
Abstract
Poor viability of mesenchymal stem cells (MSCs) at the transplanted site often hinders the efficacy of MSCs-based therapy. Platelet lysate (PL) contains rich amounts of growth factors, which benefits cell growth. This study aimed to explore how human PL benefits umbilical cord-derived MSCs (huc-MSCs), and whether they have synergistic potential in osteoarthritis (OA) treatment. As quality control, flow cytometry and specific staining were performed to identify huc-MSCs, and ELISA was used to quantify growth factors in PL. CCK-8 and flow cytometry assays were performed to evaluate the effects of PL on the cell viability and cell cycle progression of huc-MSCs. Wound healing and transwell assays were conducted to assess the migration of huc-MSCs. RNA sequencing, real time PCR, and Western blot assays were conducted to explore the growth factors-based mechanism of PL. The in vitro results showed that PL significantly promoted the proliferation, cell cycle, and migration of huc-MSCs by upregulating relevant genes/proteins and activating beclin1-dependent autophagy via the AMPK/mTOR signaling pathway. The main growth factors (PDGF-AA, IGF-1, TGF-β, EGF, and FGF) contributed to the effects of PL in varying degrees. The in vivo data showed that combined PL and huc-MSCs exerted significant synergistic effect against OA. The overall study determined the beneficial effects and mechanism of PL on huc-MSCs and indicated PL as an adjuvant for huc-MSCs in treating OA. This is the first report on the growth factors-based mechanism of PL on huc-MSCs and their synergistic application. It provides novel knowledge of PLʹs roles and offers a promising strategy for stem cell-based OA therapy by combining PL and huc-MSCs.
Similar content being viewed by others


Introduction
Mesenchymal stem cells (MSCs) become an attractive and promising therapeutic tool for cell therapy and regenerative medicine, due to their self-renewal capacity, multilineage differentiation potential, paracrine effects, and immunomodulatory properties, which can promote all regenerative stages of damaged tissues. MSCs were firstly identified from bone marrow and thereafter were isolated from various tissues1. Bone marrow has been considered as the gold standard for MSCs isolation, but its acquisition is invasive and painful for donors2. Recently, new sources such as umbilical cord are preferred2. Umbilical cord is a noninvasive source with higher accessibility and fewer ethical constraints than bone marrow and other sources3. Human umbilical cord-derived MSCs (huc-MSCs) are noncontroversial compared to embryonic stem cells and induced pluripotent stem cells2,4. Moreover, huc-MSCs possess higher proliferative potential than MSCs from other sources5. Besides, huc-MSCs have an advantages of availability in large quantities, great expansion capability, no formation of teratomas or tumors, and strong immunomodulatory capacities6. It is known that MSCs can be self-protected from immunological defense during allogeneic transplantation, due to the expression of major histocompatibility complex (MHC) class I but not II7. Huc-MSCs are less immunogenic than other MSCs, making huc-MSCs a better candidate for allogeneic transplantation3. Currently, the FDA has registered dozens of clinical trials on the transplantation of huc-MSCs for treating refractory diseases, such as knee osteoarthritis (OA)2,8.
Under physiological conditions, platelets play an important roles not only in primary hemostasis but also in wound healing and tissue regeneration via the release of growth factors during activation. Abundant growth factors in platelet granules can be artificially obtained by freeze/thaw lysis, producing platelet lysates (PL). The primary factors in PL include platelet derived growth factor (PDGF), insulin-like growth factor (IGF), basic epidermal growth factor (EGF), fibroblast growth factor (FGF), and transforming growth factor-β (TGF-β), which can benefit cell proliferation, chemotaxis, and extracellular matrix production9,10,11. Most clinical applications of MSCs require sufficient ex vivo expansion to reach a meaningful cell number, and PL has a great potential to enable large-scale expansion of MSCs owing to these growth factors12,13,14. A positive dose-response relationship has been confirmed between platelet concentrations and MSCs proliferation15, indicating a correlation between PL and MSCs activities. Up to our knowledge, most studies have focused on the use of PL in MSCs culture as medium supplement to substitute fetal bovine serum (FBS), due to the safety concern of FBS about immune reactions and zoonotic infections14. The scientific rationale for the use of PL is the presence of the growth factors16. However, which factors contribute to the effects of PL and what mechanism they have, remains unknown.
A major technical impairment to MSCs-based cell therapy is the difficulty to isolate MSCs from tissue sources in which MSCs are present at low levels as well as the difficulty to culture these cells with sufficient quality and quantity. Successful isolation and rapid expansion of MSCs require a large amount of complete media containing bioactive supplement, such as FBS. Therefore, PL may not be suitable for MSCs culture as FBS substitute, because human blood is not usually available as routine source, although it gains advantages over FBS in safety. Given the rich growth factors in PL, we hypothesized that PL can benefit and strengthen MSCs in cell therapy as an adjuvant. To verify this hypothesis, we evaluated the multifaceted effects of PL on huc-MSCs at the cellular and molecular levels. Then, the roles of each growth factor in PL were explored. Given the known efficacy of MSCs on knee OA17, we employed a rat model of OA to evaluate the adjuvant role of PL in MSCs-based cell therapy. Recently, several reports have shown that PL can stimulate the proliferation of huc-MSCs and enhanced huc-MSCs-based bone tissue regeneration, but the underlying mechanism is unclear18,19. Also, the combination of PL and huc-MSCs for OA treatment has never been attempted. Therefore, this study would provide new information to the action mechanism of PL on MSCs and explore the synergistic application of combined PL and MSCs for OA therapy.
Results
Huc-MSCs identification
As shown in Fig. 1A, the umbilical cord-isolated cells showed fibroblast-like morphology with plastic adherent properties and expressed the surface markers CD73 (>99%), CD90 (>99%), and CD105 (>98%), but not CD14 (<0.5%), CD19 (<0.5%), CD34 (<0.5%), or CD45 (<0.5%), which complied with the international standard of MSCs20. Figure 1B showed that the cells have the ability of three-line differentiation (osteogenesis, chondrogenesis, and adipogenesis) when cultured in appropriate induction medium. The expression pattern and specific staining results indicated the cultured cells as typical MSCs.

The immunophenotype of huc-MSCs surface markers determined by flow cytometry (A). Specific staining of the cells after inducing three-line differentiation (scale bar = 100 μm): osteogenic differentiation (left) was examined by Alizarin red staining, chondrogenic differentiation (middle) was examined by Alcian blue staining, and adipogenic differentiation (right) was examined by Oil Red O staining (B). Flow cytometric pattern of CD41a expression on PRP (C). ELISA-tested concentration of PDGF-AA, PDGF-AB, PDGF-BB, TGF-β, IGF-1, EGF, FGF in PL (D). Cell viability of huc-MSCs upon treatment of PL and PRP for 24 and 48h (E). Data are mean values ± SD.
PL characterization
CD41a is a specific surface marker for human platelets. As shown in Fig. 1C, flow cytometrical analyses showed more than 99% positive expression of CD41a in PL production before freeze-thaw lysis. As shown in Fig. 1D, PL was obtained after the lysis, containing 26.25 ± 0.48 ng/ml PDGF-AA, 18.87 ± 0.74 ng/ml PDGF-AB, 11.31 ± 0.49 ng/ml PDGF-BB, 7.62 ± 1.29 ng/ml TGF-β, 5.05 ± 0.15 ng/ml IGF-1, 1.39 ± 0.08 ng/ml EGF, and 0.33 ± 0.01 ng/ml FGF.
Proliferative effect of PL on huc-MSCs
The proliferative effects of PL and its precursor PRP on huc-MSCs were evaluated by CCK-8 assay. As shown in Fig. 1E, PL at a dilution rate range from 1/200 to 1/10 and PRP at a dilution rate range from 1/100 to 1/10 both exerted significant proliferative effects on huc-MSCs in a dose-dependent manner after 24 and 48 h treatment. The effect of PL at 1/50, 1/25, and 1/10 dilution rate was significant after 24 h and that at 1/100, 1/50, 1/25, and 1/10 dilution rate was significant after 48 h. The effect of PRP was only significant at dilution rates from 1/50 to 1/10 after 48 h treatment. Therefore, the effect of PL was better than that of PRP on huc-MSCs.
RNAseq screening of molecular targets of PL on huc-MSCs
About 53–55 million reads with 80–83 hundred million bases of huc-MSCs were obtained. The error rate for each sample was less than 0.02%. More than 97.7% of total reads mapped to reference database genes. Statistical analysis revealed 774 differentially expressed genes (P < 0.01) out of 58,233 reference genes between PL-H-treated huc-MSCs and untreated huc-MSCs. The significantly upregulated and downregulated genes were functionally analyzed using GO and KEGG databases. As shown in Fig. 2B, the most significant and relevant pathways annotated by KEGG analysis were the cell cycle and cell proliferation (DNA replication) activated by PL. As shown in Fig. 2C, GO analysis showed a similar result that many of the PL-activated biological processes were closely related to the cell cycle and cell proliferation, such as DNA replication initiation, cell cycle phase transition, mitotic cell cycle, cell division, spindle organization, regulation of cell cycle process, DNA replication, and cell cycle. Moreover, cell migration-related processes, such as regulation of microtubule depolymerization, regulation of microtubule-based process, and microtubule cytoskeleton organization, were found to be activated by PL through the GO analysis. Therefore, the RNAseq analysis preliminarily revealed PL′s mechanism associated with the activation of cell cycle, cell proliferation, and cell migration of huc-MSCs. The representative huc-MSCs genes upregulated by PL were shown in Table 1.

Volcano plot (A), KEGG analysis (B), and GO analysis (C) for the comparison between PL-H treated huc-MSCs and untreated huc-MSCs as assayed by RNAseq analysis. Cell cycle progression in huc-MSCs determined by flow cytometry (D), and the relative mRNA expressions of genes of interest in huc-MSCs detected by qPCR (E).
Effect and relevant molecular action of PL on cell cycle progression of huc-MSCs
According to the RNAseq result, cell cycle progression was the primary action, by which PL affects huc-MSCs. To verify this effect, flow cytometry was performed. As shown in Fig. 2D, PL induced cell cycle progression of huc-MSCs from G0/G1 phase to S and G2/M phases. With increasing doses of PL, the G0/G1 phase percentage was progressively decreased, and the S and G2/M phases were increased, indicating a dose-dependent manner of PL. PRP also induced the cell cycle progression, but its effect was much less than PL-L. To elucidate the molecular action of PL, mRNA expressions of the cell cycle-related genes were detected by real time PCR (qPCR) assay. As shown in Fig. 2E, PL-M significantly upregulated the expressions of CDC25A, CCNA2, CCNB1, CCND1, CDK2, E2F1, and E2F2 and downregulated the expressions of P15 and P16 in huc-MSCs (each P < 0.01 vs. NC).
Effect and relevant molecular action of PL on cell migration capacity of huc-MSCs
Wound healing assay was conducted to evaluate the horizontal migration of huc-MSCs. As shown in Fig. 3A, the ratio of wound area with PL treatment at 12, 24, and 36 h to the area without treatment (0 h) was significantly decreased with elevated concentrations of PL (each P < 0.01 vs. NC), indicating that PL promoted wound closure of huc-MSCs by enhancing huc-MSCs migration in a dose-dependent manner. In particular, PL-H almost completed huc-MSCs wound closure after 36 h treatment. Transwell assay was conducted to detect the chemotactic migration of huc-MSCs. As shown in Fig. 3B, the migrated number of huc-MSCs to the bottom of chamber was significantly increased with the elevated concentrations of PL (each P < 0.01 vs. NC), indicating a dose-dependent manner of PL. However, there was no significant difference in migrated cell number between PRP-treated and untreated huc-MSCs. The result revealed that PL other than PRP raised huc-MSCs migration. qPCR assay was performed to detect mRNA expressions of the cell migration-related genes. As shown in Fig. 3C, PL-H significantly upregulated the expressions of MMP2, FIBRONECTIN (FN1), VEGF-A, VEGF-C, VIMENTIN (VIM), and TWIST1 in huc-MSCs (each P < 0.01 vs. NC).

Wounding healing assay of huc-MSCs with PL treatment at 24, 36, and 48h (A). Cell migration is represented as the ratio of the scratched wound area of huc-MSCs with 12, 24, and 36h treatment to the area without treatment (0h). Transwell migration assay of huc-MSCs with PL treatment (B), and the relative mRNA expressions of genes of interest in huc-MSCs detected by qPCR (C). Data are mean values ± SD. Different symbols (a, b, c, and d) indicate significant difference among groups [Fisher’s least significant difference (LSD), P<0.05], and the values decrease with the order from a to d.
Autophagy-related action mechanism of PL on huc-MSCs
Western blot (WB) assay was performed to explore the molecular actions of PL on cell cycle progression and cell proliferation of huc-MSCs. As shown in Fig. 4A, after 24 h treatment, PL significantly upregulated the protein expressions of cell proliferation (PCNA and Mki67) and cell cycle progression (CCNB1, CCNE1, and CDK4) (each P < 0.001 vs. NC). Moreover, PL significantly upregulated the expressions of phosphorylated AMPK (p-AMPK), phosphorylated mTOR (p-mTOR Ser2481/Ser2488), phosphorylated P79 S6K (p-P70 S6K), ATG5, ATG7, Beclin1, and LC3-II (each P < 0.001 vs. NC), indicating activation of the AMPK–mTOR–beclin1 signaling-related autophagy of huc-MSCs.

Protein bands and protein expressions of huc-MSCs with PL treatment (A) and with PL plus BECLIN1-siRNA (siBeclin1) treatment (B). Cell viability of huc-MSCs with PL and siBeclin1 treatment at 24 and 48h (C). siNC: nontargeting control siRNA-treated group; siBeclin1: BECN1-siRNA-treated group. Data are mean values ± SD. Different symbols (a, b, and c) indicate significant difference among groups [Fisher’s least significant difference (LSD), P < 0.05], and the values decrease with the order from a to c.
To verify the autophagy-related mechanism of PL, BECN1-siRNA was applied to block the autophagy of huc-MSCs by knocking down the beclin1 expression. As shown in Fig. 4B, BECN1-siRNA successfully blocked the beclin1 expression and significantly suppressed the expression of PCNA of huc-MSCs in the presence of PL treatment (each P < 0.001 vs. siNC). As shown in Fig. 4C, cell viability result showed that BECN1-siRNA significantly counteracted the PL-induced proliferation of huc-MSCs (P < 0.001 vs. siNC). These results verified that the beclin1-dependent autophagy participated in the proliferative mechanism of PL.
Effect of PL-contained growth factors on huc-MSCs
CCK-8 assay, cell cycle flow cytometry, and transwell assay were performed to evaluate the individual effects of each growth factor contained by PL. The concentrations of recombinant EGF (1.39 ng/ml), FGF (0.33 ng/ml), IGF-1 (5.05 ng/ml), PDGF (26.25 ng/ml), and TGF-β (7.62 ng/ml) were applied as same as those of the corresponding growth factors in PL. As shown in Fig. 5A, proliferate rate of huc-MSCs was significantly increased by PL and by each growth factor after 24 and 48 h treatment (each P < 0.05 vs. NC). Among those growth factors, EGF was most effective, followed by IGF-1 as the second most effective one, while FGF, PDGF, and TGF-β exerted lesser effects. The proliferative rate produced by EGF was approaching the highest level achieved by PL, indicating EGF as a main contributor to the proliferative effect of PL. As shown in Fig. 5B, cell cycle progression of huc-MSCs was significantly promoted by PL, EGF, IGF-1, PDGF, and TGF-β. EGF induced significant decrease of G0/G1 phase percentage and significant increase of S phase percentage as PL (each P < 0.05 vs. NC). FGF, IGF-1, PDGF, and TGF-β induced slight decreases of G0/G1 phase percentage (P > 0.05 vs. NC), followed by significant increases of S phase percentage induced by IGF-1, PDGF, and TGF-β (each P < 0.05 vs. NC). As shown in Fig. 5C, the migrated huc-MSCs number was significantly increased by PL, EGF, IGF-1, PDGF, and TGF-β (each P < 0.05 vs. NC). PL achieved the highest level, followed by IGF-1, PDGF, TGF-β, EGF, and FGF in the order of decreasing effects, indicating IGF-1 as a main contributor to the pro-migrative effect of PL.

Cell viability (A), cell cycle (B) and cell migration (C) of huc-MSCs with treatment of PL and PL-contained growth factors. Data are mean values ± SD. Different symbols (a, ab, b, bc, c, cd, d, e, and f) indicate significant difference among groups [Fisher’s least significant difference (LSD), P < 0.05], and the values decrease with the order from a to f.
Molecular actions of PL-contained growth factors on huc-MSCs
qPCR and WB assays were conducted to elucidate the PL- and growth factors-induced molecular changes in huc-MSCs. As shown in Fig. 6A, PL significantly up-regulated the mRNA expressions of CDC25A, CCNA2, CCNB1, CCND1, E2F1, E2F2, VEGFA, and VEGFC, while the growth factors performed various actions on these genes. CDC25A was significantly upregulated by PDGF, IGF-1, EGF, TGF-β, and FGF in a descending order (each P < 0.05 vs. NC). CCNA2 was significantly upregulated by PDGF (P < 0.05 vs. NC) and statistically upregulated by IGF-1, EGF, and TGF-β in a descending order. CCNB1 was significantly upregulated by IGF-1 (P < 0.05 vs. NC) and statistically upregulated by TGF-β, EGF, PDGF, and FGF in a descending order. CCND1 was significantly upregulated by IGF-1, EGF, PDGF, TGF-β, and FGF in a descending order (each P < 0.05 vs. NC). E2F1 was significantly upregulated by IGF-1 (P < 0.05 vs. NC) and statistically upregulated by TGF-β, PDGF, FGF, and EGF in a descending order. E2F2 was significantly upregulated by IGF-1 and PDGF (each P < 0.05 vs. NC) and statistically upregulated by TGF-β, EGF, and FGF in a descending order. VEGFA was significantly upregulated by IGF-1, FGF, and TGF-β in a descending order (each P < 0.05 vs. NC) and statistically upregulated by EGF. VEGFC was statistically upregulated by IGF-1, PDGF, EGF, and TGF-β in a descending order. As shown in Fig. 6B, PL significantly upregulated protein expression of PCNA and Mki67, while the growth factors performed various actions on those proteins. The expression of PCNA was significantly increased by EGF, TGF-β, IGF-1, FGF, and PDGF in a descending order (each P < 0.05 vs. NC), and that of Mki67 was significantly increased by EGF, IGF-1, PDGF, TGF-β, and FGF in a descending order (each P < 0.05 vs. NC). The above data indicated that none of the growth factors exerted effects as strong as PL, but each one contributed to the effects of PL (Table 2).

qPCR analysis of mRNA expression (A) and Western blot analysis of protein expression (B) of huc-MSCs upon treatment of PL and PL-contained growth factors as well as a sketch diagram related to the effects and molecular actions of PL-contained growth factors (C). The thickness of size line positively represents the degree of effect of each growth factor. Data are mean values ± SD. Different symbols (a, ab, b, bc, c, cd, d, e, and f) indicate significant difference among groups [Fisher’s least significant difference (LSD), P < 0.05], and the values decrease with the order from a to f.
In vivo evaluation of adjuvant role of PL on huc-MSCs
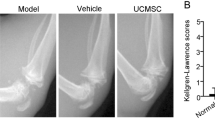
According to the above results, PL improved cell proliferation, cell cycle transition, and cell migration of huc-MSCs through actions of PL-contained growth factors, indicating its adjuvant role on huc-MSCs. An OA rat model was employed to evaluate such adjuvant role in vivo. As shown in Fig. 7A, cartilage degeneration, characterized by chondrocyte hypertrophy and loss, collagen disruption, and subchondral bone cyst, was observed in the OA rat joints, while the degeneration was reversed, respectively by PL, MSC, and PL + MSC. With these treatments, the number of chondrocytes and mass of collagen were obviously increased, and the chondrocyte hypertrophy and subchondral bone cyst disappeared. Nevertheless, there remained several areas of slight collagen disruption in the PL-treated and MSC-treated samples, while no sign of degeneration was seen in the PL+MSC-treated samples. The OARSI scoring demonstrated that PL+MSC, MSC, and PL significantly decreased the histopathological score in a descending order (each P < 0.05 vs. model), indicating their anti-OA effects in the following order: PL+MSC > MSC > PL. TWL and MWT were classic pain behavior indices that reflect mechanical allodynia and thermal hyperalgesia respectively. As shown in Fig. 7B, TWL and MWT were significantly decreased with OA modeling (each P < 0.05 vs. NC) and significantly increased by PL+MSC, MSC, and PL (each P < 0.05 vs. Model). The effects of PL+MSC, MSC, and PL on TWL were similar to each other, and the effect of PL+MSC on MWT was similar to MSCs but better than PL. As shown in Fig. 7C, the mRNA expressions of Col2 and Sox9 were significantly downregulated and that of Mmp13, Adamts4, and Adamts9 were significantly upregulated in the OA rat joints (each P < 0.01 vs. NC). These pathological changes were statistically reversed by PL, MSC, and PL+MSC in an order of increasing effects. PL+MSC exerted significant effects on all of these genes (each P < 0.01 vs. Model), while that of PL and MSC were significant only on Mmp13, Adamts4, and Adamts9 (each P < 0.01 vs. Model). As shown in Fig. 7D, the protein expression pattern in joints was similar to the mRNA expression pattern that PL+MSC exerted the strongest reversal effects than that of PL or MSC on the abnormal expressions of Col2, Mmp13, and Adamts4 (each P < 0.01 vs. Model).

Histopathological observation on cartilage (black asterisk: chondrocyte loss; orange asterisk: collagen disruption; red arrow: chondrocyte hypertrophy) and subchondral bone (green arrow: subchondral bone cyst) by using SO staining, and OARSI scoring of rat knee joints (A). Pain behavior observation (TWL and MWT) of rats (B). qPCR analysis of mRNA expression of OA pathological genes in cartilage tissue with treatment of PL, MSC, and PL+MSC (C). Western blot analysis of protein expression of OA pathological molecules in cartilage tissue with treatment of PL, MSC, and PL+MSC (D). Data are mean values ± SD. Different symbols (a, ab, b, bc, c, cd, and d) indicate significant difference among groups [Fisher’s least significant difference (LSD), P < 0.05], and the values decrease with the order from a to d. Scale bar = 100 μm.
Discussion
The main innovation of this paper is the finding of PL′s beneficial effects on huc-MSCs and its mechanism of growth factors-based action, which differs from previous studies focusing mainly on the potential of PL as substitute for FBS in MSCs expansion. For clinical applications, MSCs have to be expanded to reach a meaningful cell number, requiring a large amount of FBS-like supplement13. However, platelets lack ex vivo expanding capacity, resulting in unavailability of large-scale use of PL, especially for the autologous use. Thus, PL is more suitable for short-term use as an adjuvant for MSCs, but its adjuvant role has not yet been confirmed. A clinical study reported that the endogenous growth factors released from platelets in serum positively influenced the number of MSCs in vivo and induced proliferation of MSCs in vitro, and the effect had a direct relationship with platelets/growth factors21. Moreover, the growth factors of PL have been found to benefit the proliferation, chemotaxis, regeneration, and angiogenesis of MSCs15. Although the benefits of growth factors of PL to MSCs have been observed, the underlying mechanism remains poorly understood. To fill this gap, we conducted cellular and molecular experiments to investigate the adjuvant effect, and the growth factors-based mechanism of PL on huc-MSCs. Furthermore, an animal experiment was performed to determine the adjuvant role of PL in MSCs-based cell therapy. To our knowledge, this is the first study regarding this issue.
The platelet purity of our PL was above 99% before freeze-thaw lysis, and the contents of growth factors in PL were much higher than that released by PPR or other PL products13, making our product as growth factors-enriched PL (GF-PL). Cell viability assay showed that the GF-PL exerted stronger proliferative effect than PRP on huc-MSCs. To explore the mechanism of GF-PL, we conducted RNAseq analysis and found that GF-PL-targeted genes related to cell cycle progression, DNA replication, and microtubule-based activity of MSCs. Thus, we deduced that GF-PL exerted effects not only on proliferation, but also on cell cycle and migration of huc-MSCs. To verify this deduction, flow cytometry, wound healing, and transwell assays were conducted, and meanwhile, qPCR was employed to verify the RNAseq result. Our data demonstrated that GF-PL dose-dependently affected huc-MSCs by inducing cell cycle progression and migration. With these phenotypic changes, genes associated with cell cycle progression (CDC25A, CCNA2, CCNB1, CCND1, CDK2, CDKN2A, CDKN2B, E2F1, and E2F2) and cell migration (MMP-2, FN1, VEGF-A, VEGF-C, VIM, and TWIST1) as well as proteins associated with cell proliferation (PCNA and Mki67) and cell cycle progression (cyclin B, cyclin E, and CDK4) were all significantly regulated by GF-PL, indicating molecular actions of GF-PL on proliferation, cell cycle transition, and migration of huc-MSCs. It has been reported that self-renewal of human stem cells is controlled by the cell cycle regulatory machinery involving cyclins, CDKs, and E2F22, suggesting that GF-PL may enhance self-renewal ability of huc-MSCs. Moreover, the migration-related genes are known as metastatic genes of cancer cells, suggesting that GF-PL may have a metastasis-like promigrative mechanism, which confers strong mobility to huc-MSCs.
Autophagy is a mechanism essential for maintaining cellular characteristics and physiological functions of MSCs. Accumulating evidences have highlighted that autophagy serves to promote proliferation and inhibit apoptosis and inflammatory responses of MSCs23,24. And, activation of autophagy contributes to adaptive cell cycle response of MSCs25. Moreover, modulation of autophagy may have an impact on the regenerative and therapeutic potential of MSCs26. Beclin1 acts as a crucial modifier of autophagy involved in the activation of autophagy and formation of autophagosomes. In this study, canonical autophagy machinery (e.g., Beclin1, ATG5, ATG7, and LC3-II) of huc-MSCs was activated by GF-PL, and knock-down of beclin1 expression by BECN1-siRNA resulted in the suppression of GF-PL-induced proliferation and PCNA expression of huc-MSCs. Therefore, our data indicated an autophagy-mediated mechanism of GF-PL on the proliferation of MSCs. In future, further studies are needed to explore whether autophagy contributes to the mechanism of GF-PL on the cell cycle progression and migration of huc-MSCs and whether autophagy determines the therapeutic potential of huc-MSCs.
In GF-PL, the contents of growth factors were found in the following order: PDGF-AA > PDGF-AB > PDGF-BB > TGF-β > IGF-1 > EGF > FGF. Previous studies have demonstrated that each of these factors has positive effect on MSCs (Table 3)27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61. Of them, PDGF, IGF-1, and FGF promote proliferation and cell cycle transition of huc-MSCs, while the effects of TGF-β and EGF on huc-MSCs remain undetermined (Table 3)30,39,41,54,58. Moreover, no literature was found regarding the promigrative effect of any of these factors on huc-MSCs. In this study, we firstly compared the activities among the three forms of PDGF. As shown in Fig. S1, PDGF-AA, PDGF-AB, and PDGF-BB at 20 ng/ml induced proliferation and migration as well as PCNA expression of huc-MSCs in the following order of effects: PDGF-AA > PDGF-AB > PDGF-BB, indicating PDGF-AA as the most effective form on huc-MSCs. Therefore, PDGF-AA was selected as the representative PDGF for the formal experiments. Then, we found that not only PDGF, IGF-1 and FGF, but also TGF-β and EGF promoted the proliferation and cell cycle transition of huc-MSCs. For the first time, each of these factors was found to stimulate the migration of huc-MSCs. The effects of these factors were all significant, except that of FGF on cell cycle and migration of huc-MSCs. It might attribute to the low content of FGF in GF-PL. As shown in Table 2, we concluded that PDGF, TGF-β, IGF-1, and EGF affected all the GF-PLʹs cellular and molecular targets with significant effects respectively on 10, 8, 11, and 7 of them, while FGF affected 12 targets of GF-PL with significant effects on 5 of them. It indicates that all these factors have contributed to the effects of GF-PL in varying degrees. The content and property of each factor determine the degree of the contribution, which warrants further investigations.
OA is a prevalent joint disease still lacking effective therapy. Intra-articular injection of MSCs possesses efficacy of pain relief and cartilage repair, providing a therapeutic potential in OA treatment62. To our knowledge, little study has been conducted to demonstrate the in vivo therapeutic efficacy of huc-MSCs on knee OA. There is nonetheless a common concern on MSCs-based therapies that the survival and function of MSCs at the transplanted site are poor, often resulting in a decreased therapeutic efficacy63. Thus, it is necessary to improve the viability of MSCs and enhance their biological functions during the treatment. Taking advantage of the benefits of GF-PL on huc-MSCs, we combined them in this study to reach the synergistic outcome for OA treatment. For the first time, our data not only demonstrated the in vivo therapeutic activity of huc-MSCs on OA, but also revealed significant synergistic effect of combined GF-PL and huc-MSCs, according to the histopathological scoring and pain behavior (mechanical allodynia). Moreover, the combined treatment significantly reversed the OA pathological changes of anabolic biomarkers (Col2, Sox9) and catabolic biomarkers (Mmp13, Adamts4, and Adamts9), which was stronger than the single treatment of GF-PL or huc-MSCs, testifying their synergistic effect in the molecular level. Although this study revealed the adjuvant role and mechanism of GF-PL on huc-MSCs, it is not sufficient to show the synergistic relationship between them in OA treatment. Further studies are needed to explore, how GF-PL and huc-MSCs work in joint for OA cartilage repair and which mechanism is responsible for this synergism.
Materials and methods
Reagents and materials
Minimum essential medium-alpha modification (α-MEM) with Glutamax™-1 was purchased from Gibco BRL (NY, USA). Trypsin (0.25%) was purchased from Thermo Fisher Scientific (MA, USA). Fetal bovine serum (FBS) was purchased from CellMax (Beijing, China). Cell counting kit-8 (CCK-8) kit was purchased from Bimake (TX, USA). Cell cycle kit was purchased from BD Biosciences (CA, USA). Cell culture plates were purchased from Eppendorf (Hamburg, Germany) and Transwell chambers purchased from Corning (NY, USA). All ELISA kits were purchased from Multi Sciences (Lianke) Biotech Co., Ltd (Hangzhou, China). All human recombinant cytokines were purchased from Peprotech (London, UK). TRIzol reagent and DNase I kits were obtained from TaKaRa Biotechnology Co. Ltd. (Dalian, China). All-in-One cDNA Synthesis SuperMix kit was purchased from Biotool (TX, USA). 2× SYBR Green qPCR Master Mix (low ROX) kit was obtained from Bimake (TX, USA). The antibodies were purchased from Cell Signaling Technology Inc. (MA, USA). Monoiodoacetate (MIA) was purchased from Sigma-Aldrich (MO, USA). Human Umbilical Cord Mesenchymal Stem Cell Osteogenic Differentiation Medium kit, Human Umbilical Cord Mesenchymal Stem Cell Chondrogenic Differentiation Medium kit, and Human Umbilical Cord Mesenchymal Stem Cell Osteogenic Differentiation Medium kit were purchased from Cyagen Biosciences (Suzhou, China).
Human PL extraction
PL was obtained using a two-step procedure consisting of double-spin centrifugation followed by freeze-thawing lysis, as described in our previous report64. All blood specimens used in this study were approved by the Ethical Committee of the Zhejiang Chinese Medical University. Briefly, after obtaining informed consent from healthy adult donors, whole blood was collected in tubes containing sodium citrate anticoagulant (3.2%, w/v; blood:citrate = 9:1). To obtain platelet-containing plasma separate from erythrocytes and leukocytes, each 50 ml of whole blood was centrifuged at 210×g for 10 min. Then the yellow plasma with buffy coat, mainly containing platelets, was collected in a new tube and centrifuged at 210×g for 5 min. The residual erythrocytes were discarded, before the supernatant plasma and platelet pellet were collected as platelet-rich plasma (PRP).
To measure the positive rate of platelet in our PRP, the expression of human platelet surface marker (CD41a) was analyzed as previously described64. Briefly, the PRP was incubated with the antibody against CD41a. After 1 h of incubation, the sample was washed with PBS and the expression of CD41a was analyzed by flow cytometer (BD FACS Calibur, BD Biosciences, CA, USA) in triplicates. Fluorescent signal intensity was recorded and analyzed by CellQuest software. The platelet number of the PRP was measured by Mindray BC-3000plus blood cell analyzer (Shenzhen, China) and standardized to 1 × 108 platelets/ml, followed by repeating freeze-thaw (−80 to 37 °C) lysis for three times. The residual platelet fragments were removed by centrifugation and the obtained supernatant was collected as PL and stored at −80 °C before use.
Immunophenotypic analysis of platelet and quantitative analysis of growth factors
The concentrations of platelet-derived growth factor (PDGF), insulin-like growth factor (IGF-1), transforming growth factor β (TGF-β), epidermal growth factor (EGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF) in PL were measured in triplicates using commercially available enzyme-linked immunosorbent assay kits in accordance with each manufacturer’s instructions (Lianke Biotech Co., Hangzhou, China). The absorbance was measured using a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Huc-MSCs isolation and identification
The section of human umbilical cord was obtained from the first affiliated hospital of Zhejiang Chinese Medical University with the permission of donors and was freshly processed within 4 h. The umbilical cord was cut into pieces with <2 cm3 volume and washed with phosphate buffered saline (PBS) (1×, pH 7.4) for three times. After removal of epidermal tissue and vessel endothelium, samples were immersed in α-MEM containing 10% FBS at 37 °C and 5% CO2 for incubation. The culture medium was replaced every 2 or 3 days. The isolated cells were collected as huc-MSCs and resuspended in α-MEM containing 10% FBS, with passage being carried out after confluence had occurred.
Characterization of the huc-MSCs phenotype was performed by flow cytometry. In passage 3–8, the cells were resuspended in PBS at a density of 106 cells/ml and incubated with CD19, CD34, CD45, CD73, CD90, CD105, CD11b, and HLA-DR mouse antihuman antibodies, respectively. After incubation for 30 min at room temperature (RT), each cell suspension was centrifuged at 2000 rpm for 5 min. The supernatant was removed and 100 μl PBS were added to resuspend the cell pellet for flow cytometry analysis (BD FACSVerse, NJ, USA).
Three-line differentiation ability of the huc-MSCs was test by specific staining. The cells were respectively incubated in osteogenic (HUXUC-90021, Cyagen), chondrogenic (HUXUC-90041, Cyagen), and adipogenic (HUXUC-90031, Cyagen) induction medium for 14 days, according to the manufacturer’s instructions. The medium was replaced every 3 days. After confirming the morphological manifestations of differentiation, the cells were fixed with 4% polyformaldehyde for 30 min. After washing twice with PBS, osteogenic differentiation was examined with Alizarin red staining, chondrogenic differentiation was examined with Alcian blue staining, and adipogenic differentiation was examined with Oil Red O staining. All images were obtained using an inverted phase microscope (Carl Zeiss, Göttingen, Germany).
Cell viability assay
The cell viability of huc-MSCs was determined by CCK-8 assay at 24 and 48 h. Cells were seeded on 96-well plates at a density of 4 × 103 cells/well in 200 μl medium for 24 h, followed by the treatment of PL at dilution rates from 1/10 to 1/1000 and PRP at gradient concentrations corresponding to PL′s. Aliquots of each 20 μl CCK-8 solution were added to each well and incubated at 37 °C for 2 h, until the color turned to orange. The optical density (OD) value was measured at 450 nm with a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proliferative rate (%) = (EAA-treated OD/untreated OD) × 100. PL at dilution rates of 1/50, 1/100 and 1/200 were used as high-dose PL (PL-H), middle-dose PL (PL-M), and low-dose PL (PL-L), respectively. PRP at a concentration corresponding to PL-M was used as positive control.
RNA sequencing analysis
Total RNA of huc-MSCs was extracted using TRIzol reagent and genomic DNA was removed using Dnase I kit. The RNA was qualitatively determined using 2100 Bioanalyzer (Agilent Technologies, CA, USA) and quantified using NanoDrop ND-2000 (NanoDrop Technologies, DE, USA). RNA purification, reverse transcription, library construction and sequencing were conducted by Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China) according to the manufacturer′s instructions (Illumina, CA, USA). RNAseq transcriptome library was prepared following TruSeqTM RNA sample preparation Kit from Illumina (CA, USA). After cDNA synthesis and quantification, paired-end RNA sequencing library was sequenced using Illumina Novaseq 6000 platform (Illumina, CA, USA). To identify differential expression genes (DEGs) between two different samples, the expression level of each transcript was calculated according to the fragments per kilobase of exon per million mapped reads (FPKM) method. Functional-enrichment analysis including GO and KEGG were performed to identify which DEGs were enriched in GO terms and KEGG pathways at P-value < 0.05 compared with the whole-transcriptome background.
Cell cycle analysis
Cell cycle progression of huc-MSCs was analyzed using flow cytometry as described in our previous report20. Briefly, cells in the logarithmic growth phase were collected, inoculated in 6-well plates (cell density = 3 × 105/well) and divided into five groups: normal control group (treated with saline), PL-L group (treated with PL-L), PL-M group (treated with PL-M), PL-H group (treated with PL-H), and PRP group (treated with PRP). After the treatment for 24 h, the huc-MSCs were harvested by centrifugation, washed with PBS, and re-suspended in cold ethanol solution (70%) at 4 °C overnight. Then, the cells were centrifuged to remove ethanol and stained with PI/RNase staining solution (10 μl of 50 μg/ml PI) at RT for 30 min. The cell cycle progression was analyzed in three replicates using flow cytometry (BD FACSVerse, NJ, USA).
Wound healing assay
To conduct the wound healing assay, huc-MSCs in the logarithmic growth phase were inoculated in 6-well plates (3 × 105/well) and artificially scratched in cross form, followed by treatment of PL with serum-free medium. The cells were observed and imaged at four different time points (0, 12, 24, and 36 h) under an inverted microscope (CarlZeiss, Göttingen, Germany). The wound area was calculated with Image J 1.47 software. Each experiment was conducted in triplicate.
Transwell migration assay
A transwell assay was performed in 24-well transwell chambers with 8 μm nitrocellulose pore filters (Corning Costar, NY, USA) to evaluate the effect of PL on huc-MSCs migration. huc-MSCs in the logarithmic growth phase were collected, washed with PBS, and resuspended (2 × 105/ml) in serum-free α-MEM medium. Aliquots of 200 µl of the huc-MSCs suspension were loaded into the upper chamber, and 600 µl serum-free α-MEM medium with PL (PL-L, PL-M or PL-H) were added into the lower chamber as PL group. The NC group applied only serum-free α-MEM medium in the lower chamber, while the PRP group applied serum-free α-MEM medium with PRP in the lower chamber. The concentration of PRP was similar to that of PL-M and can be estimated as 1% (v/v). After incubation for 18 h, the cells that passed through the membranes of the upper chambers were fixed with 4% paraformaldehyde (Servicebio, Wuhan, China) and stained with 1% crystal violet dye solution (Beyotime Biotechnology, Shanghai, China), before images of each group were captured using a microscope in different areas in three independent repeated experiments (CarlZeiss, Göttingen, Germany).
Real time PCR
The mRNA expression of targeted genes in huc-MSCs was measured using a real time PCR (qPCR) assay on an ABI QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems; Thermo Scientific, USA). Total RNA was extracted with TRIzol reagent and quality controlled by NanoDrop2000 spectrophotometer (Thermo Scientific, USA). cDNA reverse transcription was performed by using All-in-One cDNA Synthesis SuperMix. The PCR reaction system was 20 μl, including 10 μl SYBR Green qPCR Master Mix (low ROX), 0.4 μl PCR Forward Primer, 0.4 μl PCR Reverse Primer, 1 μl template cDNA and 8.2 μl ddH2O, with the following reaction conditions: initial denaturation at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 3 s, and annealing and extension at 60 °C for 30 s. β-Actin was used as reference gene and the 2−Δ∆CT method was used to analyze the relative mRNA expressions (Table 4).
Western blot analysis
Total protein of huc-MSCs was extracted with lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton and 0.1% SDS) containing proteinase inhibitor cocktail (Bimake, Houston, TX, USA) for 30 min on ice. The targeted protein was separated by denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 6–12%) and transferred onto a nitrocellulose membrane (Sartorius Stedim, Göttingen, Germany). The membrane was blocked with 5% nonfat milk for 2 h, which was followed by overnight incubation at 4 °C with the following primary antibodies against β-actin, PCNA, Mki67, CCNB1, CCNE1, and CDK4. Following incubation with peroxidase-conjugated goat antirabbit/mouse IgG at room temperature for 2 h, each protein was visualized using Western Lightning® Plus ECL (Perkin Elmer, Inc., Waltham, MA, USA), detected using X-ray film (Kodak, Tokyo, Japan) and scanned.
Molecular and cellular verification by small interfering RNA (siRNA) treatment
For transient knockdown of human BECLIN1 gene, siRNA of BECLIN1 (BECN1-siRNA) and nontargeting control siRNA was designed (GenePharma, Shanghai, China) to transfect huc-MSCs by using lipofectamine RNAiMAX Transfection Reagent (Thermo, Waltham, USA), according to the manufacturerʹs instruction. Western blot (WB) and cell viability assays were conducted to verify the siRNA knockdown of BECLIN1 gene and the function of beclin1-related mechanism in the proliferative effect of PL on huc-MSCs.
Animals and OA modeling
Male Sprague-Dawley (SD) rats weighing 180–220 g were provided by Shanghai Super B&K Laboratory Animal Co. Ltd. (Grade SPF II, SCXK (Shanghai): 2013-0016) and housed under a controlled pathogen-free condition with 12 h light/dark cycle and allowed ad libitum food and water. All rats were treated in strict accordance with the China legislation on the use and care of laboratory animals and all experiments on the rats were approved by the Medical Norms and Ethics Committee of Zhejiang Chinese Medical University (Approval number: ZSLL-2017-091). Osteoarthritis (OA) model was established applying monoiodoacetate (MIA) method as described previously64. Briefly, rats were intra-articularly injected with 50 μl of 30 mg/ml MIA for 7 days, followed by therapeutic treatment.
Animal experiment
Fifty rats were randomly divided into five groups for ten in each: NC as normal control group, model as OA model group, PL as PL-M treated model group, MSC as huc-MSCs (106 cells/ml) treated model group, and PL+MSC as PL-M and huc-MSCs together treated model group. Rats in the NC group were intra-articularly injected with 50 μl of saline, while all other groups were modeled. All treatments were weekly performed and lasted for 4 weeks, while the NC group was intra-articularlly given an equal volume of saline. At the end, the pain behavior parameters TWL (thermal withdrawal latency) and MWT (mechanical withdrawal threshold) were tested in triplication after the final treatment. Then all rats were sacrificed under anesthesia and their knee joints were immediately taken for histopathological analysis. The joints were resected, fixed in 10% formalin for 24 h, decalcified with 10% EDTA in PBS for 8 weeks, embedded in paraffin, sectioned into 2–3 μm thickness, and stained with SO (safranin-O). The slides were examined under microscopy and statistically graded on a scale of 0–13 by double-blind observation, according to the OARSI scoring systems65. The rat knee joint samples from each group were ground into powder with liquid nitrogen using a mortar and a pestle. An aliquot of the powder was treated with TRIzol reagent for total RNA extraction, and another was treated with T-PER™ Tissue Protein Extraction Reagent (Thermal Scientific) for total protein extraction. Then, qPCR and Western blot assays were conducted as described above.
Data analysis
Data were expressed as mean values ± standard deviation (SD). Data from different groups were compared using one-way ANOVA followed by Fisher’s least significant difference (LSD) comparison. A P-value < 0.05 was considered to indicate a significant difference and P-value < 0.01 considered to indicate a very significant difference. All analyses were performed using an updated version of DPS software66.
References
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999).
Ding, D. C., Chang, Y. H., Shyu, W. C. & Lin, S. Z. Human umbilical cord mesenchymal stem cells: a new era for stem cell therapy. Cell Transpl. 24, 339–347 (2015).
El Omar, R. et al. Umbilical cord mesenchymal stem cells: the new gold standard for mesenchymal stem cell-based therapies? Tissue Eng. Part B 20, 523–544 (2014).
Gore, A. et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67 (2011).
Arutyunyan, I., Elchaninov, A., Makarov, A. & Fatkhudinov, T. Umbilical cord as prospective source for mesenchymal stem cell-based therapy. Stem Cells Int. 2016, 6901286 (2016).
Fong, C. Y. et al. Human Wharton’s jelly stem cells have unique transcriptome profiles compared to human embryonic stem cells and other mesenchymal stem cells. Stem Cell Rev. Rep. 7, 1–16 (2011).
Ryan, J. M., Barry, F. P., Murphy, J. M. & Mahon, B. P. Mesenchymal stem cells avoid allogeneic rejection. J. Inflamm. 2, 8 (2005).
Fan, C. G., Zhang, Q. J. & Zhou, J. R. Therapeutic potentials of mesenchymal stem cells derived from human umbilical cord. Stem Cell Rev. Rep. 7, 195–207 (2011).
Burnouf, T. et al. Blood-derived biomaterials and platelet growth factors in regenerative medicine. Blood Rev. 27, 77–89 (2013).
Hara, Y., Steiner, M. & Baldini, M. G. Platelets as a source of growth-promoting factor(s) for tumor cells. Cancer Res. 40, 1212–1216 (1980).
Umeno, Y., Okuda, A. & Kimura, G. Proliferative behaviour of fibroblasts in plasma-rich culture medium. J. Cell Sci. 94, 567–575 (1989).
Doucet, C. et al. Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell-based therapy applications. J. Cell Physiol. 205, 228–236 (2005).
Shih, D. T. B. & Burnouf, T. Preparation, quality criteria, and properties of human blood platelet lysate supplements for ex vivo stem cell expansion. New Biotechnol. 32, 199–211 (2015).
Burnouf, T., Strunk, D., Koh, M. B. C. & Schallmoser, K. Human platelet lysate: replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials 76, 371–387 (2016).
Liu, Y., Kalén, A., Risto, O. & Wahlström, O. Fibroblast proliferation due to exposure to a platelet concentrate in vitro is pH dependent. Wound Repair Regen. 10, 336–340 (2002).
Blair, P. & Flaumenhaft, R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 23, 177–189 (2009).
Shariatzadeh, M., Song, J. & Wilson, S. L. The efficacy of different sources of mesenchymal stem cells for the treatment of knee osteoarthritis. Cell Tissue Res. 378, 399–410 (2019).
Murphy, M. B. et al. Adult and umbilical cord blood-derived platelet-rich plasma for mesenchymal stem cell proliferation, chemotaxis, and cryo-preservation. Biomaterials 33, 5308–5316 (2012).
Wen, Y. et al. Platelet-rich plasma enhanced umbilical cord mesenchymal stem cells-based bone tissue regeneration. Arch. Oral Biol. 59, 1146–1154 (2014).
Li, N. et al. Lysophosphatidic acid enhances human umbilical cord mesenchymal stem cell viability without differentiation via LPA receptor mediating manner. Apoptosis 22, 1296–1309 (2017).
Tan, H. B., Giannoudis, P. V., Boxall, S. A., Mcgonagle, D. & Jones, E. The systemic influence of platelet-derived growth factors on bone marrow mesenchymal stem cells in fracture patients. BMC Med. 13, 6 (2015).
Becker, K. A. et al. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell Physiol. 209, 883–893 (2006).
Bo, Y. et al. Autophagy alleviates the decrease in proliferation of amyloid β1-42-treated bone marrow mesenchymal stem cells via the AKT/mTOR signaling pathway. Mol. Med. Rep. 19, 4091–4100 (2019).
Sun, Z. et al. VX-765 enhances autophagy of human umbilical cord mesenchymal stem cells against stroke‐induced apoptosis and inflammatory responses via AMPK/mTOR signaling pathway. CNS Neurosci. Ther. 26, 952–961 (2020).
Maria, L. et al. Autophagy mediates cell cycle response by regulating nucleocytoplasmic transport of PAX6 in limbal stem cells under ultraviolet-A stress. PLoS ONE 12, e0180868 (2017).
Ceccariglia, S., Cargnoni, A., Silini, A. R. & Parolini, O. Autophagy: a potential key contributor to the therapeutic action of mesenchymal stem cells. Autophagy 16, 28–37 (2019).
Hu, Y., Cheng, P., Ma, J. C., Xue, Y. X. & Liu, Y. H. Platelet-derived growth factor BB mediates the glioma-induced migration of bone marrow-derived mesenchymal stem cells by promoting the expression of vascular cell adhesion molecule-1 through the PI3K, P38 MAPK and NF-κB pathways. Oncol. Rep. 30, 2755–2764 (2013).
Kang, Y. J. et al. Role of c-Jun N-terminal kinase in the PDGF-induced proliferation and migration of human adipose tissue-derived mesenchymal stem cells. J. Cell Biochem. 95, 1135–1145 (2005).
Mizuno, M. et al. Platelet-derived growth factor (PDGF)-AA/AB in human serum are potential indicators of the proliferative capacity of human synovial mesenchymal stem cells. Stem Cell Res. Ther. 6, 243 (2015).
Qiu, P. et al. Platelet-derived growth factor promotes the proliferation of human umbilical cord-derived mesenchymal stem cells. Cell Biochem. Funct. 31, 159–165 (2013).
Zhang, M. et al. The effects of platelet-derived growth factor-BB on human dental pulp stem cells mediated dentin-pulp complex regeneration. Stem Cells Transl. Med. 6, 2126–2134 (2017).
Josh, F. et al. Concentration of PDGF-AB, BB and TGF-β1 as valuable human serum parameters in adipose-derived stem cell proliferation. J. Nippon Med. School 80, 140–147 (2013).
Morscheid, S. et al. Therapeutic effects of rAAV-mediated concomittant gene transfer and overexpression of TGF-β and IGF-I on the chondrogenesis of human bone-marrow-derived mesenchymal stem cells. Int. J. Mol. Sci. 20, 2591 (2019).
Yun, C., Mendelson, J., Blake, T., Mishra, L. & Mishra, B. TGF-beta signaling in neuronal stem cells. Dis. Markers 24, 251–255 (2008).
Lv, T. et al. Insulin-like growth factor 1 promotes the proliferation and committed differentiation of human dental pulp stem cells through MAPK pathways. Arch. Oral Biol. 72, 116–123 (2016).
Qu, J. et al. Insulin-like growth factor 1 promotes the proliferation and differentiation of human periodontal ligament stem cells into periodontal ligament fibroblasts. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 33, 471–476 (2017).
Supeno, N. E. et al. IGF-1 acts as controlling switch for long-term proliferation and maintenance of EGF/FGF-responsive striatal neural stem cells. Int. J. Med. Sci. 10, 522–531 (2013).
Youssef, A. & Han, V. K. Low oxygen tension modulates the insulin-like growth factor-1 or -2 Signaling via both insulin-like growth factor-1 receptor and insulin receptor to maintain stem cell identity in placental mesenchymal stem cells. Endocrinology 157, 1163–1174 (2016).
Zhao, L. et al. Effects of IGF-1 on neural differentiation of human umbilical cord derived mesenchymal stem cells. Life Sci. 151, 93–101 (2016).
Zhu, M. et al. Human cerebrospinal fluid regulates proliferation and migration of stem cells through insulin-like growth factor-1. Stem Cells Dev. 24, 160–171 (2015).
Wang, Q., Zhang, F. & Hong, Y. Blocking of autocrine IGF-1 reduces viability of human umbilical cord mesenchymal stem cells via inhibition of the Akt/Gsk-3β signaling pathway. Mol. Med. Rep. 17, 4681–4687 (2018).
Ai, G. et al. Epidermal growth factor promotes proliferation and maintains multipotency of continuous cultured adipose stem cells via activating STAT signal pathway in vitro. Medicine 96, e7607 (2017).
Baer, P. C., Schubert, R., Bereiter-Hahn, J., Plösser, M. & Geiger, H. Expression of a functional epidermal growth factor receptor on human adipose-derived mesenchymal stem cells and its signaling mechanism. Eur. J. Cell Biol. 88, 273–283 (2009).
Bai, T. et al. Epidermal growth factor induces proliferation of hair follicle-derived mesenchymal stem cells through epidermal growth factor receptor-mediated activation of ERK and AKT signaling pathways associated with upregulation of cyclin d1 and downregulation of p16. Stem Cells Dev. 26, 113–122 (2017).
Li, Y. et al. The novel miRNA N-72 regulates EGF-induced migration of human amnion mesenchymal stem cells by targeting MMP2. Int. J. Mol. Sci. 19, 1363 (2018).
Mogi, A. et al. Entrainment of the circadian clock in neural stem cells by epidermal growth factor is closely associated with ERK1/2-mediated induction of multiple clock-related genes. Neuroscience 379, 45–66 (2018).
Nakaji-Hirabayashi, T., Kato, K. & Iwata, H. Surface-anchoring of spontaneously dimerized epidermal growth factor for highly selective expansion of neural stem cells. Bioconjug. Chem. 20, 102–110 (2009).
Tian, H., Guo, M., Zhuang, Y., Chu, J. & Zhang, S. Enhanced proliferation of bone marrow mesenchymal stem cells by co-culture with TM4 mouse Sertoli cells: involvement of the EGF/PI3K/AKT pathway. Mol. Cell Biochem. 393, 155–164 (2014).
Cai, Y. & Wu, J. A study on effect of basic fibroblast growth factor on human limbal stem cell proliferation cultured in low calcium medium. Zhonghua Yan Ke Za Zhi 37, 259–262 (2001).
Duan, S. R. et al. Influences of bFGF and EGF on the proliferation and differentiation of endogenous neural stem cells after cerebral infarction in human. Zhonghua Yi Xue Za Zhi 88, 3337–3341 (2008).
Hebert, T. L. et al. Culture effects of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) on cryopreserved human adipose-derived stromal/stem cell proliferation and adipogenesis. J. Tissue Eng. Regen. Med. 3, 553–561 (2009).
Kang, W. et al. Sequential application of bFGF and BMP-2 facilitates osteogenic differentiation of human periodontal ligament stem cells. J. Periodont. Res. 54, 424–434 (2019).
Kim, J. H., Lee, M. C., Seong, S. C., Park, K. H. & Lee, S. Enhanced proliferation and chondrogenic differentiation of human synovium-derived stem cells expanded with basic fibroblast growth factor. Tissue Eng. Part A 17, 991–1002 (2011).
Liu, L. Y. et al. Basic fibroblast growth factor/vascular endothelial growth factor in the serum from severe burn patients stimulates the proliferation of cultured human umbilical cord mesenchymal stem cells via activation of Notch signaling pathways. J. Trauma Acute Care Surg. 75, 789–797 (2013).
Liu, Y. et al. A novel chemical-defined medium with bFGF and N2B27 supplements supports undifferentiated growth in human embryonic stem cells. Biochem. Biophys. Res. Commun. 346, 131–139 (2006).
Morito, A. et al. Effects of basic fibroblast growth factor on the development of the stem cell properties of human dental pulp cells. Arch. Histol. Cytol. 72, 51–64 (2009).
Nedeau, A. E. et al. A CXCL5- and bFGF-dependent effect of PDGF-B-activated fibroblasts in promoting trafficking and differentiation of bone marrow-derived mesenchymal stem cells. Exp. Cell Res. 314, 2176–2186 (2008).
Ramasamy, R. et al. Basic fibroblast growth factor modulates cell cycle of human umbilical cord-derived mesenchymal stem cells. Cell Prolif. 45, 132–139 (2012).
Sukarawan, W., Nowwarote, N., Kerdpon, P., Pavasant, P. & Osathanon, T. Effect of basic fibroblast growth factor on pluripotent marker expression and colony forming unit capacity of stem cells isolated from human exfoliated deciduous teeth. Odontology 102, 160–166 (2014).
Zhang, R. et al. [Effects of basic fibroblast growth factor and vascular endothelial growth factor on the proliferation, migration and adhesion of human periodontal ligament stem cells in vitro]. Zhonghua Kou Qiang Yi Xue Za Zhi 48, 278–284 (2013).
Wang, R. et al. The differential effect of basic fibroblast growth factor and stromal cell-derived factor-1 pretreatment on bone morrow mesenchymal stem cells osteogenic differentiation potency. Mol. Med. Rep. 17, 3715–3721 (2018).
Ha, C. W., Park, Y. B., Kim, S. H. & Lee, H. J. Intra-articular mesenchymal stem cells in osteoarthritis of the knee: a systematic review of clinical outcomes and evidence of cartilage repair. Arthroscopy 35, 277–288 (2019).
Nakamura, Y., Ishikawa, H., Kawai, K., Tabata, Y. & Suzuki, S. Enhanced wound healing by topical administration of mesenchymal stem cells transfected with stromal cell-derived factor-1. Biomaterials 34, 9393–9400 (2013).
Yan, L. et al. Chondroprotective effects of platelet lysate towards monoiodoacetate-induced arthritis by suppression of TNF-α-induced activation of NF-ĸB pathway in chondrocytes. Aging 11, 2797–2811 (2019).
Glasson, S. S., Chambers, M. G., Van Den Berg, W. B. & Little, C. B. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr. Cartil. 18, S17–S23 (2010).
Tang, Q. Y. & Zhang, C. X. Data processing system (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 20, 254–260 (2013).
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81774331, 82074464, 81973873 and 81873049), Zhejiang Provincial Natural Science Foundation of China (Grant No. LY18H270004), Zhejiang Provincial Science and Technology Project of Traditional Chinese Medicine of China (Grant No. 2016ZZ011), Zhejiang Provincial Key Construction University Superiority Characteristic Discipline (Traditional Chinese Pharmacology) Opening Foundation of China (Grant No. ZYX2018006).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Y. Shi
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, L., Zhou, L., Yan, B. et al. Growth factors-based beneficial effects of platelet lysate on umbilical cord-derived stem cells and their synergistic use in osteoarthritis treatment. Cell Death Dis 11, 857 (2020). https://doi.org/10.1038/s41419-020-03045-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03045-0
This article is cited by
-
Human umbilical cord mesenchymal stem cells promoting knee joint chondrogenesis for the treatment of knee osteoarthritis: a systematic review
Journal of Orthopaedic Surgery and Research (2023)
-
Enhancement of Immunosuppressive Activity of Mesenchymal Stromal Cells by Platelet-Derived Factors is Accompanied by Apoptotic Priming
Stem Cell Reviews and Reports (2023)
-
Platelet Lysate as a Promising Medium for Nanocarriers in the Management and Treatment of Ocular Diseases
Current Ophthalmology Reports (2022)
-
Pain relief and cartilage repair by Nanofat against osteoarthritis: preclinical and clinical evidence
Stem Cell Research & Therapy (2021)

{kind=link}