
Abstract
Neonatal mice and adult zebrafish can fully regenerate their hearts through proliferation of pre-existing cardiomyocytes. Previous studies have revealed that p53 signalling is activated during cardiac regeneration in neonatal mice and that hydrogen peroxide (H2O2) generated near the wound site acts as a novel signal to promote zebrafish heart regeneration. We recently demonstrated that the expression of the p53 isoform Δ133p53 is highly induced upon stimulation by low-level reactive oxygen species (ROS) and that Δ133p53 coordinates with full-length p53 to promote cell survival by enhancing the expression of antioxidant genes. However, the function of p53 signalling in heart regeneration remains uncharacterised. Here, we found that the expression of Δ113p53 is activated in cardiomyocytes at the resection site in the zebrafish heart in a full-length p53- and ROS signalling-dependent manner. Cell lineage tracing showed that Δ113p53-positive cardiomyocytes undergo cell proliferation and contribute to myocardial regeneration. More importantly, heart regeneration is impaired in Δ113p53M/M mutant zebrafish. Depletion of Δ113p53 significantly decreases the proliferation frequency of cardiomyocytes but has little effect on the activation of gata4-positive cells, their migration to the edge of the wound site, or apoptotic activity. Live imaging of intact hearts showed that induction of H2O2 at the resection site is significantly higher in Δ113p53M/M mutants than in wild-type zebrafish, which may be the result of reduced induction of antioxidant genes in Δ113p53M/M mutants. Our findings demonstrate that induction of Δ113p53 in cardiomyocytes at the resection site functions to promote heart regeneration by increasing the expression of antioxidant genes to maintain redox homeostasis.
Similar content being viewed by others


Introduction
The adult mammalian heart has limited regenerative capability following cardiac damage, and this is the main reason that cardiac infarction is one of the leading causes of death worldwide1. In contrast, the hearts of adult zebrafish and neonatal mice exhibit full cardiac regeneration capacity following ventricular resection or cryoinjury through robust cardiomyocyte proliferation2,3,4. In zebrafish, cardiomyocytes from the subepicardial ventricular layer dedifferentiate into gata4-positive cardiomyocytes to proliferate and invade the area of injury, and this is the major process underlying heart regeneration5,6.
A number of signalling pathways, including the Notch, BMP, PDGF, RA, Nrg1 and Brg1 pathways, have been documented to regulate zebrafish cardiac regeneration7,8,9,10,11,12,13,14,15. Reactive oxygen species (ROS), specifically H2O2, produced in the epicardium and adjacent myocardium near the wound site have also been found to promote the proliferation of cardiomyocytes16. ROS, including superoxide anion (O2•–), hydroxyl radical (OH•) and the non-radical species hydrogen peroxide (H2O2), play a dual role in cell fate determination. At moderate levels, ROS can function as signals that promote cell growth and division17,18,19. In contrast, when ROS are overproduced beyond a cell’s capacity to maintain redox homeostasis, they can lead to oxidation of macromolecules such as proteins, membrane lipids and mitochondria and genomic DNA20,21. The harmful accumulation of ROS eventually results in abnormal cell death and senescence.
To maintain redox homeostasis, organisms have evolutionarily developed numerous antioxidant defence systems, including both enzymatic and non-enzymatic antioxidant mechanisms that can either scavenge ROS or prevent their formation22. In response to oxidative stress, the signalling pathway of the tumour repressor p53 plays important and complex roles23,24,25,26. Under physiological conditions and during low levels of oxidative stress, p53 functions to maintain oxidative homeostasis and promote cell survival through transcriptionally expressing antioxidant genes27,28,29,30,31,32. However, p53 triggers apoptotic activity by upregulating the expression of pro-oxidative genes and apoptotic genes in response to high levels of oxidative stress30,33,34,35. Zebrafish Δ113p53 and its human counterpart Δ133p53, N-terminal truncated isoforms of p53, are both transcribed by an alternative p53 promoter in intron 436,37. Full-length p53 can directly transactivate the transcription of these isoforms in response to both developmental and DNA damage stresses38,39,40. In turn, the induction of Δ113p53/Δ133p53 inhibits p53-dependent apoptosis by differentially modulating the expression of p53 target genes36,37,40. Δ113p53/Δ133p53 can form a complex with p53 both in vitro and in vivo, and this interaction is essential for its anti-apoptotic activity41. The basal expression of Δ133p53 prevents normal human fibroblasts, T-lymphocytes and astrocytes from p53-mediated replicative senescence by repressing miR-34a expression42,43. In response to γ-irradiation, Δ113p53/Δ133p53 not only represses cell apoptosis but also coordinates with p73 to promote DNA DSB repair by upregulating the transcription of repair genes44,45. Interestingly, our recent study revealed that upon treatment with sub-toxic ROS stresses, Δ133p53 does not antagonise the activity of p53 but coordinates with p53 to promote cell survival by promoting antioxidant gene expression46.
A study in mice showed that p53 signalling is activated in cardiomyocytes during neonatal mouse heart regeneration47. However, the roles p53 signalling plays and whether its isoforms are activated in heart regeneration are unknown. In this report, we reveal that Δ113p53 is induced in cardiomyocytes at the resection site in the zebrafish heart and that this induction is dependent on full-length p53 and ROS signalling. Furthermore, Δ113p53 promotes heart regeneration through upregulating the expression of antioxidant genes. Our results demonstrate that activation of the p53 signalling pathway is required for heart regeneration by maintaining redox homeostasis.
Results
The expression of Δ113p53 is induced in cardiomyocytes at the resection site in the zebrafish heart
To investigate whether the p53 signalling pathway is also activated during zebrafish heart regeneration as in neonatal mice, we surgically removed ~15% of ventricular cardiomyocytes from tg(Δ113p53:GFP) transgenic zebrafish, in which the expression of GFP faithfully mimics the transcription of endogenous Δ113p5340. Interestingly, we found that the GFP signal was co-localised with MHC (the myosin heavy chain of cardiomyocytes) at the resection site beginning 7 days post-amputation (dpa; Fig. 1c), reached a peak at 21 dpa and decreased at 30 dpa (Fig. 1d–f), whereas the green fluorescent signal was merely observed in the ventricles of both the sham hearts and the resected hearts at 4 dpa (Fig. 1a, b, g).

a–f Cryosections of Tg(Δ113p53:GFP) hearts at sham (a, a′), 4 (b, b′), 7(c, c′), 14 (d, d′), 21 (e, e′) and 30dpa (f, f′) were immunostained by anti-GFP (in green) and anti-MHC (MF20) (in red) antibodies. The nucleus were stained with DAPI (in blue). Scale bar, 50μm. g Average size of GFP+ cardiomyocytes on heart sections of Tg(Δ113p53:GFP) at sham, 4, 7, 14, 21 and 30dpa, was presented as the percentage of the total ventricular area. Each dot presented an individual heart. Data are means of 3 sections/heart from 3 hearts/time point. h, i RNA in situ hybridisation was performed with the DIG-labelled probe to detect both p53 and Δ113p53 on cryosections of WT hearts at sham (h) and 14dpa (i). The representative picture was taken from three hearts in each group. Scale bar, 50μm. j Relative mRNA expression of p53, Δ113p53 and p21 in the WT injury hearts at sham and 7dpa. The total RNA was extracted from a pool of at least 10 hearts in each group. k, l Cryosections of Tg(Δ113p53:GFP) hearts of p53+/+ sibling (k) and p53M214K mutant (l) at 14dpa were immunostained by anti-GFP antibody. The representative picture was taken from three hearts in each group. The white arrow heads indicate wounding site. Scale bar, 50μm. The experiments were repeated independently for at least three times with similar results. Statistical analysis was performed on relevant data using Student’s two-tailed t test in GraphPad Prism 5. The p values were represented by n.s. and asterisks. n.s., p>0.05. *p<0.05. **p<0.01. ***p<0.001.
To confirm the activation of the p53 signalling pathway, we performed an in situ hybridisation assay with a probe that detects both full-length p53 and Δ113p53. Positive signals were observed in cells near the resection site in wild-type (WT) hearts at 14 dpa (Fig. 1i) but not in the ventricles in sham hearts (Fig. 1h). Quantitative reverse transcription PCR (qRT-PCR) showed that the expression of Δ113p53 and p21 (also a p53 target gene), but not full-length p53, was significantly increased in the resected hearts compared to the sham hearts at 7 dpa (Fig. 1j).
As Δ113p53 is a p53 target gene, we asked whether the induction of the transgene was p53-dependent. For this purpose, the tg(Δ113p53:GFP) transgene was crossed into the p53M214K mutant background, in which the transcriptional activity of mutant p53 is lost48. Unlike in the resected hearts of WT fish, GFP was not detectable in the resected hearts of p53M214K mutant fish at 14 dpa (Figs. S1 and 1k, l). Taken together, the results suggest that full-length p53 was post-transcriptionally activated to upregulate the expression of its downstream genes, including Δ113p53, during heart regeneration.
Δ113p53-positive cardiomyocytes undergo cell proliferation and contribute to heart regeneration
To explore whether the induction of Δ113p53 is related to the proliferation of cardiomyocytes, we subjected tg(Δ113p53:GFP) zebrafish to EdU (5-ethynyl-2′-deoxyuridine)-labelling from 5 to 7 dpa. At 7 dpa, ~4.3% of cardiomyocytes (MF20-positive cells) in the wound area were labelled with Edu (Fig. 2b–d), whereas up to 10.2% of Δ113p53-positive cardiomyocytes were labelled with Edu (Fig. 2e). The Edu-labelled Δ113p53-positive cardiomyocytes accounted for 24.4% of total Edu-labelled cardiomyocytes (Fig. S2). The Edu-labelled cardiomyocytes or Edu-labelled Δ113p53-positive cardiomyocytes were rarely observed in the sham hearts (Fig. 2a). The results demonstrate that many Δ113p53:GFP+ cells near the lateral edges of the wound have newly undergone DNA synthesis.

a–e Cryosections of Edu-labelled Tg(Δ113p53:GFP) hearts at sham (a) and 7 dpa (b, c) were immunostained by anti-GFP (in green) and anti-MF20 (in red) antibodies. The nucleus were stained with DAPI (in blue). Framed area in b was magnified in c. The representative picture was taken from 3 to 7 hearts. Scale bar, 50 μm. Yellow arrows: Edu+/GFP−/MF20− cells; white arrows: Edu+/GFP-/MF20+ cells; white arrow head: Edu+/GFP+/MF20+ cells. The number of Edu+/MF20+ cells on heart sections of Tg(Δ113p53:GFP) at sham and 7 dpa, was presented as the percentage of the total MF20+ cells at the wound site (d). The number of Edu+/GFP+/MF20+ cells on heart sections of Tg(Δ113p53:GFP) at sham and 7 dpa, was presented as the percentage of the total GFP+/MF20+ cells at the wound site (e). Data are means of 4–6 sections/heart with the largest wound area from 3 to 7 hearts in different treatments. Scale bar, 50 μm. Each dot represents an individual heart. f A schematic diagram representing the 4HT-based Cre-LoxP system driven by Δ113p53 promoter. Δ113p53-P (blue arrow): the 3.6-kb DNA fragment from the upstream of Δ113p53 transcription start site; β-act2-P (blue arrow): the promoter of β-actin2; CreER (Brown bar): the coding region of tamoxifen-inducible Cre recombinase–oestrogen receptor fusion protein; LoxP (blue bar): the site of LoxP; DsRed (red bar): the coding region of DsRed; Stop (black bar): the translation stop codon; EGFP (light green bar): the coding region of EGFP gene; 4HT: the treatment of 4-hydroxytamoxifen. g Schematics of the cell lineage tracing experiment. Either sham or surgical Tg(Δ113p53:CreER; β-act2:RSG) zebrafish were treated with 4HT at 7 and 10 dpa as indicated. The treated surgical zebrafish were sampled at 14 and 30 dpa, while all of the treated sham zebrafish were sampled at 30 dpa. h–k Red and green fluorescence on the cryosections of Tg(Δ113p53:CreErt2;β-actin:RSG) hearts at sham (i), 14 (j) and 30 dpa (k) were from the en vivo DsRed and EGFP protein respectively. The nuclei were stained with DAPI (blue). Scale bar, 50 μm. The number of EGFP+ cells on heart sections of Tg(Δ113p53:CreER; β-act2:RSG) at 14 and 30 dpa, was presented as the percentage of the total DsRed+ cells at the resection site (h). Data are from the biggest section with most EGFP+ cells of every heart. Each dot represents an individual heart. l–n Fibrin clot stained with Masson’s trichrome on the crysections of Δ113p53+/+ (l) and Δ113p53M/M mutant hearts (m) at 30 dpa. Yellow dotted lines indicate the approximate injury area. Scale bar, 50 μm. Average injury area with fibrin clots on sections of Δ113p53+/+ and Δ113p53M/M mutant hearts at 30 dpa was presented as the percentage of the total ventricular area (n). Data are means of three sections/heart. Each dot represents the average injury area of an individual heart. The experiments were repeated independently for at least three times with similar results. Statistical analysis was performed on relevant data using Student’s two-tailed t test in GraphPad Prism 5. The p values were represented by n.s. and asterisks. n.s., p > 0.05. *p < 0.05. **p < 0.01. ***p < 0.001.
To investigate the dynamics of Δ113p53-positive cardiomyocytes in heart regeneration, a cell lineage tracing assay was performed. We generated tg(Δ113p53:CreER) transgenic zebrafish using a 3.6-kb fragment of the Δ113p53 promoter to drive CreER (tamoxifen-inducible Cre recombinase–oestrogen receptor fusion protein) expression and crossed them with tg(β-act2:RSG) zebrafish to generate tg(Δ113p53:CreER; β-act2:RSG) double transgenic fish (Fig. 2f). Our previous study revealed that the expression of Δ113p53 is strongly induced upon treatment with DNA-damaging drugs40. To verify the utility of the double transgenic fish, the transgenic embryos were treated with either camptothecin (Campt, a DNA-damaging drug), 4-hydroxytamoxifen (4HT) or a combination of both. Western blot analysis showed that the expression of endogenous Δ113p53 was induced by Campt but not by 4HT (Fig. S3a). Green fluorescence appeared in the transgenic embryos treated with the combination of Campt and 4HT (Fig. S3e) but not in the untreated embryos or the embryos treated with either drug alone (Fig. S3b–d). The results demonstrated that the double transgenic fish could be used to trace the induction of Δ113p53.
Next, we treated the sham zebrafish and adult double transgenic zebrafish subjected to surgery with 4HT at 7 and 10 dpa (Fig. 2g), the time points preceding detectable of Δ113p53-driven GFP fluorescence in the injury site. At 14 dpa, a small number of EGFP+ cardiomyocytes (2.6%) were detected near the border of the wound in the 4HT-treated tg(Δ113p53:CreER;β-act2:RSG) animals (Fig. 2j, h) but not in the sham controls (Fig. 2i). Moreover, the number of EGFP+ cardiomyocytes significantly increased to 13.5% at 30 dpa (Fig. 2k, h). These results indicate that Δ113p53-positive cardiomyocytes undergo cell proliferation and contribute to heart regeneration.
Heart regeneration is impaired in Δ113p53M/M mutant zebrafish
During zebrafish heart regeneration, a large clot of blood cells (most of them being erythrocytes) forms in the resection site after a few seconds of profuse bleeding from the ventricular lumen; these blood cells are replaced by fibrin beginning 2 dpa. Cardiomyocytes surround, penetrate and finally replace the fibrin clot from 9 to 30 dpa2. The area of the injury containing the fibrin clot is a critical parameter for evaluating the quality of heart regeneration49. To investigate the role of Δ113p53 in heart regeneration, we performed Masson’s staining to compare the area of the injury containing the fibrin clot between the resected hearts of WT zebrafish and those of Δ113p53M/M mutant zebrafish. The Δ113p53M/M mutant generated in our previous study exhibits relatively normal development and carries an 11-bp deletion in a p53 responsive element in the Δ113p53 promoter located in the 4th intron of p53, which abolishes the expression of Δ113p53 but does not influence the expression of full-length p5344. The results showed that there were no visible differences between uninjured Δ113p53M/M mutant and WT hearts (Fig. S4), which suggests that the expression of Δ113p53 in heart development is weak and that Δ113p53 plays a small role in heart development. However, the percentage of the injury area containing the fibrin clot was significantly larger in Δ113p53M/M mutant hearts (4.94%) (Fig. 2m, n) than in WT hearts (2.67%) (Fig. 2l, n) at 30 dpa. These results demonstrate that Δ113p53 is induced to promote heart regeneration.
Δ113p53 has little effect on the activation of gata4-positive cardiomyocytes and their migration to the edge of the wound site
The gata4-positive cardiomyocytes dedifferentiated from cardiomyocytes in the subepicardial ventricular layer migrate to the injury site and proliferate to contribute to zebrafish heart regeneration5,6. To investigate whether Δ113p53-positive cells were dedifferentiated cardiomyocytes, we generated tg(Δ113p53:mCherry) transgenic zebrafish by using a 3.6-kb fragment of the Δ113p53 promoter to drive mCherry expression (Fig. S5) and crossed them with Tg(gata4:EGFP) zebrafish to obtain tg(Δ113p53:mCherry; gata4:EGFP) double transgenic fish. Immunostaining assays showed that mCherry was co-expressed with EGFP in some EGFP+ cardiomyocytes near the wound site at 14 dpa (Fig. 3a). These results demonstrate that Δ113p53+ cells are dedifferentiated cardiomyocytes.

a Co-localisation of red (Δ113p53+ cells) (a′) and green fluorescence (gata4+ cells) (a′′) on the cryosections of Tg(Δ113p53:mCherry; gata4:EGFP) hearts at 14 dpa. The white arrow heads indicate co-labelling. The nucleus were stained with DAPI (in blue). The representative picture was taken from three hearts. Scale bar, 50 μm. b–e Cryosections of Tg(gata4:EGFP); Δ113p53+/+ (b, b′, d, d′) and Tg(gata4:EGFP); Δ113p53M/M hearts (c, c′, e, e′) at 7 and 14 dpa, were immunostained by anti-GFP (in green) and anti-MF20 (in red) antibodies. The nucleus were stained with DAPI (in blue). Scale bar, 50 μm. f Average size of EGFP+ cardiomyocytes on the edge of wound site in Tg(gata4:EGFP); Δ113p53+/+ and Tg(gata4:EGFP); Δ113p53M/M mutant zebrafish at 7 and 14 dpa, was presented as the percentage of the ventricular area at the resection site. Data are means of three sections/heart from six hearts/time point. Scale bar, 50 μm. Each dot represents an individual heart. The experiments were repeated independently for at least three times with similar results. Statistical analysis was performed on relevant data using Student’s two-tailed t test in GraphPad Prism 5. The p values were represented by n.s. and asterisks. n.s., p > 0.05. *p < 0.05. **p < 0.01. ***p < 0.001.
Next, Tg(gata4:EGFP) transgenic reporter zebrafish were used to track newly regenerated cardiomyocytes in injured Δ113p53M/M mutant hearts. We found that there were no visible differences in the location or percentage of gata4-positive cardiomyocytes in WT and Δ113p53M/M mutant hearts at 7 dpa (Fig. 3b, c, f). Similar to those in the WT hearts, gata4-positive cardiomyocytes in Δ113p53M/M mutant hearts migrated to the edge of the wound site at 14 dpa (Fig. 3d, e), although the percentage of gata4-positive cardiomyocytes at the edge of the wound site was slightly lower in Δ113p53M/M mutant hearts than in WT hearts at 14 dpa (Fig. 3d–f). However, unlike in WT hearts, gata4-positive cardiomyocytes were rarely observed in the intermediate zone of the wound area in Δ113p53M/M mutant hearts (Fig. 3d, e); it is unclear whether this phenomenon resulted from cardiomyocyte proliferation or from the penetration of gata4-positive cells. These results suggest that Δ113p53 does not play a critical role in cardiomyocyte dedifferentiation or the migration of gata4-positive cardiomyocytes from the outer compact layer of the ventricle to the edge of the wound site.
Δ113p53 promotes heart regeneration by enhancing cardiomyocyte proliferation, but not by inhibiting cardiomyocyte apoptosis
A recent study showed that cryoinjury triggers the DNA damage response during zebrafish heart regeneration49. Our previous studies revealed that Δ133p53 is induced during cell reprogramming to promote reprogramming efficiency through its anti-apoptotic activity and ensure the genomic integrity of induced pluripotent stem cells by increasing DNA DSB repair50. To compare apoptotic activity and the DNA damage response between the ventricles of WT and Δ113p53M/M mutant hearts during regeneration, the tg(myl7:nDsRed) transgenic line (in which the promoter of zebrafish myosin light chain 7 drives the expression of nuclear DsRed) was crossed onto the Δ113p53M/M mutant background. The TUNEL assay and immunostaining for γ-H2AX (an early marker of the DNA damage response) were performed to analyse apoptotic cells and the DNA damage response, respectively. We found that there were only a few apoptotic cardiomyocytes and γ-H2AX-positive cardiomyocytes (co-stained with nDsRed) in the wound site in both WT and Δ113p53M/M mutant hearts at 14 dpa (Fig. S6). These results suggest that 15% resection of the ventricle does not trigger a strong DNA damage response in cardiomyocytes during heart regeneration.
To compare myocardial proliferation in the ventricles of WT and Δ113p53M/M mutant hearts during regeneration, we quantified injury-induced cardiomyocyte proliferation by counting EdU+/Myl7+ or PCNA+ (the DNA replication marker proliferating cell nuclear antigen)/Myl7+ double-positive cardiomyocytes during heart regeneration. Compared to WT hearts, Δ113p53M/M mutant hearts harboured significantly fewer proliferating cardiomyocytes labelled with EdU+/Myl7+ at 14 dpa (54% of the number in WT hearts) (Fig. 4a, b, e) and with PCNA+/Myl7+ at 7 dpa (85% of the number in WT hearts) (Fig. 4c, d, f). These data reveal that Δ113p53 is required for cardiomyocyte proliferation following injury.

a, b The DsRed+ nucleus of cardiomyocytes (in red) at the resection site in Tg(myl7:nDsRed); Δ113p53+/+(a) and Tg(myl7:nDsRed); Δ113p53M/M (b) hearts were labelled by EdU (in green) at 14 dpa. Framed areas were magnified in a′ and b′. nDsRed: nuclear DsRed. The white arrow heads indicate co-labelling. Scale bar, 50 μm. c, d Cryosections of Tg(myl7:nDsRed); Δ113p53+/+ (c) and Tg(myl7:nDsRed); Δ113p53M/M (d) hearts at 7 dpa were co-stained by anti-DsRed and anti-PCNA antibodies. The nucleus were stained with DAPI (in blue). Framed areas were magnified in c′ and c″, or d′ and d″. The white arrow heads indicate co-labelling. Scale bar, 50 μm. e, f The number of co-labelled nDsRed+ nucleus of cardiomyocytes with either EdU (e) or PCNA (f) in Tg(myl7:nDsRed); Δ113p53+/+ and Tg(myl7:nDsRed); Δ113p53M/M hearts at 14 or 7 dpa, was presented as the percentage of the total nDsRed+ nucleus at the resection site. Data are means of three sections/heart. Each dot represents the average number of co-labelled nDsRed+ nucleus of cardiomyocytes with either EdU or PCNA in an individual heart. The experiments were repeated independently for at least three times with similar results. Statistical analysis was performed on relevant data using Student’s two-tailed t test in GraphPad Prism 5. The p values were represented by n.s. and asterisks. n.s., p > 0.05. *p < 0.05. **p < 0.01. ***p < 0.001.
Δ113p53 upregulates the expression of antioxidant genes to maintain redox homeostasis during heart regeneration
A recent study revealed that H2O2 is produced near the wound site of ventricles to promote heart regeneration16. Our previous study demonstrated that the human orthologue Δ133p53 is induced in response to sub-toxic levels of ROS to promote cell proliferation by upregulating the expression of antioxidant genes46. Therefore, we investigated whether the induction of Δ113p53 is related to maintaining redox homeostasis during heart regeneration. For this purpose, we treated tg(Δ113p53:GFP) zebrafish with diphenylene iodonium (DPI), an NADPH oxidase (Duox/Nox enzymes) inhibitor, to block the production of H2O216 after amputation. The results showed that compared to control treatment, DPI treatment significantly reduced the percentage of Δ113p53+ cardiomyocytes near the wound site at 7 and 14 dpa (Fig. 5a–e), suggesting that the induction of Δ113p53 depends on elevation of ROS levels during heart regeneration.

(a–d) After surgery, Tg(Δ113p53:GFP) animals were treated with either DMSO (a, a′, c, c′) or DPI (b, b′, d, d′) daily at 3–7 or 7–14 dpa. The treated animals were sampled at 7 and 14 dpa and subjected to cryosection. Cryosections of hearts were immunostained by anti-GFP (in green) and anti-MHC (MF20) (in red) antibodies. The nuclei were stained with DAPI (in blue). Scale bar, 50 μm. e Average size of GFP+ cardiomyocytes on heart sections of Tg(Δ113p53:GFP) treated with DMSO or DPI at 7 and 14 dpa, was presented as the percentage of the ventricular area at the resection site. Data are means of three sections/heart. Each dot represents the average size of GFP+ cardiomyocytes in an individual heart. Scale bar, 50 μm. f–l Ex vivo HyPer heart images of either Δ113p53+/+ (f, h, j) or Δ113p53M/M mutant hearts (g, i, k) at sham, 10.5 and 21 dpa. Spatially resolved H2O2 image, indexed by the ratio between the F488 and F405 images of HyPer (below), is presented in pseudocolor. Ratiometric HyPer signals (F488/F405) averaged over the regenerative zone of injured heart at 3.5, 7, 10.5, 14, 17.5 and 21 dpa were presented as the difference to the average F488/F405 ratio at the apex of respective sham hearts (l). Each dot represents the ratiometric HyPer signal in an individual heart. Statistical analyses were performed on data from Δ113p53+/+ and Δ113p53M/M mutant hearts at the same time point. The experiments were repeated independently for at least three times with similar results. Statistical analysis was performed on relevant data using Student’s two-tailed t test in GraphPad Prism 5. The p values were represented by n.s. and asterisks. n.s., p > 0.05. *p < 0.05. **p < 0.01. ***p < 0.001.
Next, we determined the status of ROS in the injured hearts of both WT and Δ113p53M/M mutant zebrafish at different time points with the tg(myl7:HyPer) transgene (in which the promoter of myl7 drives the expression of HyPer, a fluorescent protein-based H2O2 sensor)16. Similar to a previous study16, the level of H2O2 in the injured WT hearts started to increase at 3 dpa, decreased beginning at 14 dpa and reached the basal level at 21 dpa (Fig. 5f, h, j, l), whereas the H2O2 levels in the injured Δ113p53M/M mutant hearts were significantly higher than those in the injured WT hearts at 10.5 and 21 dpa (Fig. 5g, i, k, l). These results suggest that depletion of Δ113p53 results in elevated levels of intracellular H2O2 during heart regeneration.
To investigate whether elevated ROS levels in the injured Δ113p53M/M mutant hearts were related to antioxidant genes, we examined the expression of six antioxidant genes (p53 target genes), including gpx1a, sesn2, aldh4, sesn1, sod1 and sod2, by using qRT-PCR. The expression of gpx1a and sesn2, as well as the expression of Δ113p53 was significantly upregulated in the injured WT hearts (Fig. 6a–c) compared to the sham hearts at 14 dpa, whereas the expression of the remaining 4 genes was not significantly changed (Fig. S7). Interestingly, the expression of all six antioxidant genes in sham Δ113p53M/M mutant hearts was lower than that in sham WT hearts (Figs. 6b, c andS7). Furthermore, the induction of gpx1a was not triggered in injured Δ113p53M/M mutant hearts at 14 dpa (Fig. 6b), while the induction of sesn2 was significantly lower in injured Δ113p53M/M mutant hearts than in injured WT hearts at 14 dpa; however, the expression of sesn2 was increased in injured Δ113p53M/M mutant hearts compared to sham Δ113p53M/M mutant hearts (Fig. 6c). These results demonstrate that the antioxidant response is triggered in wounded hearts and that Δ113p53 promotes the expression of antioxidant genes. This result also implies that the elevation of ROS levels in the injured Δ113p53M/M mutant hearts is due to lower expression of antioxidant genes.

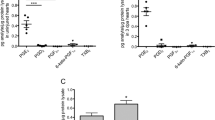
a–f Relative mRNA expression of Δ113p53 (a), gpx1a (b), sesn2 (c), hif1al2 (d), jak2a (e) and pim2 (f) in the Δ113p53+/+ and Δ113p53M/M hearts at sham and 14 dpa. The total RNA was extracted from a pool of at least 10 hearts in each group. g Western blot was performed to analyse the induction of zebrafish Hif1α in different samples as indicated. The Δ113p53+/+ and Δ113p53M/M mutant zebrafish with heart resection were treated with DPI from 3 to 7 dpa. Total protein was isolated from four hearts/treatment at 7 dpa and subjected to western blot analysis. Gapdh was used as the protein loading control. The experiments were repeated independently for at least three times with similar results. Statistical analysis was performed on relevant data using Student’s two-tailed t test in GraphPad Prism 5. The p values were represented by n.s. and asterisks. n.s., p > 0.05. *p < 0.05. **p < 0.01. ***p < 0.001.
Finally, we tried to gain insight into the role of elevated ROS levels in cardiomyocyte proliferation. ROS stress elicits the ATM-homodimer-Chk2 pathway to trigger the DNA damage response51. However, our results showed that apoptotic activity and the DNA damage response was rarely induced by 15% ventricular resection in both WT and Δ113p53M/M mutant hearts (Fig. S6). A previous study revealed that cardiac injury induces the hypoxia response in zebrafish ventricles, resulting in activation of Hif1α signalling, which promotes cardiomyocyte proliferation by upregulating the expression of numerous proproliferative genes, including many components of the Jak-STAT pathway52. A number of studies have also documented that an increase in ROS levels can downregulate Hif1α signalling53,54,55. Therefore, we evaluated the expression of three genes in the Hif1α signalling pathway, including hif1al2 (hypoxia inducible factor 1 subunit alpha, like 2), jak2a and pim2 (two HIF1α downstream genes), by qRT-PCR. Consistent with a previous study52, the expression of these three genes was upregulated in injured WT hearts (Fig. 6d–f) compared to sham hearts at 14 dpa. The expression of all three genes in sham Δ113p53M/M mutant hearts was lower than that in sham WT hearts (Fig. 6d–f). Although the expression of hif1al2 was also upregulated in injured Δ113p53M/M mutant hearts at 14 dpa (Fig. 6d), the induction of two downstream genes, jak2a and pim2, was abolished in injured Δ113p53M/M mutant hearts at 14 dpa.(Fig. 6e, f). These results suggest that the elevation of ROS levels may repress cardiomyocyte proliferation through inactivating the Hif1α signalling pathway.
To verify the activation of hif1α in heart regeneration and address if the hif1α activation is dependent on the ROS signal, we treated WT and Δ113p53M/M mutant zebrafish with DPI to block the production of H2O2 after amputation and analysed the level of Hif1α protein at 7 dpa. The western blot analysis confirmed that the expression of Hif1α was induced in both WT and Δ113p53M/M mutant resected hearts, compared to that in respective sham hearts (Fig. 6g). Interestingly, DPI treatment observably reduced the activation of Hif1α protein in both WT and Δ113p53M/M mutant resected hearts at 7 dpa (Fig. 6g), suggesting that the induction of Hif1α depends on elevation of ROS levels during heart regeneration.
Discussion
It is well documented that ROS are produced after tissue injury and play an important role in wound healing by initiating acute inflammation, clarifying infection and dead tissue, and mediating various intracellular signal transduction pathways56,57,58. However, when the level of ROS is beyond a cell’s capacity to maintain redox homeostasis, oxidative stress occurs, which results in direct or indirect ROS-mediated damage to nucleic acids, proteins and lipids20,21. Therefore, ROS levels in cells are tightly controlled by antioxidant systems22. P53 and its isoform Δ133p53/Δ113p53 play a critical role in the maintenance of redox homeostasis by regulating the expression of antioxidant genes46. Interestingly, ROS are also generated during zebrafish heart regeneration to promote cardiomyocyte proliferation16, and the p53 signalling pathway is activated during cardiac regeneration in neonatal mice47. However, how redox homeostasis is maintained and whether p53 signalling plays a role in heart regeneration remain unclear.
In this report, we applied partial zebrafish ventricular resection to investigate the function of Δ113p53 in heart regeneration. Based on a p53-based genetic tracing system involving the insertion of a CreER cassette immediately after the first ATG of the full-length mouse p53 BAC clone (located in the second exon of p53), a previous study revealed that full-length p53-positive cardiomyocytes are activated by injury in neonatal mice and undergo proliferation to contribute to heart regeneration47. In contrast, using Δ113p53 transgenic reporter fish, in situ hybridisation and qRT-PCR, we found that the transcription of Δ113p53, but not full-length p53, was induced in cardiomyocytes near the injury site in zebrafish ventricles (Fig. 1a–j). The induction of Δ113p53 was not observed in injured p53M214K mutant hearts (Fig. 1k, l), which is consistent with Δ113p53 being a p53 target gene40. The discrepancy between the two studies in mouse and zebrafish may be explained by the fact that the mouse p53 reporter system contains the first exon of p5347, which might be the promoter for the mouse Δ113p53/Δ133p53 orthologue. Next, we explored the function of Δ113p53 in heart regeneration in Δ113p53M/M mutants. Masson’s staining showed that the area of the injury containing the fibrin clot was significantly increased in the wound site in Δ113p53M/M mutant hearts (Fig. 2l–n) compared to WT hearts at 30 dpa, which demonstrates that heart regeneration is impaired in the Δ113p53M/M mutants. Although there were no observable differences in dedifferentiation to gata4-positive cardiomyocytes (Fig. 3b–f) or cardiomyocyte apoptosis between injured WT and Δ113p53M/M mutant hearts (Fig. S6c, d), the percentages of EDU-labelled cardiomyocytes and PCNA-labelled cardiomyocytes were significantly lower in injured Δ113p53M/M mutant hearts than in injured WT hearts (Fig. 4). These results reveal that Δ113p53 promotes heart regeneration by increasing cardiomyocyte proliferation. Further analysis showed that H2O2 levels in the injured Δ113p53M/M mutant hearts were significantly higher than those in the injured WT hearts (Fig. 5f–l) and that the increase in H2O2 levels was coincident with a decrease in antioxidant gene expression in the injured Δ113p53M/M mutant hearts (Fig. 6b, c). These results suggest that Δ113p53 promotes cardiomyocyte proliferation by maintaining redox homeostasis.
Taken together, our findings demonstrate that although ROS signalling plays an important role in promoting heart regeneration16, the level of ROS should be tightly controlled. The induction of Δ113p53 functions to maintain redox homeostasis by promoting antioxidant gene expression.
Oxidative stress has been implicated in human cardiac diseases, including ischaemia-reperfusion (IR), myocardial infarction (MI) and heart failure57,59. ROS are produced in two stages, namely, ischaemia and reperfusion, at low and high levels, respectively60. ROS play a dual role in tissue injuries, as massive amounts of mitochondrial ROS induce apoptosis and necrosis of cells61, whereas moderate levels of ROS promote cell survival and proliferation16,62,63. Therefore, maintaining redox homeostasis plays an important role in the mechanisms of and therapeutic strategies for cardiac diseases. It has also been reported that during pressure overload, the activation of full-length p53 has a crucial function in the transition from cardiac hypertrophy to heart failure by repressing Hif1 activity64. Here, we demonstrate that Δ113p53 is induced by ROS during zebrafish heart regeneration and functions to promote cardiomyocyte proliferation by maintaining redox homeostasis and Hif1α activity. Our results suggest that the expression of Δ133p53 may also be activated during IR and protect patients from IR-induced heart failure.
Methods and materials
Zebrafish lines
Zebrafish were raised and maintained in standard zebrafish units at Zhejiang University as described previously44. The Tg(Δ113p53:GFP) transgenic line and Δ113p53M/M mutant zebrafish were generated in our previous studies40,44. A 3.6-kb fragment of the Δ113p53 promoter40 was used to create the Tg(Δ113p53:CreER) and tg(Δ113p53:mCherry) transgenic lines on the AB genetic background through Tol2-based transgenesis40. p53M214K mutant48, Tg(β-act2:RSG)5, Tg(gata4:EGFP)65, Tg(myl7:nDsRed)66 and Tg(myl7:HyPer)16 zebrafish were generated by different labs as previously reported.
Ethics statement
All animal procedures were performed in full accordance with the requirements of the Regulation for the Use of Experimental Animals of Zhejiang Province. This work was specifically approved by the Animal Ethics Committee of the School of Medicine, Zhejiang University (ethics code permit no. ZJU20190012).
Adult zebrafish heart resection
Ventricular surgery was performed on 5- to 10-month-old zebrafish according to previously described procedures2. Briefly, zebrafish were anaesthetised with 0.02% Tricaine and then subjected to ~15% ventricular amputation at the apex with scissors.
Quantitative real-time reverse transcriptional PCR
Hearts were freshly isolated from anaesthetised zebrafish subjected to sham surgery or resection at different time points. The outflow tracts and atriums were removed from the isolated hearts. Total RNA was extracted from ~10 isolated ventricles from each group using a homogeniser (JXFSTPRP-24, Shanghai Jingxin) in Invitrogen TRIzol reagent (Cat No. 15596026). Isolated RNA was treated with DNaseI (NEB, M0303S) prior to reverse transcription and purified through lithium chloride. First-strand cDNA was synthesised using M-MLV Reverse Transcriptase (Invitrogen, C28025021). The reaction was performed using a CFX96TM Real-Time System (Bio-Rad) with AceQ qPCR SYBR Green (Vazyme, Q111-02) according to the manufacturer’s instructions. Total RNA levels were normalised to the level of β-actin. Statistics were obtained from three repeats. The primer sequences of the analysed genes are listed in Table S1.
In situ hybridisation
For the in situ hybridisation assay, isolated zebrafish hearts were fixed in 4% PFA for 2 days before cryosectioned. The probes were generated by NEB T7 RNA Polymerase (M0251S) and Roche DIG RNA Labelling Mix (11277073910) from a Δ113p53-pCS2+ plasmid constructed in our previous study44. Staining was performed with Anti-Digoxigenin-AP (Roche, 11093274910) and the BCIP/NBT Alkaline Phosphatase Colour Development Kit (Beyotime Biotechnology, C3206).
Ex vivo intact heart imaging
Ex vivo Tg(myl7:HyPer) heart imaging and image processing were performed according to previously described procedures16. Briefly, images were taken under an Olympus FV1000 upright confocal microscope, and the HyPer 488/405 ratio was calculated based on the integrated optic density using Adobe Photoshop CS5.
EdU incorporation assay and small-molecule treatment
For the EdU incorporation assay, 15 μL of 100 mM EdU (Invitrogen, A10044) was injected once daily into the abdominal cavity of each animal that underwent surgery for 3 or 7 days until the hearts were collected at 7 or 14 dpa. The hearts were then fixed for cryosectioning. EdU staining was performed using Azide Alexa Fluor 647 (Invitrogen, A10277).
For DPI treatment, 50 μL of 10 μM DPI (Sigma, D2926) was injected daily into the thoracic cavity of each animal that underwent surgery beginning 3 or 7 dpa until the hearts were collected at 7 or 14 dpa16.
For the cell lineage tracing assay, tg(Δ113p53:CreER; β-act2:RSG) fish subjected to sham surgery or surgery were bathed in 3 μM 4HT (Sigma, H7904) for 24 h at 7 and 10 dpa as previously described15.
Western blot, immunostaining and histological methods
For the western blot assay, a zebrafish p53 monoclonal antibody was generated by HuaAn Biotechnology (Hangzhou, China) as previously described67. A human HIF1α antibody (BOSTER, A00013-1) was used to detect zebrafish Hif1a. A β-actin antibody (Huabio, R1207-1) was used as the protein loading control for the experiments in embryonic stages. A Gapdh antibody (HuaBio, R1208-3) was used as the protein loading control for the experiments in zebrafish heart regeneration. The secondary antibodies were HRP-conjugated goat anti-mouse IgG (Huabio, HA1006) and HRP-conjugated goat anti-rabbit IgG (Huabio, HA1001).
Zebrafish hearts were fixed, cryosectioned (14 μm) as described previously2 and then subjected to immunostaining. The primary antibodies were anti-GFP (Abcam, ab13970), anti-MYH1E (MF20; Developmental Studies Hybridoma Bank, AB 2147781), anti-PCNA (Sigma, P8825), anti-DsRed (Clontech, 632496) and anti-H2A.XS139ph (Genetex, GTX127340). The secondary antibodies were Alexa Fluor 488-conjugated anti-chicken IgY H&L (Abcam, ab150169), Alexa Fluor 647-conjugated anti-mouse IgG H&L (Abcam, ab150115), Dylight 549-conjugated anti-rabbit IgG H&L (EarthOx, E032320) and Alexa Fluor 647-conjugated anti-rabbit IgG H&L (Abcam, ab150143). Nuclei were stained with DAPI (BYT, C1002).
Masson’s staining was performed on cryosections using trichrome Masson’s staining solution from Servicebio (G1006). Myosin was visualised as red, and fibrin was visualised as blue. Images were captured under an Olympus BX53 microscope with a camera from Qimaging MicroPublisher 5.0 RTV.
TUNEL assay
The TUNEL assay was performed on freshly prepared cryosections of tg(myl7:nDsRed) zebrafish hearts using a fluorescein-based Roche In Situ Cell Death Detection Kit (Cat No. 11684795910).
Quantification and statistical analysis
Sample sizes were designed based on routine genetic analysis in zebrafish studies. The investigators were blinded to group allocation during data collection and analysis. No data were excluded from the analyses. All samples were randomly selected.
References
Lallukka, T., Millear, A., Pain, A., Cortinovis, M. & Giussani, G. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specifi c mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015 (vol 388, pg 1459, 2016). Lancet389, E1–E1 (2017).
Poss, K. D., Wilson, L. G. & Keating, M. T. Heart regeneration in zebrafish. Science298, 2188–2190 (2002).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science331, 1078–1080 (2011).
Gonzalez-Rosa, J. M., Martin, V., Peralta, M., Torres, M. & Mercader, N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development138, 1663–1674 (2011).
Kikuchi, K. et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature464, 601–605 (2010).
Gupta, V. et al. An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr. Biol.23, 1221–1227 (2013).
Raya, A. et al. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc. Natl Acad. Sci. USA100, 11889–11895 (2003).
Zhao, L. et al. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl Acad. Sci. USA111, 1403–1408 (2014).
Wu, C. C. et al. Spatially resolved genome-wide transcriptional profiling identifies BMP signaling as essential regulator of zebrafish cardiomyocyte regeneration. Dev. Cell36, 36–49 (2016).
Lien, C. L., Schebesta, M., Makino, S., Weber, G. J. & Keating, M. T. Gene expression analysis of zebrafish heart regeneration. PLoS Biol.4, e260 (2006).
Kim, J. et al. PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc. Natl Acad. Sci. USA107, 17206–17210 (2010).
Lepilina, A. et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell127, 607–619 (2006).
Kikuchi, K. et al. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell20, 397–404 (2011).
Gemberling, M., Karra, R., Dickson, A. L. & Poss, K. D. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife4, e05871 (2015).
Xiao, C. et al. Chromatin-remodelling factor Brg1 regulates myocardial proliferation and regeneration in zebrafish. Nat. Commun.7, 13787 (2016).
Han, P. et al. Hydrogen peroxide primes heart regeneration with a derepression mechanism. Cell Res.24, 1091–1107 (2014).
Terada, L. S. Specificity in reactive oxidant signaling: think globally, act locally. J. Cell Biol.174, 615–623 (2006).
Takahashi, A. et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol.8, 1291–1297 (2006).
Weinberg, F. & Chandel, N. S. Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci.66, 3663–3673 (2009).
Valko, M. et al. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol.39, 44–84 (2007).
Cooke, M. S., Evans, M. D., Dizdaroglu, M. & Lunec, J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J.17, 1195–1214 (2003).
Ladelfa, M. F., Toledo, M. F., Laiseca, J. E. & Monte, M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid. Redox Signal.15, 1749–1761 (2011).
Hafsi, H. & Hainaut, P. Redox control and interplay between p53 isoforms: roles in the regulation of basal p53 levels, cell fate, and senescence. Antioxid. Redox Signal.15, 1655–1667 (2011).
Vigneron, A. & Vousden, K. H. p53, ROS and senescence in the control of aging. Aging2, 471–474 (2010).
Holley, A. K., Dhar, S. K. & St Clair, D. K. Manganese superoxide dismutase vs p53 Regulation of mitochondrial ROS. Mitochondrion10, 649–661 (2010).
Liu, B., Chen, Y. M. & Clair, D. K. S. ROS and p53: a versatile partnership. Free Radic. Biol. Med.44, 1529–1535 (2008).
Tomko, R. J., Bansal, P. & Lazo, J. S. Airing out an antioxidant role for the tumor suppressor p53. Mol. Inter.6, 23–25 (2006).
Stambolsky, P. et al. Regulation of AIF expression by p53. Cell Death Differ.13, 2140–2149 (2006).
Matoba, S. et al. p53 regulates mitochondrial respiration. Science312, 1650–1653 (2006).
Sablina, A. A. et al. The antioxidant function of the p53 tumor suppressor. Nat. Med.11, 1306–1313 (2005).
Brand, K. A. & Hermfisse, U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J.11, 388–395 (1997).
Chance, B., Sies, H. & Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev.59, 527–605 (1979).
Pinton, P. et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science315, 659–663 (2007).
Liochev, S. I. & Fridovich, I. The effects of superoxide dismutase on H2O2 formation. Free Radic. Biol. Med.42, 1465–1469 (2007).
Dhar, S. K., Xu, Y., Chen, Y. & St Clair, D. K. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. J. Biol. Chem.281, 21698–21709 (2006).
Chen, J. et al. Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev.19, 2900–2911 (2005).
Bourdon, J. C. et al. p53 isoforms can regulate p53 transcriptional activity. Gene Dev.19, 2122–2137 (2005).
Aoubala, M. et al. p53 directly transactivates Delta133p53alpha, regulating cell fate outcome in response to DNA damage. Cell Death Differ.18, 248–258 (2011).
Marcel, V. et al. p53 regulates the transcription of its Delta 133p53 isoform through specific response elements contained within the TP53 P2 internal promoter. Oncogene29, 2691–2700 (2010).
Chen, J. et al. p53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via BclxL activation in zebrafish. Genes Dev.23, 278–290 (2009).
Ou, Z., Yin, L., Chang, C. Q., Peng, J. R. & Chen, J. Protein interaction between p53 and delta 113p53 is required for the anti-apoptotic function of delta 113p53. J. Genet. Genomics41, 53–62 (2014).
Fujita, K. et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol.11, 1135–1142 (2009).
Mondal, A. M. et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Invest.123, 5247–5257 (2013).
Gong, L. et al. p53 isoform Delta113p53/Delta133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res.25, 351–369 (2015).
Gong, H. J. et al. p73 coordinates with Delta 133p53 to promote DNA double-strand break repair. Cell Death Differ.25, 1063–1079 (2018).
Gong, L., Pan, X., Yuan, Z. M., Peng, J. & Chen, J. p53 coordinates with Delta133p53 isoform to promote cell survival under low-level oxidative stress. J. Mol. Cell Biol.8, 88–90 (2016).
Xiao, Q. et al. A p53-based genetic tracing system to follow postnatal cardiomyocyte expansion in heart regeneration. Development144, 580–589 (2017).
Berghmans, S. et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl Acad. Sci. USA102, 407–412 (2005).
Bednarek, D. et al. Telomerase is essential for zebrafish heart regeneration. Cell Rep.12, 1691–1703 (2015).
Gong, L. et al. p53 isoform Delta 133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep.6, 37281 (2016).
Ray, P. D., Huang, B. W. & Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal.24, 981–990 (2012).
Jopling, C., Sune, G., Faucherre, A., Fabregat, C. & Belmonte, J. C. I. Hypoxia induces myocardial regeneration in zebrafish. Circulation126, 3017–U3435 (2012).
Callapina, M., Zhou, J., Schmid, T., Kohl, R. & Brune, B. NO restores HIF-1 alpha hydroxylation during hypoxia: role of reactive oxygen species. Free Radic. Biol. Med.39, 925–936 (2005).
Kohl, R., Zhou, J. & Brune, B. Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1 alpha stabilization. Free Radic. Biol. Med.40, 1430–1442 (2006).
Chang, T. C. et al. Stabilization of hypoxia-inducible factor-1 alpha by prostacyclin under prolonged hypoxia via reducing reactive oxygen species level in endothelial cells. J. Biol. Chem.280, 36567–36574 (2005).
Niethammer, P., Grabher, C., Look, A. T. & Mitchison, T. J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature459, 996–999 (2009).
Bagheri, F. et al. Reactive oxygen species-mediated cardiac-reperfusion injury: Mechanisms and therapies. Life Sci.165, 43–55 (2016).
Dunnill, C. et al. Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int. Wound J.14, 89–96 (2017).
Braunersreuther, V. & Jaquet, V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol.13, 97–114 (2012).
Kevin, L. G., Novalija, E. & Stowe, D. F. Reactive oxygen species as mediators of cardiac injury and protection: the relevance to anesthesia practice. Anesth. Analg.101, 1275–1287 (2005).
Murphy, E. & Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev.88, 581–609 (2008).
Alizadeh, A. M., Faghihi, M., Sadeghipour, H. R., Mohammadghasemi, F. & Khori, V. Role of endogenous oxytocin in cardiac ischemic preconditioning. Regul. Pept.167, 86–90 (2011).
Bernardi, P. & Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol.78, 100–106 (2015).
Sano, M. et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature446, 444–448 (2007).
Heicklen-Klein, A. & Evans, T. T-box binding sites are required for activity of a cardiac GATA-4 enhancer. Dev. Biol.267, 490–504 (2004).
Mably, J. D., Mohideen, M. A., Burns, C. G., Chen, J. N. & Fishman, M. C. heart of glass regulates the concentric growth of the heart in zebrafish. Curr. Biol.13, 2138–2147 (2003).
Tao, T. et al. Def defines a conserved nucleolar pathway that leads p53 to proteasome-independent degradation. Cell Res.23, 620–634 (2013).
Acknowledgements
We thank Dr. Peidong Han for his kind help with processing images of ex vivo HyPer hearts. We also thank Drs. Chenglu Xiao, Nannan Chang, Meijun Pang, Mengmeng Huang and Yu Zhang for their generous gifts of protocols and/or transgenic zebrafish lines. This work was supported by the National Key R&D Program of China (2018YFA0801000, 2018YFA0800500 and 2017YFA0504501), the National Natural Science Foundation of China (31871500) and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by I. Amelio
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, S., Zhao, T., Zhang, W. et al. p53 isoform Δ113p53 promotes zebrafish heart regeneration by maintaining redox homeostasis. Cell Death Dis 11, 568 (2020). https://doi.org/10.1038/s41419-020-02781-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-02781-7
This article is cited by
-
A novel gene-trap line reveals the dynamic patterns and essential roles of cysteine and glycine-rich protein 3 in zebrafish heart development and regeneration
Cellular and Molecular Life Sciences (2024)
-
Wdr5-mediated H3K4me3 coordinately regulates cell differentiation, proliferation termination, and survival in digestive organogenesis
Cell Death Discovery (2023)
-
Loss-of-function of p53 isoform Δ113p53 accelerates brain aging in zebrafish
Cell Death & Disease (2021)
