spheroid diameter at 7 days by 64.2–72.9% (Fig. 4e, Suppl. Fig. 4D). We also performed soft agar colony formation and found that ERK1/2 shRNA or U0126 reduced colony formation of CD44(+)-HNSCC cells by 64.6–75.1% (Fig. 4f, Suppl. Fig. 4E). JNK1/2 inhibitor, SP600125 had little effect on colony formation, and no effects were observed after p38 inhibition with SB202190 in CD44(+) cells (Suppl. Fig. 4E). Western blot also indicated that JNK and p38 inhibitors did not alter the expression levels of CD44 and Nanog in CD44(+) cells (Suppl. Fig. 4F,G). Furthermore, ERK1/2 inhibition reduced CD44(+) HNSCC cell migration and invasion by 75.4–92.3% and 80.1–87.5%, respectively (Fig. 4g). Expression of N-cadherin and Snail were also decreased after ERK1/2 inhibition in CD44(+) HNSCC cells (Fig. 4h). Thus, our results indicate that CD44(+)-HNSCC cells are relatively malignant, which can be overcome by inhibiting the ERK1/2-Nanog pathway.
ERK1/2 signaling increases EMT and acquisition of CSC phenotypes in xenograft mouse models
To confirm the possible role of the ERK1/2 pathway in the stemness of HNSCC, CD44(+)-SCC-15 cells with sh.ERK1/2 or sh.Scr were injected into the flanks of immunodeficient mice; knockdown was confirmed by western blotting (Fig. 5a). Once tumors reached a volume of 100 mm3, the mice were randomized into IR-treated (8 Gy) or untreated groups. Tumors of untreated mice grew to over 936–1030 mm3 in just 20 days following randomization. Tumors of mice injected with sh.ERK1/2 in CD44(+)-SCC-15 cells or treated with 8 Gy grew to an average size of 583.2 mm3 and 566.4 mm3, respectively. The combination of sh.ERK1/2 and 8 Gy further inhibited tumor growth, with tumors growing to an average of 213.6 mm3 (79.1% less) (Fig. 5b). After 20 days, the xenografts were analyzed (Fig. 5c) and we found that combining sh.ERK1/2 and IR treatment resulted in a marked decrease in CD44, Nanog, Snail, and an increase in the apoptosis marker cleaved caspase-3 (Fig. 5d, Suppl. Fig. 5A). Moreover, we confirmed that ERK1/2 signaling induces resistance to cisplatin or 5-fluorouracil (5-FU) in CD44(+) cells. However, there was a more than additive effect when ERK1/2 inhibition and cisplatin or 5-FU were added to CD44(+) cells, with decreases in cell viability ranging from 52.1% to 68.4% (Suppl. Fig. 5B). Altogether, these in vivo results also indicate that HNSCC stem-like cells are relatively malignant and that this malignancy can be overcome by inhibiting the ERK1/2-Nanog pathway.

a Western blotting of ERK1/2, ERK1, and ERK2 in CD44(+) cells transduced with sh.ERK1/2 or sh.Scr. b Tumor growth curves of CD44(+) SCC-15 cell xenografts after treatment with sh.ERK1/2, sh.Scr, or 8 Gy. c Representative images of tumors from each treatment group on the 20th day. d IF analysis of treated tumors for CD44 (green), Nanog (red), and Snail (white) in SCC-15 xenografts treated with sh.ERK1/2, sh.Scr, or 8 Gy. Scale bar = 50 µm. Error bars represent standard deviation. *p < 0.05.
The ERK1/2-Nanog signaling pathway controls stemness and metastasis in mouse models
To further examine the involvement of the ERK1/2 signaling pathway on tumor growth, stemness, and invasiveness in vivo, we created flank tumor xenografts using 5000 CD44(+)-SCC-25 cells transduced with sh.ERK1/2 or sh.Scr and injected them into the immunodeficient mice; knockdown was confirmed by western blot (Suppl. Fig. 6A). All five mice implanted with CD44(+)-SCC-25 cells grew flank tumors, whereas one of five mice that received ERK1/2 shRNA-transduced CD44(+) cells grew a flank tumor. (Fig. 6a, Suppl. Fig. 6B). Control- CD44(+) cells formed tumors with an infiltrating-leading edge, whereas ERK1/2-knockdown CD44(+) cells exhibited a well-defined border between the tumor and normal surrounding tissues, as evidenced by H&E and IF staining (Fig. 6b). ERK1/2 knockdown also reduced the number of infiltrating CD44(+) cells expressing Snail from 14.3 per mm2 to 3.4 per mm2 (Fig. 6b). We next examined the effect of ERK1/2 knockdown on the ability of CD44(+) cells to metastasize using an athymic nude mouse model. CD44(+)-SCC-25 cells injected into the tail vein of mice led to significantly more lung metastases compared with ERK1/2-knockdown cells. The average number of lung metastatic nodules was 9.5 nodules on day 30 in control group and 1.2 nodules on 35 day in ERK1/2 shRNA group (Fig. 6c). When lung metastases were examined for CD44 and Nanog expression, ERK1/2-knockdown in CD44(+)-SCC-25 cells group showed dramatically decreased CD44 and Nanog expression levels compared with that of CD44(+)-SCC-25 cells group (Fig. 6d). To further investigate the role of phosphorylated ERK1/2 and Nanog expression in HNSCC patients, we examined the expression of phosphorylated ERK1/2, CD44, Nanog, and Snail in the tumor tissues of five patients (Fig. 6e). All patient samples exhibited high levels of phosphorylated ERK1/2, CD44, Nanog, and Snail in tumor tissues. Taken together, our findings indicate that the ERK1/2-Nanog signaling pathway is involved the stemness and metastasis in HNSCCs.

a Photos of immunodeficient mice injected with 5000 CD44(+) SCC-25 cells stably transduced with sh.Scr, or sh.ERK1/2. b Hematoxylin and Eosin (H&E) and IF staining of infiltrating cells for DAPI (blue), CD44 (green), and Snail (red). Dashed line indicates tumor border. The graph shows the number of CD44+/Snail+ infiltrating cells. c Representative images and quantitation of detectable lung nodules on the surface of whole lungs. d Lung metastasis from immunodeficient mice following H&E and IF staining for DAPI (blue), CD44 (red), and Nanog (green). e Representative images of patient tumor tissues following IF staining for DAPI (blue), CD44 (green), Nanog (red), p-ERK1/2 (yellow), and Snail (white). Error bars represent standard deviation. *p < 0.05.
Discussion
This study is the first to demonstrate the central role of the ERK1/2 and Nanog signaling pathway in the maintenance of stemness and radiotherapy resistance in a subset of HNSCC cells with CD44 expression. We grew three different head and neck cancer cell lines as spheroids and found enrichment of the CSC marker CD44 as well as increased levels of Nanog protein and phosphorylation of ERK1/2. The spheroid cell lines exhibited increased CD44 and Nanog expression compared with monolayer cells in HNSCC. Inhibiting either ERK1/2 signaling or Nanog in spheroid or CD44(+) cells using shRNA or inhibitor can reduce Nanog expression, spheroid formation, and anchorage-independent growth in soft agar. CD44(+)-HNSCC spheroids were also highly resistant to radiation therapy, which was reversed with ERK1/2 or Nanog inhibition. In mouse xenografts, inhibition of Nanog or ERK1/2 pathway acted synergistically with radiotherapy to block tumor growth, and histologic examination of treated tumors found synergistic increases in tumor cell apoptosis and depletion of CD44(+) cells. Moreover, ERK1/2 inhibition in CD44(+) cells reduced the expression of N-cadherin and Snail, greatly diminished migratory and invasive capabilities, attenuated the infiltrative nature of xenografts, and reduced lung metastases.
CD44 as cell surface receptor has an important role in cell communications and is involved in cell motility, proliferation, and survival18. CD44(+) subpopulation of cells with CSC properties have been reported in HNSCC and a high frequency of CD44+ cells has been correlated with known poor prognostic factors such as advanced T classification and recurrence30. CD44 overexpression appears to reflect cellular invasiveness and leads to increased aggressiveness of tumors in the head and neck31. Understanding CD44 is important in the study of tumor progression and invasiveness because invasive tumors attack the extracellular matrix of surrounding tissues to expand; moreover, the interaction between CD44 and hyaluronic acid has a decisive role in various cellular pathways. In addition, CD44 is the most frequently used marker for SCC CSCs31,32. This study showed that the FACS-sorted CD44(+)-HNSCC population increased spheroid formation, migration, and invasion compared with CD44(−) cells. In addition, CD44(+) cells were resistant to the cytotoxicity induced by radiation therapy. Thus, CD44 can be used as a marker for the CSC-enriched fraction in HNSCCs.
Although the relationship between ERK activation and CSC function has not been extensively studied in HNSCC. It has been reported that the ERK1/2 or Wnt/β-catenin signaling pathway is vital for CSC tumorigenicity and enhances the tumorigenicity of colon cancers and gastric cancer33,34. Moreover, in prostate cancer, it has been demonstrated that the MEK-ERK pathway contributes to the role of EGFR in maintaining prostate CSC stemness35. In rhabdomyosarcoma, the U0126 dramatically prevented rhabdo-sphere formation and downregulated the stem cell markers CD133, CXCR4, and Nanog expression36. In this study, we used a human phosphokinase array with CD44(−) and CD44(+)-SSC-15 spheroids and identified several activated kinases, including phosphorylation of ERK1/2, AKT, and JNK, among others. We also found that ERK1/2-β-catenin signaling pathway promotes Nanog expression in head and neck CSCs (Suppl. Fig. 7). Inhibition of ERK1/2 signaling and Nanog led to decreased spheroid formation, migration, invasion, and EMT proteins. The ERK1/2 signaling was found to have a critical role in the maintenance of the malignant CSC population in HNSCC. Thus, ERK1/2-Nanog signaling may be a main pathway supporting CSC function in HNSCC.
EMT is associated with the acquisition and maintenance of stem cell-like characteristics and is sufficient to endow differentiated normal and cancer cells with stem cell properties; moreover, CSCs often exhibit EMT properties37. The link between EMT and CSCs has been examined in numerous studies, but the recently uncovered link between EMT activation and the acquisition of stem cell-like properties suggests that EMT may be a mechanism for generating CSCs38,39. Snail is a master regulator that promotes EMT by repressing epithelial markers and upregulating mesenchymal markers, it also mediates invasiveness and metastasis in many different types of malignant tumors including HNSCC40,41. Furthermore, other groups demonstrated that Snail expression may regulate treatment resistance and the CSC-like properties of HNSCC42,43. It has also been reported that Snail is an independent marker of tumor metastasis in HNSCC patients41. In this study, spheroids and CD44(+) cells showed elevated expression of the EMT-associated proteins Snail and N-cadherin and exhibited significantly enhanced migration and invasion. Inhibition of Nanog or the ERK1/2 signaling pathway in HNSCC CSCs downregulated Snail and N-cadherin and reduced migration and invasion.
HNSCC patients are at risk for both lung metastases and secondary primary cancers of the lung44. In this study, the increased motility seen for CD44(+) cells is characteristic of cells undergoing EMT, which may explain why head and neck CSCs metastasized in vivo, whereas CD44(+) cells with sh.ERK1/2 did not. Indeed, Takahashi et al.45 showed that in tumor necrosis factor-induced EMT, the interaction between CD44 and hyaluronan-mediated cell–cell dissociation and actin remodeling, thereby enhancing motility. These findings, in conjunction with our own, suggest that cell motility and the ability to undergo EMT are the most important characteristics of a metastatic cell, and it appears that CSCs possess these capabilities. Thus, our findings show that targeting Nanog or ERK1/2 signaling pathway can affect both CSCs and EMT-associated phenotypes in different cancer types, including HNSCC.
Radiation therapy is important for the treatment of HNSCC; however, standard therapies possess several limitations as they exhibit side effects and relapse in >50% of HNSCC patients. Recently, CSCs were demonstrated to have important implications regarding the treatment of many cancers, including HNSCC. CSCs are reported to endow resistance to chemo and radiotherapy46,47. But it remains unclear whether head and neck CSCs may also be resistant to targeted therapies. Other group demonstrated that resistance to radiotherapy frequently occurs in HNSCC CSCs and is associated with poor outcomes44. Furthermore, it has been reported that targeting molecules or signaling pathways is important for curing cancers including HNSCC48. It has been established that the PI3K/AKT pathway contributes to radio-resistance in many cancers48,49. In this study, we observed that the stemness factor Nanog was involved in radio-resistance. Our findings thus support a potential therapeutic strategy to inhibit the ERK1/2-Nanog pathway in the CD44(+) subgroup of HNC cells. Such therapies are urgently needed to overcome the challenge of radiotherapy resistance.
In conclusion, our findings indicate that HNSCCs maintain a CSC population via the ERK1/2 signaling pathway and Nanog. ERK1/2 activation is a critical pathway for self-renewal of the HNSCC CSC population, and likely acts through regulation of Nanog expression. These findings may suggest a new therapeutic target in HNSCC CSCs.
Change history
15 March 2024
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41419-024-06608-7
References
Wang, C. et al. Targeting head and neck cancer by vaccination. Front. Immunol. 9, 830 (2018).
Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386, 743–800 (2015).
GBD 2013 Mortality and Cause of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385, 117–171 (2015).
Baillie, R., Tan, S. T. & Itinteang, T. Cancer stem cells in oral cavity squamous cell carcinoma: a review. Front. Oncol. 7, 112 (2017).
Quintana, E. et al. Efficient tumour formation by single human melanoma cells. Nature 456, 593–598 (2008).
Jaworska, D., Krόl, W. & Szliszka, E. Prostate cancer stem cells: research advances. Int J. Mol. Sci. 11, 27433–27449 (2015).
Kumar, D., Gorain, M., Kundu, G. & Gopal, C. K. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 16, 7 (2017).
Sullivan, J. P., Minna, J. D. & Shay, J. W. Evidence for self-renewing lung cancer stem cells and their implications in tumor initiation, progression, and targeted therapy. Cancer Metastasis Rev. 29, 61–72 (2010).
Nio, K., Yamashita, T. & Kaneko, S. The evolving concept of liver cancer stem cells. Mol. Cancer 16, 4 (2017).
Wenjun, G. Concise review: breast cancer stem cells: regulatory network, stem cell niches, and disease relevance. Stem Cells Transl. Med 3, 942–948 (2014).
Abou-Antoun, T. J., Hale, J. S., Lathia, J. D. & Dombrowski, S. M. Brain cancer stem cells in adults and children: cell biology and therapeutic implications. Neurotherapeutics 14, 372–384 (2017).
Giacomo, L. et al. Clinical programs of stem cell therapies for liver and pancreas. Stem Cells 31, 2047–2060 (2013).
Lupia, M. & Cavallaro, U. Ovarian cancer stem cells: still an elusive entity? Mol. Cancer 16, 64 (2017).
Wu, C. L. et al. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 67, 8216–8222 (2007).
Birkeland, A. C., Owen, J. H. & Prince, M. E. Targeting head and neck cancer stem cells: current advances and future challenges. J. Dent. Res 11, 1516–1523 (2015).
Prince, M. E. et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl Acad. Sci. USA 104, 973–978 (2007).
Hu, J., Li, G., Zhang, P., Zhuang, X. & Hu, G. A CD44v+ subpopulation of breast cancer stem-like cells with enhanced lung metastasis capacity. Cell Death Dis. 8, e2679 (2017).
Chen, C., Zhao, S., Karnad, A. & James, W. F. The biology and role of CD44 in cancer progression: therapeutic implications. J. Hematol. Oncol. 11, 64 (2018).
UKCCCR guidelines for the use of cell lines in cancer research. Br. J. Cancer. 82, 1495–1509 (2000).
Yoon, C. et al. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 20, 3974–3988 (2014).
Bisson, I. & Prowse, D. M. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 19, 683–697 (2009).
Huang, Z. et al. Responsive nematic gels from the self-assembly of aqueous nanofibres. Nat. Commun. 2, 459 (2011).
Yoon, C. et al. Role of Rac1 pathway in epithelial-to-mesenchymal transition and cancer stem-like cell phenotypes in gastric adenocarcinoma. Mol. Cancer Res. 15, 1106–1116 (2017).
Lamouille, S., Xu, J. & Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014).
Jun, T. et al. Dasatinib sensitises triple negative breast cancer cells to chemotherapy by targeting breast cancer stem cells. Br. J. Cancer 119, 1495–1507 (2018).
Pingting, Z. et al. The epithelial to mesenchymal transition (EMT) and cancer stem cells: implication for treatment resistance in pancreatic cancer. Mol. Cancer 16, 52 (2017).
Gasch, C., Ffrench, B., O’Leary, J. J. & Gallagher, M. F. Catching moving targets: cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol. Cancer 16, 43 (2017).
Kazuhito, N. et al. Dipeptide species regulate p38MAPK-Smad3 signalling to maintain chronic myelogenous leukaemia stem cells. Nat. Commun. 6, 8039 (2015).
Mei, X. et al. Role of p38γMAPK in regulation of EMT and cancer stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3605–3617 (2018).
Kawano, T. et al. Soluble CD44 standard, CD44 variant 5 and CD44 variant 6 and their relation to staging in head and neck cancer. Acta Otolaryngol. 125, 392–397 (2005).
Joshua, B. et al. Frequency of cells expressing CD44, a head and neck cancer stem cell marker: correlation with tumor aggressiveness. Head Neck. 34, 42–49 (2012).
Khammanivong, A., Gopalakrishnan, R. & Dickerson, E. B. SMURF1 silencing diminishes a CD44-high cancer stem cell-like population in head and neck squamous cell carcinoma. Mol. Cancer 13, 260 (2014).
Wang, Y. K. et al. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis 31, 1376–1380 (2010).
Yong, X. et al. Helicobacter pylori upregulates Nanog and Oct 4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 374, 292–303 (2016).
Rybak, A. P., Ingram, A. J. & Tang, D. Propagation of human prostate cancer stem-like cells occurs through EGFR-mediated ERK activation. PLos ONE 19, e61716 (2013).
Ciccarelli, C. et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol. Cancer 15, 16 (2016).
Liu, X. & Fan, D. The epithelial-mesenchymal transition and cancer stem cells: functional and mechanistic links. Curr. Pharm. Des. 21, 1279–1291 (2015).
Pradella, D., Naro, C., Sette, C. & Ghigna, C. EMT and stemness: flexible processes turned by alternative splicing in development and cancer progression. Mol. Cancer 16, 8 (2017).
Shibue, T. & Weinberg, R. A. EMT, CSC, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14, 611–629 (2017).
St. John, M. A. et al. Proinflammatory mediators up-regulate snail in head and neck squamous cell carcinoma. Clin. Cancer Res. 15, 6018–6027 (2009).
Mendelsohn, A. H. et al. Snail as a novel marker for regional metastasis in head and neck squamous cell carcinoma. Am. J. Otolaryngol. 33, 6–13 (2012).
Chen, Y. C. et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 385, 307–313 (2009).
Li, L. & Neaves, W. B. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 66, 4553–4557 (2006).
Yotsukura, M. et al. Survival predictors after resection of lung metastases of head or neck cancers. Thorac. Cancer 6, 579–583 (2015).
Takahashi, E. et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J. Biol. Chem. 285, 4060–4073 (2010).
Prieto-Vila, M., Takahashi, R.-u., Usuba, W., Kohama, I. & Ochiya, T. Drug resistance driven by cancer stem cells and their niche. Int J. Mol. Sci. 18, 2574 (2017).
Krause, M., Dubrovska, A., Linge, A. & Baumann, M. Cancer stem cells: radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 109, 63–73 (2017).
Perri, F. et al. Radioresistance in head and neck squamous cell carcinoma: biological bases and therapeutic implications. Head Neck. 37, 763–770 (2015).
Toulany, M. & Rodemann, H. P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 35, 180–190 (2015).
Acknowledgements
This study was supported by the National Natural Science Foundation grants of China (grant no. 81972942), the Performance Motivation and Guidance Special Project of Chongqing Science & Technology Commission (Grant No. cstc2018jxjl10004), the Natural Science Foundation grants of Chongqing (grant no. cstc2018jcyjAX0136) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (2015R1D1A1A01058771).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Peschiaroli
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41419-024-06608-7
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, C., Yoon, C., Zhou, XH. et al. RETRACTED ARTICLE: ERK1/2-Nanog signaling pathway enhances CD44(+) cancer stem-like cell phenotypes and epithelial-to-mesenchymal transition in head and neck squamous cell carcinomas. Cell Death Dis 11, 266 (2020). https://doi.org/10.1038/s41419-020-2448-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-2448-6
This article is cited by
-
SALL4 promotes cancer stem-like cell phenotype and radioresistance in oral squamous cell carcinomas via methyltransferase-like 3-mediated m6A modification
Cell Death & Disease (2024)
-
Retraction Note: PIK3R3, part of the regulatory domain of PI3K, is upregulated in sarcoma stem-like cells and promotes invasion, migration, and chemotherapy resistance
Cell Death & Disease (2024)
-
A novel 3-miRNA network regulates tumour progression in oral squamous cell carcinoma
Biomarker Research (2023)
-
Metronomic and single high-dose paclitaxel treatments produce distinct heterogenous chemoresistant cancer cell populations
Scientific Reports (2023)
-
Targeting cancer stem cells as a strategy for reducing chemotherapy resistance in head and neck cancers
Journal of Cancer Research and Clinical Oncology (2023)
