
Abstract
By targeting the tumor microenvironment to stimulate antitumor immunity, immunotherapies have revolutionized cancer treatment. However, many patients do not respond initially or develop secondary resistance. Based on the limited resources in the tumor microenvironment and competition between tumor and immune cells, the field of immune metabolism has produced extensive knowledge showing that targeting metabolism could help to modulate antitumor immunity. However, among all the different potentially targetable metabolic pathways, it remains unclear which have more potential to overcome resistance to immune checkpoint inhibitors. Here, we explore metabolic reprogramming in cancer cells, which might inhibit antitumor immunity, and strategies that can be used to favor the antitumor response.
Similar content being viewed by others
Facts
-
Immunotherapy targeting the tumor microenvironment (TME) has changed the paradigm of cancer treatment.
-
Cancer cells and antitumor effector cells share metabolic dependencies.
-
Targeting cancer cell metabolism could be key to bypassing immune checkpoint blockade (ICB) resistance
Open questions
-
Which pathway(s) target to favor anti-tumor immune response?
-
How monitored metabolic competition between cancer and immune cells in patients to determine when is time to interfere?
Introduction
During the last decade, the paradigm of cancer therapy has been revolutionized by the development of immunotherapies. Indeed, for the first time, the goal is to stimulate the host immune system to attack tumor cells, while previous treatments were designed to directly target cancer cells. Historically, after the proof of concept1, the first evidence of the efficiency of immunotherapy was in the treatment of cutaneous melanoma with antibodies targeting cytotoxic T lymphocyte-associated protein 4 (CTLA-4). CTLA4 is a negative checkpoint protein expressed at the plasma membrane of resting T cells after T cell receptor (TCR) engagement and costimulatory signaling through CD28. CTLA4 competes with CD28 for the binding of CD80 and CD86 and induces the inhibition of T cell activation. However, even if some durable responses were observed to antibodies targeting CTLA4 (ipilizumab) in patients with metastatic melanoma, the overall response was modest and unfortunately associated with frequent toxicities resulting from tissue-specific inflammation2,3.
Then, a second strategy was developed based on the targeting of the interaction between PD1 and PD-L14. The overexpression of PD-L1 is an adaptive resistance mechanism, which tumor cells utilize to escape the antitumor immune response via PD-1–mediated T cell exhaustion. The first evidence of the antitumor activity of antibodies targeting PD1 (e.g., nivolumab and pembrolizumab) was obtained in patients with melanoma and non-small cell lung cancer (NSCLC). Interestingly, adverse events are less frequent for anti-PD1 treatment than for anti-CTLA4 treatment, and durable responses are observed in ~30% of patients treated with anti-PD15,6. Currently, there are five anti–PD-1 or anti-PD-L1 antibodies approved by regulatory agencies for the treatment of 11 different cancer types7. Finally, a combination strategy was developed utilizing a combination of anti-CTLA4 and anti-PD1 antibodies. However, even if the efficiency is greatly enhanced, unfortunately, the frequency and intensity of adverse events increase dramatically, limiting the utilization of this combination8.
It seems that cancer treatment with immunotherapy has now reached a plateau, which cannot be crossed without a better understanding of the adaptive mechanisms underlying the resistance of cancer cells to treatment.
Cancer cells are characterized by their plasticity. Indeed, cancer cells will alter many different cellular processes to adapt to stress conditions and to continue to proliferate. Concerning the specifics of cell metabolism, since the 1920s, it has been known that metabolic reprogramming is a hallmark of transformation9. However, due to the important needs for energy and building blocks, we know now that the reality of metabolic reprogramming in cancer cells is infinitely more complex than that implied by this first observation10.
Due to the consumption of resources by cancer cells and vascularization impairments, the tumor microenvironment is frequently poor in nutrients and oxygen, establishing competition between cancer and stromal cells. Because immune checkpoint therapies target immune effector cells and do not directly target cancer cells, metabolic crosstalk between these two cell populations appears to be a determinant of the effects of immunotherapy. Here, we have decided to focus our interest on some metabolic adaptations in cancer cells that can interfere directly or indirectly with T cell effector and immune checkpoint blockade efficiency.
Tumor cells starve T cells to block antitumor immunity
Even though numerous studies have shown that oxidative phosphorylation (OXPHOS) is intact in many different tumors, the first described metabolic characteristic of cancer cells was their preference for conversion to the production of ATP through anaerobic glycolysis11 to adapt to hypoxic conditions prevailing during tumor development12. The high demand for glucose of cancer cells creates competition in the tumor microenvironment that has a negative impact on neighboring cells, such as immune cells13,14. Interestingly, during their activation, proliferation, and differentiation, T cells alter their metabolism. In the quiescent state, naïve T cells rely mainly on OXPHOS and fatty acid oxidation (FAO) to support their needs. After activation through the T cell receptor (TCR) and costimulatory receptor engagement, T cells alter their metabolism to support proliferation and effector functions15. In particular, CD28 costimulatory engagement activates the PI3K/AKT pathway and increases glycolytic flux16,17. The tremendous increase in glycolysis flux supports the pentose phosphate pathway (PPP), serine biosynthesis, and fatty acid synthesis pathway and produces intermediates for nucleotide synthesis18. Thus, because the availability of glucose in the tumor microenvironment is limited, competition between cancer cells and T cells for glucose appears to be a potential key determinant of whether the overall antitumor immune response will lead to tumor elimination or growth and resistance to antitumoral immune surveillance. In support of this idea, melanoma cells isolated from patients with high levels of glycolysis showed a reduced response to adoptive T cell therapy19. However, recently a study has shown that a transitory glucose restriction can enhance CD8 T cell effector functions highlighting the complexity of the crosstalk between metabolic and effector functions20.
As amino acids are protein building blocks, the high availability of amino acids is essential for tumor growth. However, amino acids are also required by immune cells to differentiate and develop their effector functions and ultimately control tumor development. Considering this, a better understanding of the utilization of amino acids by each population of cells in the tumor microenvironment appears essential to be able to stimulate antitumor immunity efficiently.
Glutamine, through glutaminolysis, fuels the tricarboxylic acid (TCA) cycle to provide metabolic intermediates that serve as building blocks for lipids, proteins, and nucleic acids, which are necessary for cancer cell proliferation21,22. Interestingly, this same metabolic pathway has been shown to be essential for T cell activation and proliferation18,23. More specifically, during T cell activation, the MAPK/ERK pathway coordinates the upregulation of glutamine uptake and glutaminolysis23. Due to the activation of oncogenic signaling pathways, such as the MAPK/ERK pathway, cancer cells increase their glutamine uptake and utilization24. Thus, competition for glutamine between cancer cells and activated T cells can have a negative impact on T cell and antitumor immune responses, but it can also present therapeutic opportunities. Even though targeting glutamine metabolism has shown a modest effect globally in vivo25,26, Leone et al. have recently shown that glutamine metabolism inhibition using an analog of the broad spectrum inhibitor 6-diazo-5-oxo-L-norleucine (DON) can enhance immune checkpoint inhibitor efficiency due to glutaminolysis blockade, which can be compensated for in CD8+ T cells but not in cancer cells27. To survive DON treatment, cells need to adapt their metabolic flux. However, cancer cells, that accumulate metabolic vulnerabilities, appear less flexible from a metabolic point of view compare to CD8 + T cells. Even if further studies will be necessary to fully understand the dichotomic effect in cancer cells and CD8 + T cells, we can speculate that which could explain the anti-tumor immune response observed.
Glutamine blockade can also have an indirect immunostimulatory antitumor effect, as illustrated by the fact that a small-molecule inhibitor of glutamine metabolism inhibited the generation and recruitment of myeloid-derived suppressor cells (MDSCs) via the inhibition of Colony Stimulating Factor 3 (CSF3) production28. Interestingly, in this study, the authors showed that glutamine metabolism inhibition also has an effect on tryptophan catabolism via IDO suppression. However, Ma et al. demonstrated that in renal cancer, glutamine deprivation induced the expression of PD-L129, showing the difficulty of targeting glutamine in the context of immunotherapy.
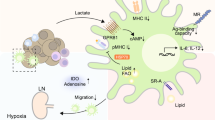
Tryptophan is an essential amino acid that cells utilize through the kynurenine pathway30. Because tumor cancer cells and T cells need to import tryptophan from the microenvironment for their needs, there is competition for this resource. Specifically, in T cells, tryptophan availability is critical for proliferation31. After stimulation, T cells upregulate the expression of several amino acid transporters, including SLC7A5. Interestingly, SLC7A5 knockdown blocks T cell clonal expansion and effector differentiation by activating mTOR and inducing the expression of c-Myc32. Thus, tryptophan catabolism is an important mechanism by which cancer cells can inhibit the antitumor immune response. The most studied pathway of tryptophan catabolism involves indoleamine-2,3-dioxygenase (IDO), an enzyme that catalyzes the conversion of tryptophan to kynurenine. Interestingly, IDO expression can be stimulated by type I interferon (interferon-αβ) and type II interferon (interferon-γ)33 because IDO1 gene promoters possess IFN-stimulated response elements (ISREs) and IFN-activated sites (GAS). Interferon-γ is one of the hallmarks of activated CD8 + T cells, and IDO expression, like PD-L1 expression, by cancer cells can be seen as an adaptive mechanism that cells adopt in response to CD8 + T cell infiltration in tumors. Two other enzymes have been shown to be IDO-related enzymes, indoleamine-2,3-dioxygenase 2 (IDO2) and tryptophan-2,3-dioxygenase (TDO), but they show different patterns of expression34,35,36. TDO seems to be more interesting in the cancer context, since it has been shown to be able to sustain tryptophan catabolism and induce inhibition of the antitumor immune response37,38.
Tryptophan starvation inhibits CD8 + T cell effector functions and stimulates CD4 + regulatory T (Treg) cell functions, creating robust immunosuppression that can affect immune checkpoints mediated by the CTLA4 and PD1/PD-L1 pathways to create a tolerogenic tumor microenvironment. Mechanistically, the effects are mediated through the activation of the stress response kinase GCN2, which inhibits mTORC2 and downstream AKT39,40. Initially, it was proposed that IDO mediated the immunosuppressive effect directly through local tryptophan starvation41. In support of these findings, IDO1 expression has been associated with poor prognosis in multiple tumor types42,43. However, it has been shown in B16 melanoma model studies that the suppression of the antitumor T cell response induced by tryptophan catabolism can be mediated independently of the effect of GCN244.
Interestingly, numerous studies have shown that metabolites generated through tryptophan catabolism, such as kynurenine, kynurenic acid, 3-hydroxy-kynurenine, and 3-hydroxy-anthranilic acid, can suppress T cell-mediated antitumor immunity45. Kynurenine, the first metabolite product in the IDO-dependent tryptophan degradation pathway, can be exported to the tumor microenvironment by cancer cells to inhibit antitumor immunity and prevent tumor clearance37,46,47. Several studies have identified that kynurenine can activate the aryl hydrocarbon receptor (AhR)37,48. Functionally, AhR is a cytosolic protein that will translocate to the nucleus after interaction with its ligands. In the nucleus, AhR binds the promoter regions of target genes containing sequences called “aryl hydrocarbon response elements” (AHREs) as well as dioxin-response elements (DREs)49,50,51. Interestingly, AhR expression is enriched in interleukin 17 (IL-17)-producing CD4 + T cells and is implicated in the generation of regulatory T cells (Tregs)52,53,54. Another study has also shown that the immunosuppressive effect of AhR after its interaction with kynurenine can be mediated by the alteration of CD8 + T-cell function37.
The interconnection between the inhibition of antitumor immunity mediated via GCN2 activation that is induced by tryptophan depletion and kynurenine-mediated AhR translocation needs to be studied more deeply, but one study has already shown that these two pathways can cooperate to induce a regulatory T cell phenotype and permit tumor development55. One possible connection between these two mechanisms could be the amino-acid transporter LAT1, which has been shown to import tryptophan into the cell and, at the same time, to function as an antiport system for kynurenine56.
Altogether, the evidence suggests that tryptophan catabolism appears to be one of the major metabolic mechanisms driving the inhibition of the antitumor immune response and is therefore a major therapeutic target that we will discuss later in this review.
Arginine is another amino acid that has limited availability in the tumor microenvironment. A recent study showed that amino acids are the most depleted substances in tumors57. Due to the common need for arginine of tumor cells and immune cells, arginine starvation in immune cells appears to be another road used by cancer cells. Indeed, L-arginine uptake has been shown to be necessary for CD8+ T cell proliferation, memory response formation, and finally antitumor responses58. The absence of arginine availability has been shown to interfere with glycolysis in T cells, leading to the inhibition of cytokine production and T cell proliferation59,60.
However, the case of arginine is particularly interesting because even if some studies have shown that cancer cells produce arginases, which comprise the enzyme family responsible for arginine degradation61,62,63, a large proportion of the literature shows that the majority of arginases come from the tumor stroma. Indeed, it is principally the MDSCs, whose accumulation in the tumor stroma is a hallmark of tumor development, that are responsible for arginine deprivation. Indeed, during its development, the tumor corrupts the myeloid compartment, leading to the development of MDSCs64 that express arginases and inhibit antitumor immunity65,66.
Taken together, these studies highlight the importance of arginine availability for antitumor immunity efficiency and point to potential actionable mechanisms that could be used to target MDSC-dependent antitumor immunity suppression (Fig. 1).

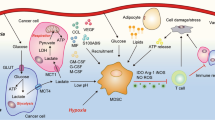
Tumors cells and effector T cells share dependencies for glucose, glutamine, tryptophan, or arginine. a Tumor cells, by their extensive need of glucose, glutamine, tryptophan of arginine will impoverish the tumor area. This resource consumption from tumor cells will directly impact CD8 T cell metabolism leading to exhausted phenotype and indirectly, through metabolite secretion like for example lactate, stimulate the development of an immunosuppressive tumor micro-environment. b At the opposite, if tumor cells have moderate metabolic demand, due to intrinsic characteristics or therapeutic manipulations, a fully functional anti-tumor immune response will mediated tumor elimination.
Tumor cell metabolism creates a hostile tumor microenvironment
Interestingly, not only the limitation of glucose availability for T cells but also the increase in glycolysis flux, which will produce immunosuppressive metabolites such as lactate, can reduce tumor immune rejection and responses to immunotherapy. Indeed, lactate is produced from pyruvate by the glycolytic enzyme LDH. Interestingly, a high LDH level is associated with poor clinical outcomes for various tumor types67. Even if it is commonly admitted that the level of LDH is a reflection of the tumoral mass, the reason for the predictive value of LDH remains controversial. The accumulation of lactate in the tumor microenvironment is associated with acidification of the tumor area. Indeed, monocarboxylate transporters (MCT1 and 4) cotransport lactate and protons from the cytoplasm to the extracellular space68,69. This production of lactate in tumor cells impacts interferon-gamma production by tumor-infiltrating T cells, NK activation, and the proportion of myeloid-derived suppressor cells. This will result in decreased immune surveillance and thus support tumor growth70,71. Lactate can also impact dendritic cells by modulating their differentiation. Indeed, lactate promotes a tumor-associated dendritic cell phenotype characterized by a decrease in major histocompatibility complex I molecules and the maintenance of a tolerogenic phenotype72,73. Concerning the effect of lactic acid on macrophages, the results of different studies are more contradictory, even though Colegio et al. have shown that a high concentration of lactate promotes M2 macrophage polarization through the stabilization of HIF1α74. Recently, lactate has been also implicated in lactate-derived lactylation of histone lysine residues. This epigenetic modification stimulates gene transcription of M2 marker genes, such as IL6 and ARG175.
Apart from the direct effects of lactate, the fact that the export of lactate from cancer cells induces a pH decrease in the tumor microenvironment will also impact immune cells76. For example, pH decreases impair T cell cytotoxicity and the natural cytotoxicity of NK cells77,78,79. Acidosis induced by tumors can also promote macrophage polarization toward a tumor-associated macrophage phenotype that will sustain tumor growth80. More specifically, in the context of immunotherapy, Bosticardo et al. have shown that in addition to interfering with activation (via interleukin-2 and interferon-gamma secretion) and proliferation, a low pH also upregulates CTLA-4 expression on T lymphocytes81. Mechanistically, Pilon-Thomas et al. suggested that the inhibition of the immunotherapy effects mediated by tumor could be mediated by specific acid-sensing receptor(s)82,83, such as G-proteins, T-cell inhibitory receptor and T-cell death–associated gene-8, which have been shown to suppress MYC translation in lymphocytes84,85.
Altogether, these studies highlight the fact that tumor cells, apart from their direct starvation effect, can use glycolysis and related processes to model a hostile tumor microenvironment to escape antitumor immunity and continue to proliferate (Fig. 2).

Tumor cells, through aerobic glycolysis, produce lactate that will be exported in the tumor micro-environment via MCT1 and MCT4 transporters. Lactate has a direct tolerogenic effect by modulating T cells and NK cells activation as well as tumor-associated dendritic cells differentiation and M2 macrophages polarization. Lactate export also impacts micro-environment pH by inducing acidosis that will participate in the impairment of T cells and NK cells cytotoxicity, M2 polarization, and upregulation of immune checkpoints.
Targeting tumor cell metabolism increases immune checkpoint blockade efficiency
There is an extensive number of studies that have tried to target different metabolic pathways to increase immune checkpoint blockade efficiency or bypass resistance. However, cancer cells and immune cells from the tumor stroma, especially activated CD8 + T cells, share metabolic dependencies, making it difficult to obtain combinatory effects with drugs targeting metabolic processes and immune checkpoint blockade.
A perfect illustration of this complexity can be highlighted by the targeting of fatty acid metabolism. Indeed, even if lipid and cholesterol accumulation has been shown to correlate with cancer aggressiveness86, opening therapeutic opportunities especially via targets such as fatty acid synthase (FASN)87, the consequences on the different immune cell populations can be opposing. For example, fatty acid oxidation (FAO) is essential for the development of CD8 + memory cells88 as well as the function of immunosuppressive regulator T cells (Treg)89 and immunosuppressive M2 macrophages90.
Because resources are limited in the tumor microenvironment, cells are subjected to energetic stress. AMP-activated protein kinase (AMPK) is a serine/threonine kinase that functions to sense energy homeostasis through detecting the AMP/ATP ratio91. Even if AMPK activation has been shown to modulate various functions of cells composing the tumor stroma, including MDSCs and T cells, we have decided to focus our interest on the consequences of AMPK activation in tumor cells, which can impact the antitumor immune response. Targeting AMPK has been extensively demonstrated to be a potential antitumor strategy. The most well-characterized molecule that activates AMPK is probably the anti-diabetic drug metformin92. Extensive literature exists on the potential anti-neoplasic effect of metformin on various tumor types through multiple mechanisms. Concerning antitumor immunity, the first evidence was published by Eikawa et al. in 2015. The authors have shown that metformin treatment stimulates CD8 + T cell effector functions in an AMPK-dependent manner, leading to tumor regression93. These results have been confirmed by the fact that targeting mitochondrial oxidative phosphorylation (which is also targeted by metformin) synergizes with PD-1 blockade by inducing the expansion of cytotoxic T lymphocyte effector/memory cells in tumors and the draining lymph node due to the activation of AMPK, mammalian Target Of Rapamycin (mTOR), PPAR-gamma coactivator 1α (PGC1α) and T-bet94.
However, the impact of AMPK modulation on cancer cells is controversial.
On the one hand, the expression of liver kinase B1 (LKB1), one of the kinases that phosphorylates AMPK, has been shown to be correlated with PD-L1 expression in non-small cell lung cancer. Mechanistically, LKB1 increases PD-L1 expression via AMPK and KEAP1/NRF2 signaling95. Moreover, LKB1 knockdown was demonstrated in the same study to control cytokine production via a decrease of CCL5 and CXCL12 (chemokines recruiting lymphocytes and dendritic cells) and increases of CXCL5 and CXCL7 (chemokines promoting the recruitment of neutrophils), leading to the reshaping of the tumor microenvironment towards immunosuppressive stroma.
On the other hand, activation of AMPK by metformin has been shown to induce the phosphorylation of serine 195 of PD-L1, leading to abnormal glycosylation and finally the degradation of PD-L1 through ER-associated protein degradation (ERAD)96. Thus, targeting AMPK using metformin could be an alternative strategy used in combination with anti-CTLA4 therapy to stimulate antitumor immunity. However, depending on the context, the fact that AMPK activation seems to be able to increase the expression or induce the degradation of PD-L1 is intriguing, and additional studies will be necessary prior to considering a clinical use of metformin or other AMPK activators in the context of immunotherapies.
To support the requirements of proliferation, cancer cells increase nucleotide metabolism to provide the necessary pool of nucleotides for nucleic acid and protein synthesis. Thus, nucleotide metabolism plays an essential role in cancer cell biology and constitutes a potential target for the improvement of cancer therapy. Strategies targeting nucleotide metabolism were developed a long time ago, such as the use of antimetabolites, which competitively inhibit the activity of enzymes involved in nucleotide synthesis97,98. Even if these strategies are currently used for the therapy of numerous cancer types or if emerging strategies specifically targeting purines or pyrimidines appear attractive99, there are relatively low amounts of data concerning the potential of targeting purine or pyrimidine metabolism in the context of immunotherapy. However, recent studies have shown that alteration of the urea cycle, which is the main pathway used by mammals to eliminate waste nitrogen, can modulate the response to immunotherapy100,101. Indeed, authors have demonstrated that specific alterations in the expression of urea cycle-associated enzymes induce a specific mutation signature due to an increase in the ratio of pyrimidine to purine associated with the expression of hydrophobic tumor antigens and consequently an enhanced response to immune checkpoint blockade. Even these studies highlight the fact that targeting nucleotide metabolism could be an interesting strategy to improve the immunotherapy response, additional exploration will be necessary to fully understand how nucleotides metabolism interferes with anti-tumor immunity especially because, in general, targeting nucleotides synthesis has immunosuppressive effects (Fig. 3).

Highly resources demand cancer cells necessary to sustain their proliferation opens several therapeutics opportunities to improve immune checkpoints blockade efficiency. Targeting AMPK especially with metformin of other biguanide derivatives could be an interesting combinatory strategy to increase immunotherapies efficiency. However, regarding the contradictory effects report on PD-L1 expression and degradation, additional exploration needs to fully evaluate the potential of this combination and especially with which immunotherapy (anti-PD1/PD-L1 or anti-CTLA4) targeting AMPK could be a therapeutic option. Glutamine dependency is share between a cancer cell and CD8 effector T cells highlighting glutaminolysis as a potential therapeutic target. Even if using single enzyme inhibitors have shown a modest therapeutic effect, recent results obtained with DON, a glutamine analog that inhibits a large spectrum of glutaminolysis enzyme have shown a spectacular effect by inducing a glutaminolysis that cannot be compensated by cancer cells contrariwise of CD8 T cells that increase glucose uptake to fuel PPP activity and maintain their anti-tumor activity. Nucleotides metabolism emerges as a promising therapeutic option especially with recent studies implicating nucleotide imbalance in the generation of a specific mutation pattern that predicts response to immunotherapy. However, exploration is needed to explore the consequences of nucleotides metabolism intervention, especially in the tumor stroma.
Ongoing clinical trials and future directions
While targeting metabolism theoretically offers various opportunities to improve the immune checkpoint blockade response or bypass resistance, at the moment, there is not yet a therapeutic schedule for the use of such therapies in standard care. However, several combinatory therapies are currently being or have been investigated.
The most advanced combinatory strategy for immunotherapy is probably the strategy that utilizes IDO1 inhibitors. As was already discussed, IDO1 is an enzyme responsible for tryptophan catabolism and conversion to kynurenine, which is a metabolite that will induce immunosuppression via the AhR signaling pathway. Studies showing that IDO1 is involved in the mechanism of resistance to various therapies, especially anti-CTLA4 treatment102,103, gave a proof of concept for the use of the combination. Numerous clinical trials are ongoing to test the efficiency of the combination of the IDO inhibitors Epacadostat, Indoximod, Navoximod, or BMS-986205 with anti-PD1, anti-CTLA4, or vaccine-based strategies104,105. However, even if the results of phase I and II clinical trials launched to test the efficiency of IDO1 inhibitors for the treatment of melanoma were encouraging106, the development of the drugs was stopped due to the lack of the efficiency of their combination with anti-PD1 for melanoma treatment107. The results for other cancer types will be particularly interesting as well as results of combinations with other drugs for determining the therapeutic potential of IDO1 targeting.
Concerning the amino acid arginine, inhibitors of arginase that block the myeloid cell-mediated inhibition of T cells are under investigation based on the in vivo result that showed that arginase blockade with CB-1158 synergizes with immune checkpoint blockade and adoptive T cell or NK cell therapies108. Unfortunately, the ongoing trial testing the triple combination of the arginase inhibitor INCB001158, the IDO inhibitor epacadostat, and anti-PD1 was stopped prematurely due to the negative result obtained from the combination of epacadostat and anti-PD1. Arginase plays a key role in immunosuppression in the tumor microenvironment, and it could be interesting to evaluate the efficiency of arginase inhibitors, not only in first-line treatment, but also in patients who have acquired resistance to immunotherapy, due to the importance of MDSCs in this phenomenon109,110.
Concerning the targeting of glutamine metabolism, there is one ongoing clinical trial testing the inhibition of glutaminase (GLS), the first enzyme involved in glutaminolysis, in combination with chemotherapy plus anti-PD1 for the treatment of non-small cell lung cancer. Furthermore, considering the impressive results obtained with the glutamine antagonist DON27, a clinical evaluation of the strategy appears to be essential.
Finally, the old anti-diabetic drug metformin has also found a second youth in trials testing its combination with immunotherapy. As we have seen previously in in vitro and in vivo data, targeting AMPK could be an interesting strategy to improve immune checkpoint inhibitor efficiency. Metformin is prescribed to more than 120 million people worldwide for type II diabetes mellitus. The extensive knowledge of its adverse events probably makes its combination with immunotherapy safer, and considering the numerous studies showing that patients treated with metformin have a low risk of cancer development, the results of the clinical trials will be useful (Table 1).
Conclusion
There is extensive evidence that targeting tumor cell metabolic adaptation can provide new therapeutic opportunities, especially when it is used in combination with immune checkpoint blockade strategies. The fact that PD-L1 expression in cancer cells has been demonstrated to stimulate glycolysis through AKT/mTOR activation perfectly illustrates the link that exists between the immune checkpoint and metabolism14. However, as we have seen before, cancer cells and immune effector cells, especially CD8 T cells, share various metabolic dependencies, which makes it difficult to target the metabolic adaptation of cancer cells without affecting tumor clearance induced by T cells. Thus, it is probably only by targeting essential pathways for cancer cells for which inhibition can be compensated for by immune cells that it will be possible to bypass the plateau we have reached with immunotherapies. An alternative strategy could be the use of combinations of approaches targeting cancer cell metabolism and adoptive T cell therapies. Indeed, the necessity of in vitro cell expansion provides an opportunity to genetically or pharmacologically compensate for T cell metabolic dependencies before patient transfer. In all cases, future work will be necessary to fully understand the metabolic dynamics within the tumor microenvironment, especially because the knowledge that we have accumulated via in vitro studies is hardly transferable in vivo or to patients due to the large excess of nutrients in culture media. Finally, trials to evaluate the potential of targeting the metabolic crosstalk between the tumor and the stroma should be designed more carefully and based on a deeper in vivo understanding if we wish to avoid a stinging failure like that of IDO inhibitors and be able to propose efficient combination strategies to bypass immune checkpoint inhibitor resistance.
References
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade.Science 271, 1734–1736 (1996).
Hodi, F. S. et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl Acad. Sci. USA 100, 4712–4717 (2003).
Ribas, A. et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J. Clin. Oncol. 23, 8968–8977 (2005).
Ishida, Y., Agata, Y., Shibahara, K. & Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Garon, E. B. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015).
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Pavlova, N. N. & Thompson, C. B. The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Casazza, A. et al. Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene 33, 1743–1754 (2014).
Wang, T., Liu, G. & Wang, R. The intercellular metabolic interplay between tumor and immune cells. Front. Immunol. 5, 358 (2014).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015).
O’Neill, L. A., Kishton, R. J. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Frauwirth, K. A. et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 (2002).
Powell, J. D. & Delgoffe, G. M. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33, 301–311 (2010).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Cascone, T. et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987 e974 (2018).
Klein Geltink, R. I. et al. Metabolic conditioning of CD8(+) effector T cells for adoptive cell therapy. Nat. Metab. 2, 703–716 (2020).
Altman, B. J., Stine, Z. E. & Dang, C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. 16, 619–634 (2016).
Zhang, J., Pavlova, N. N. & Thompson, C. B. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 36, 1302–1315 (2017).
Carr, E. L. et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 185, 1037–1044 (2010).
Yang, R. et al. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene 39, 2975–2986 (2020).
Davidson, S. M. et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016).
Hensley, C. T., Wasti, A. T. & DeBerardinis, R. J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Investig. 123, 3678–3684 (2013).
Leone, R. D. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019).
Oh, M. H. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig., https://doi.org/10.1172/JCI131859 (2020).
Ma, G. et al. Glutamine deprivation induces PD-L1 expression via activation of EGFR/ERK/c-Jun signaling in renal cancer. Mol. cancer Res.: MCR 18, 324–339 (2020).
Chen, Y. & Guillemin, G. J. Kynurenine pathway metabolites in humans: disease and healthy States. Int. J. tryptophan Res. 2, 1–19 (2009).
Munn, D. H. et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 189, 1363–1372 (1999).
Sinclair, L. V. et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14, 500–508 (2013).
Corm, S. et al. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients’ sera by HPLC and is inducible by IFN-gamma. Leuk. Res. 33, 490–494 (2009).
Metz, R. et al. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 67, 7082–7087 (2007).
Meininger, D. et al. Purification and kinetic characterization of human indoleamine 2,3-dioxygenases 1 and 2 (IDO1 and IDO2) and discovery of selective IDO1 inhibitors. Biochimica et. biophysica acta 1814, 1947–1954 (2011).
Kanai, M. et al. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2, 8 (2009).
Opitz, C. A. et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203 (2011).
Pilotte, L. et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl Acad. Sci. USA 109, 2497–2502 (2012).
Munn, D. H. et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642 (2005).
Sharma, M. D. et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv. 1, e1500845 (2015).
Uyttenhove, C. et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med 9, 1269–1274 (2003).
Brandacher, G. et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res. 12, 1144–1151 (2006).
Brody, J. R. et al. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle 8, 1930–1934 (2009).
Sonner, J. K. et al. The stress kinase GCN2 does not mediate suppression of antitumor T cell responses by tryptophan catabolism in experimental melanomas. Oncoimmunology 5, e1240858 (2016).
Opitz, C. A., Wick, W., Steinman, L. & Platten, M. Tryptophan degradation in autoimmune diseases. Cell Mol. Life Sci. 64, 2542–2563 (2007).
Hughes, T. et al. The transcription Factor AHR prevents the differentiation of a stage 3 innate lymphoid cell subset to natural killer cells. Cell Rep. 8, 150–162 (2014).
Liu, Y. et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer cell 33, 480–494 e487 (2018).
Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010).
Kazlauskas, A., Poellinger, L. & Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 274, 13519–13524 (1999).
Ma, Q. & Whitlock, J. P. Jr A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 272, 8878–8884 (1997).
Tomita, S. et al. T cell-specific disruption of arylhydrocarbon receptor nuclear translocator (Arnt) gene causes resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced thymic involution. J. Immunol. 171, 4113–4120 (2003).
Stockinger, B., Hirota, K., Duarte, J. & Veldhoen, M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin. Immunol. 23, 99–105 (2011).
Veldhoen, M. et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 (2008).
Quintana, F. J. et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 (2008).
Fallarino, F. et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 176, 6752–6761 (2006).
Kaper, T. et al. Nanosensor detection of an immunoregulatory tryptophan influx/kynurenine efflux cycle. PLoS Biol. 5, e257 (2007).
Sullivan, M. R. et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, https://doi.org/10.7554/eLife.44235 (2019).
Geiger, R. et al. L-Arginine Modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842 e813 (2016).
Rodriguez, P. C. et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 171, 1232–1239 (2003).
Rodriguez, P. C. et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 64, 5839–5849 (2004).
Mussai, F. et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 122, 749–758 (2013).
Mussai, F. et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 75, 3043–3053 (2015).
Gannon, P. O. et al. Androgen-regulated expression of arginase 1, arginase 2 and interleukin-8 in human prostate cancer. PLoS ONE 5, e12107 (2010).
Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012).
Raber, P., Ochoa, A. C. & Rodriguez, P. C. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunological Investig. 41, 614–634 (2012).
Fletcher, M. et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 75, 275–283 (2015).
Dhup, S., Dadhich, R. K., Porporato, P. E. & Sonveaux, P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 18, 1319–1330 (2012).
Counillon, L., Bouret, Y., Marchiq, I. & Pouyssegur, J. Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochimica et. biophysica acta 1863, 2465–2480 (2016).
Halestrap, A. P. The monocarboxylate transporter family-structure and functional characterization. IUBMB life 64, 1–9 (2012).
Husain, Z., Huang, Y., Seth, P. & Sukhatme, V. P. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 191, 1486–1495 (2013).
Brand, A. et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK Cells. Cell Metab. 24, 657–671 (2016).
Gottfried, E. et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021 (2006).
Nasi, A. et al. Dendritic cell reprogramming by endogenously produced lactic acid. J. Immunol. 191, 3090–3099 (2013).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Huber, V. et al. Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. cancer Biol. 43, 74–89 (2017).
Calcinotto, A. et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 72, 2746–2756 (2012).
Muller, B., Fischer, B. & Kreutz, W. An acidic microenvironment impairs the generation of non-major histocompatibility complex-restricted killer cells. Immunology 99, 375–384 (2000).
Fischer, B., Muller, B., Fisch, P. & Kreutz, W. An acidic microenvironment inhibits antitumoral non-major histocompatibility complex-restricted cytotoxicity: implications for cancer immunotherapy. J. Immunother. 23, 196–207 (2000).
Bohn, T. et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 19, 1319–1329 (2018).
Bosticardo, M. et al. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur. J. Immunol. 31, 2829–2838 (2001).
Pilon-Thomas, S. et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 76, 1381–1390 (2016).
Damaghi, M., Wojtkowiak, J. W. & Gillies, R. J. pH sensing and regulation in cancer. Front. Physiol. 4, 370 (2013).
Ishii, S., Kihara, Y. & Shimizu, T. Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J. Biol. Chem. 280, 9083–9087 (2005).
Li, Z., Dong, L., Dean, E. & Yang, L. V. Acidosis decreases c-Myc oncogene expression in human lymphoma cells: a role for the proton-sensing G protein-coupled receptor TDAG8. Int. J. Mol. Sci. 14, 20236–20255 (2013).
Beloribi-Djefaflia, S., Vasseur, S. & Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 5, e189 (2016).
Mullen, G. E. & Yet, L. Progress in the development of fatty acid synthase inhibitors as anticancer targets. Bioorg. Med. Chem. Lett. 25, 4363–4369 (2015).
O’Sullivan, D. et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88 (2014).
Michalek, R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011).
Zhang, Q. et al. Fatty acid oxidation contributes to IL-1beta secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 94, 27–35 (2018).
Shaw, R. J. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl Acad. Sci. USA 101, 3329–3335 (2004).
Evans, J. M., Donnelly, L. A., Emslie-Smith, A. M., Alessi, D. R. & Morris, A. D. Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305 (2005).
Eikawa, S. et al. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl Acad. Sci. USA 112, 1809–1814 (2015).
Chamoto, K. et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl Acad. Sci. USA 114, E761–E770 (2017).
Shen, X. et al. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci. 256, 117923 (2020).
Cha, J. H. et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. cell 71, 606–620 e607 (2018).
Farber, S. & Diamond, L. K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 238, 787–793 (1948).
Heidelberger, C. et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 179, 663–666 (1957).
Villa, E., Ali, E. S., Sahu, U. & Ben-Sahra, I. Cancer cells tune the signaling pathways to empower de Novo synthesis of nucleotides. Cancers 11, https://doi.org/10.3390/cancers11050688 (2019).
Lee, J. S. et al. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174, 1559–1570 e1522 (2018).
Keshet, R. et al. Targeting purine synthesis in ASS1-expressing tumors enhances the response to immune checkpoint inhibitors. Nat. Cancer, https://doi.org/10.1038/s43018-020-0106-7 (2020).
Holmgaard, R. B., Zamarin, D., Munn, D. H., Wolchok, J. D. & Allison, J. P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 210, 1389–1402 (2013).
Brown, Z. J. et al. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 67, 1305–1315 (2018).
Prendergast, G. C., Malachowski, W. P., DuHadaway, J. B. & Muller, A. J. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 77, 6795–6811 (2017).
Komiya, T. & Huang, C. H. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol. 8, 423 (2018).
Zakharia, Y. et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 36, 9512–9512 (2018).
Long, G. V. et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 20, 1083–1097 (2019).
Steggerda, S. M. et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 5, 101 (2017).
Meyer, C. et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 63, 247–257 (2014).
Santegoets, S. J. et al. Myeloid derived suppressor and dendritic cell subsets are related to clinical outcome in prostate cancer patients treated with prostate GVAX and ipilimumab. J. Immunother. Cancer 2, 31 (2014).
Acknowledgments
This work was supported by INSERM, University of Nice Sophia-Antipolis, Canceropole PACA, and SATT Sud-Est. MC is a recipient of a postdoctoral fellowship from the Fondation de France « Recherche fondamentale et translationnelle sur le cancer: étude des résistances aux traitements ».
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cerezo, M., Rocchi, S. Cancer cell metabolic reprogramming: a keystone for the response to immunotherapy. Cell Death Dis 11, 964 (2020). https://doi.org/10.1038/s41419-020-03175-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03175-5
This article is cited by
-
Metabolic interventions combined with CTLA-4 and PD-1/PD-L1 blockade for the treatment of tumors: mechanisms and strategies
Frontiers of Medicine (2023)
-
Heterogeneity of triple negative breast cancer: Current advances in subtyping and treatment implications
Journal of Experimental & Clinical Cancer Research (2022)
-
10 years of Cell Death & Disease
Cell Death & Disease (2020)
