
Abstract
Head and neck squamous cell carcinomas (HNSCCs) are an aggressive, genetically complex and difficult to treat group of cancers. In lieu of truly effective targeted therapies, surgery and radiotherapy represent the primary treatment options for most patients. But these treatments are associated with significant morbidity and a reduction in quality of life. Resistance to both radiotherapy and the only available targeted therapy, and subsequent relapse are common. Research has therefore focussed on identifying biomarkers to stratify patients into clinically meaningful groups and to develop more effective targeted therapies. However, as we are now discovering, the poor response to therapy and aggressive nature of HNSCCs is not only affected by the complex alterations in intracellular signalling pathways but is also heavily influenced by the behaviour of the extracellular microenvironment. The HNSCC tumour landscape is an environment permissive of these tumours’ aggressive nature, fostered by the actions of the immune system, the response to tumour hypoxia and the influence of the microbiome. Solving these challenges now rests on expanding our knowledge of these areas, in parallel with a greater understanding of the molecular biology of HNSCC subtypes. This update aims to build on our earlier 2014 review by bringing up to date our understanding of the molecular biology of HNSCCs and provide insights into areas of ongoing research and perspectives for the future.
Similar content being viewed by others

Facts
-
The heterogeneous nature of HNSCC at the molecular level has hindered both the identification of specific targets and development of targeted therapeutics for this group of tumours.
-
Advances in strategies in dissecting the features of the HNSCC genome, transcriptome and metabolome have revealed new altered targets. But this has not yet resulted in clinical improvements in the management of these cancers.
-
Current treatment strategies are very toxic, highlighting the need for treatment stratification using validated biomarkers to improve treatment outcome and reduce toxicity and cost of HNSCC treatment.
-
Radiotherapy resistance remains a major cause of HNSCC poor survival rates. Understanding the underlying molecular mechanism of RT resistance should significantly impact patient survival outcomes, but requires a multidisciplinary approach combining imaging and molecular profiling.
-
EGFR inhibitors, the only approved targeted drugs, have limited efficacy with the mechanisms of inherent and acquired resistance remaining unresolved.
-
There has been a significant increase in the incidence of HPV-positive HNSCC, a subgroup with more favourable prognosis. Strategies for treatment de-escalation to reduce toxicity are urgently required.
-
New, tailorable treatments such as immunotherapy have become highly valuable in the treatment of HNSCC. However, the importance of both the tumour microenvironment and the role of tumour immunity in pathogenesis and treatment response needs further understanding.
-
A possible relationship between the oral microbiome and HNSCC has been reported, warranting further research into the influence of the oral microbiome on subsequent development of HNSCC.
Open questions
-
How can HNSCC therapies, alone or in combination (radiotherapy, chemotherapy, targeted drugs and immunotherapy) be made more effective, to achieve a good prognosis while minimising undesirable treatment effects?
-
For individuals with HPV-linked HNSCC, is it safe and effective to use less aggressive treatment than the usual highly toxic therapies?
-
How can advances in precision medicine (and identification of biomarkers) help clinicians to personalise treatment and predict outcomes, based on the patient’s unique biochemistry and genetic profile?
-
Can the relationship between tumour and microenvironment (including the input of the immune system and influence of the microbiome) help us either treat, stratify patients or prevent oncogenesis?
-
To what extent is the HNSCC epigenome contributing to evolution of the tumour? Will the epigenome be the next-generation pharmacological target for HNSCC?
Introduction
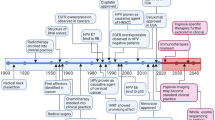
Head and neck squamous cell carcinomas (HNSCCs) are the sixth most common malignancy worldwide, accounting for over 500,000 new cases annually. Long-term tobacco use, consumption of alcohol and infection with high-risk types of Human Papilloma Virus (HPV) are considered the main oncogenic drivers1,2. Treatment involves surgical eradication, radiotherapy (RT) and chemotherapy (CT). All modalities severely reduce quality of life, and are largely ineffective. Most of the developments towards understanding this disease have occurred in the past few decades, but have fallen short of clinically meaningful discoveries (Fig. 1). Efforts to improve treatment efficacy have been largely without success, highlighting an urgent need for more effective therapies, alongside clinically relevant biomarkers to stratify patients and improve treatment outcomes.

Interestingly, the major advances in our understanding of HNSCCs have only been made in the past 20–30 years. Also, some major discoveries concerning the molecular characterisation of HNSCCs have been made almost 20 years after similar discoveries in other cancers. For example, where HPV was proven unequivocally to cause cervical cancers around 1983, the same discovery was not made for HNSCCs until around the year 2000. It is not so hard to understand therefore why therapeutic options for HNSCCs are so far behind, when our understanding of the molecular biology of these diseases only began to develop over the past 2–3 decades. Dates are not exact, and the details presented are by no means exhaustive10,11,12,13,14,15,16,17,18,19,20,21,22
HNSCCs have a high rate of genetic heterogeneity3, resulting in loss-of-function mutations in tumour-suppressor genes such as p53 and p16INK4a, and activation of oncogenes, such as the epidermal growth factor receptor (EGFR)4 and PIK3CA5. Until the recent FDA approval of immunotherapies, Cetuximab, a monoclonal antibody (mAb) targeting EGFR, has been the only targeted drug approved for both HPV-positive and -negative subtypes6. However, Cetuximab, and other therapies designed to target EGFR, has limited efficacy7 (Fig. 2).

EGFR, MET and NOTCH alterations promotes proliferation, migration and cellular survival via signalling through the RAS/RAF/ERK, PI3K and JAK/STAT pathways, all of which are regularly dysregulated in HNSCC. Disruption of the p53 pathway also leads to high levels of genomic instability. Green boxes show possible therapeutic agents either approved (*) or under investigation for clinical use in HNSCC. Information about the percentage of HNSCC cases showing either mutations or overexpression of the pathway as a whole shown in red boxes, where this data are available. Note, though activation of the JAK/Stat pathway is regularly seen in HNSCC, no mutations have yet been found1,2,3,4,5,6,7,8,9
In the absence of targeted therapies, radiotherapy remains the main treatment modality for HNSCC, and preferable for organ preservation8. HPV-positive patients respond better to RT, a trait not fully understood. The use of HPV as a biomarker for dose de-escalation has been considered, but the value of this approach has not been proven and concerns of under-treating these patients have taken precedence9.
The efficacy of RT is also substantially constrained by the presence of tumour hypoxia. Hypoxia is a biomarker of an aggressive phenotype, with higher rates of metastasis and recurrence10. Again, a full molecular understanding of this condition in HNSCC is lacking.
HNSCC subtypes are clinically, histologically and molecularly distinct. Yet, these diseases are treated uniformly, and with limited success. Despite research efforts, survival rates are at a deadlock. The lack of biomarkers for personalised treatment suggests an urgent need for better understanding of the intricate molecular biology of HNSCC, alongside an understanding of tumour–microenvironment interactions. This review will explore the current knowledge of HNSCC biology and highlights areas of ongoing research towards improving treatment outcomes.
The genetic landscape in HNSCC and its clinical implications
The TP53/RB pathway
TP53 is a tumour-suppressor gene encoding a transcription factor11 with roles in maintaining genomic stability, cell cycle, DNA repair, apoptosis and senescence12,13. p53 is a major cellular stress sensor for DNA damage or oncogene activation14,15,16. Over 80% of HPV-negative HNSCCs have p53 mutations resulting in the loss of function17,18. Mutations in TP53 occur early in carcinogenesis, and are mostly associated with HPV-negative cases, due to degradation of p53 by the HPV E6 oncoprotein19. In both subtypes, p53 mutations are associated with poor overall survival, therapy resistance and increased rate of recurrence17. In response to DNA damage, p53 is regulated in a MDM2-dependent manner20 and activated by cell cycle checkpoint kinases CHK1 and CHK2 resulting in cell-cycle arrest and apoptosis12.
Amplification or overexpression of another p53 family member, TP63, is observed in around 80% of HNSCCs21,22,23. Of the two major isoforms produced by TP63 (TAp63 and ΔNp63), ΔNp63 plays a major role in HNSCC pathogenesis, regulating key pathways, including cell survival and renewal, senescence suppression (by suppressing p16/INK4A) and growth factor signalling24,25,26. Recent evidence indicates that ΔNp63 influences the HNSCC metabolic microenvironment via a transcriptional programme involving HAS3 and HYAL genes, and the signalling of hyaluronic acid via CD44, activating pro-proliferative and pro-survival pathways in HNSCC27. Another p53 family member, p73, and its isoform TAp73 can indirectly influence growth arrest or apoptosis by regulating p53 target genes. Despite the retained DNA-binding activity of the other isoform ΔNp73, it cannot transactivate p53 reporter genes28. In HNSCC, TAp73 has been shown to suppress EGFR transcription and induce cell death in EGFR-overexpressing cell lines28. In cancers with mutant p53, proinflammatory cytokine TNF-α-induced c-REL/ΔNp63α interactions, inactivate tumour suppressor TAp73 function, promoting TNF-α resistance and survival. c-REL depletion enhanced TAp73 promoter interaction and expression of genes mediating growth arrest and apoptosis29. However, while amplification of P63 is frequent in HNSCC and the consequent oncogenic function of ΔNp63 has been sufficiently understood, the extent of p73 contributions to the disease needs further investigation.
The retinoblastoma tumour suppressor (RB) regulates cell-cycle progression at the restriction point between early and late G1. As with TP53, mutations in the RB pathway are an early alteration in HNSCC carcinogenesis. The combined mutations of p53 and RB pathways result in unlimited replication potential of cancer cells30. In HPV-positive cancers, binding of viral protein E7 to pRb results in degradation leading to E2F release and uncontrolled cellular proliferation30,31.
Several strategies for p53-targeted therapies have been developed, such as adenoviral p53 gene therapy or use of small molecules to restore TP53 function or disrupt inactivation of wild-type p53. But, these have not proved effective in clinical trials32. Other small molecules targeting p53 for reactivation of p53 are in early-stage investigations. COTI-2 (a derivative of thiosemicarbazones) showed activity in refolding mutant p53 and restoring wild-type p53 function. A clinical trial (NCT02433626) will soon test efficacy of COTI-2 in HNSCC33.
NOTCH pathway
A 2015 genomic analysis by the Cancer Genome Atlas (TCGA) showed inactivating mutations in NOTCH1-3 to be present in 17% of HPV-positive and 26% of HPV-negative HNSCCs17. These aberrations predominantly occur in NOTCH1 and include mis-sense mutations in functional regions, non-sense mutations resulting in truncated proteins and frameshift insertions or deletions34. A recent HNSCC cohort study revealed poor prognosis in NOTCH1 mutation cases, where direct downstream targets HES1 and HEY1 were overexpressed35,36. NOTCH1 signalling can contribute to the maintenance of cancer stem cell traits responsible for recurrence and metastasis through Wnt signalling37. The crosstalk between Notch and Wnt signalling has been reported in different types of cancers38. Loss of Notch signalling was shown to increase transcriptional activity of a β-catenin-responsive reporter construct in colon cancer stem and progenitor cells39. In HNSCC, concurrent NOTCH1 and FAT1 inactivating mutations drives carcinogenesis by activating β-catenin17. Importantly, loss of Notch signalling was found to promote tumorigenesis in HNSCC by upregulating ΔNp63, but the precise mechanisms of Notch-ΔNp63 regulation remain to be elucidated. Interestingly, Notch signalling in keratinocytes could be impaired by ΔNp63 expression, suggesting a reciprocal process between Notch and p63 in the epidermis34.
The data from genomic and functional studies in the lung, bladder and oesophagus support a tumour-suppressor role for Notch signalling in epithelial SCCs tumorigenesis40. Moreover, several in vivo models provide results consistent with sequencing data from patient samples that support a tumour-suppressive role of Notch. However, in vitro studies using HNSCC cell lines demonstrated that increased activity of Notch signalling is required for maintaining malignant behaviour41. It is essential to functionally validate the role of Notch signalling in HNSCC using robust in vivo models, as in vitro studies are unlikely to represent patient disease development. Collectively, whether NOTCH mutations are activating or inactivating in HNSCC remains debatable42,43. It may be that different types of mutations occur in different subtypes of HNSCC35. This therefore must be carefully considered when taking inhibitors or activators of this pathway into clinical trials44,45.
PI3K/Akt/mTOR pathway
Recent molecular characterisation showed that in HNSCC, PI3K/Akt/mTOR seems to be the most frequently deregulated pathway. In HNSCC, the PI3KCA gene harbours mutations at a rate of ~16%30. PI3Ks are a class of enzymes vital for cellular growth, differentiation and survival, activated by RTKs, such as EGFR. Other members of the pathway include the mTOR complexes (mTORC1 and mTORC2), and Akt. mTORC2 is essential for Akt phosphorylation and activation of other signalling molecules of the PI3K pathway, including SGK146.
Clinical trials have assessed the value of targeting this pathway with drugs, including rapamycin, everolimus and temsirolimus with encouraging outcomes47. Emergence of resistance to mTOR inhibitors has been encountered in HNSCC, though the mechanisms behind this resistance are still being investigated48. In one such example, a possible feedback loop between Akt and ERK/MAPK signalling by mTOR inhibition was found to act as a survival mechanism in tumour cells. Co-targeting mTOR and EGFR, thus inhibiting upstream activation of the Akt and ERK signalling pathway, has been suggested to overcome this resistance49.
Epidermal growth factor receptor (EGFR) pathway
The epidermal growth factor receptor (EGFR, HER1 or ErbB1) belongs to the HER/ErbB family of receptor tyrosine kinases (RTKs), which also includes HER2-4. EGFR is overexpressed in 80–90% of HNSCC cases and correlates with poor prognosis and treatment outcomes50. EGFR signalling is a complicated and multidimensional network, involving many individual players and overlap with other pathways. As such, the potential for therapeutic targeting of EGFR signalling is vast, representing both a daunting challenge and tantalising opportunity for HNSCC research.
EGFR is a transmembrane receptor with tyrosine kinase activity51. Ligand binding triggers homo- or hetero-dimerisation with other HER members and subsequent phosphorylation of tyrosine residues, activating downstream signalling cascades. These pathways control proliferation, differentiation, survival, angiogenesis, invasion and metastasis in cancer52. EGFR can also translocate to the nucleus where it may function as a transcription factor. This translocation has been found to be triggered by ionising radiation, resulting in radiotherapy resistance53. EGFR has also been shown to interact with other receptors, such as Axl, enhancing oncogenic potential54.
Targeting EGFR can be achieved either by blocking the ligand-binding domain using monoclonal antibodies (mAbs), or by inhibiting the activity of the tyrosine kinase domain using small-molecule tyrosine kinase inhibitors (TKIs). Cetuximab, a chimeric monoclonal antibody with high specificity and affinity to EGFR, remains the only approved targeted therapy for HNSCC in combination with RT/CT7,55,56. Despite high rates of EGFR overexpression,Cetuximab has shown limited efficacy in HNSCC. This may be due to aberrations in other HER family members and their ligands, and/or activation of other downstream signalling components57,58.
The mechanisms behind inherent and acquired resistance toCetuximab remain unsolved59. Several new targeted therapies have been developed to target EGFR or its signalling partners. So far, these have shown only modest improvements in progression-free survival (PFS), and none have been approved for treatment in HNSCC (see Table 1).
Small TKIs of EGFR have been ineffective in HNSCC although some early results from trials with combination therapies have shown promise. Erlotinib, for example, demonstrated modest improvements in PFS when used in combination with an anti VEGF antibody (Bevacizumab) in recurrent/metastatic (R/M)-HNSCC60. However, when used for locally advanced (LA)-HNSCC in combination with cisplatin and radiotherapy, the study group demonstrated no improvements in disease progression. Afatinib, a pan-HER TKI, has also been shown to have positive anti-tumour activity comparable with Cetuximab61,62. In a study comparing Afatinib to methotrexate, it was noted that patients who experienced improvements in disease outcomes had EGFR amplification, low HER3, no expression of p16 and high PTEN63, highlighting the importance of identification of biomarkers in patients who show favourable response.
MET pathway
One proposed mechanism for resistance to EGFR-targeted therapies is upregulation or activation of other RTKs, such as c-MET (hepatocyte growth factor receptor). C-MET encodes mesenchymal–epithelial transition factor associated with increased migration, invasion and metastasis in cancer64. C-MET mutations are reportedly rare in HNSCC (2–13%), while gain in MET copy number and overexpression of its ligand hepatocyte growth factor (HGF) is common in HNSCCs65. The roles of HGF/c-MET in HNSCC invasion and metastasis have been investigated in many studies. C-MET has been found overexpressed in lymph node metastasis, and HGF has been shown to promote anoikis resistance in HNSCC, an essential step for nodal metastasis66. In patients, overexpression is associated with worse prognosis and lower overall survival67.
EGFR and c-MET share common downstream pathways, including the RAS-RAF-MAPK and PI3K-AKT-mTOR pathways. Therefore, the MET-HGF axis could represent a valuable therapeutic target in HNSCC, of particular relevance to patients with resistance to EGFR-targeted therapies64. Studies on dual blockade of EGFR and c-MET have reported promising anti-tumor activity of combined treatment67. Co-targeting both receptors has demonstrated ability to sensitise cells to EGFR-targeted therapies68. Capmatinib (INC-280) is a c-MET inhibitor with anti-tumour activity in mouse models. A phase I trial assessing Capmatinib safety in advanced solid tumours (NCT01324479) is completed and awaiting results69. Ficlatuzumab, a mAB that targets the HGF/c-MET axis, is also being tested in clinical trials for HNSCC in combination with Cetuximab in R/M-HNSCC (NCT02277197). Promising results from pre-clinical models of HNSCC showed inhibition of proliferation, migration, invasion and EMT70.
JAK/STAT pathway
In both HPV-positive and HPV-negative HNSCCs, aberrant regulation of the signal transducer and activator of transcription (STAT) family has been reported. Upregulation of STAT3 and its gene targets is thought to contribute to the malignant behaviour of HNSCCs, resistance to chemo/radiotherapy and EGFR-targeted therapy71,72,73,74.
STAT3 signalling is considered immunosuppressive and may protect cancer cells from recognition and lysis by cytotoxic T lymphocytes, achieved by triggering production of cytokines, including IL-6, IL-10, VEGF and TGF- β175. STAT3 is activated in response to upstream signals of the IL-6 cytokine receptor family, RTKs such as EGFR, VEGFR, Jenus-activated kinases (JAK) and Src family kinases (SFK)76. Following activation, nuclear phospho-STAT3 promotes expression of target genes including pro-survival factors, such as cyclin D1, survivin and Bcl-xL71.
In the context of STAT3 targeting, Ruxolitinib is an approved JAK inhibitor for myelofibrosis. A clinical trial currently in the recruitment phase aims to test efficacy of Ruxolitinib in HNSCC (NCT03153982). AZD9150, a synthetic anti-sense oligonucleotide molecule targeting STAT3 by inhibiting mRNA translation, has demonstrated anti-tumour activity in xenograft models. It is currently being tested in clinical trials of metastatic HNSCC cases as a monotherapy or combined with MED14736, an immunotherapy blocking the interaction of PD-1 and PD-L1 (NCT02499328)77.
RAS/RAF/MAPK pathway
The mitogen-activated protein kinase (MAPK) pathway regulates expression of proteins involved in cell proliferation, differentiation, apoptosis, angiogenesis, invasion and metastasis78. It comprises four sub-pathways of which the Erk1/2 pathway has received most attention in HNSSCC. Upon binding of growth factors (such as EGF), a signalling cascade results in activated Erk1/2 that dissociates from the Ras-Raf-MEK-ERK1/2 complex and phosphorylates a number of cytoskeletal proteins, kinases and transcription factors, including NF-κB, AP-1, ETS-1 and c-Myc79. Mutations in the MAPK pathway have been implicated in other cancers, however, in HNSCC mutations only represent 4% of cases17,80. MEK inhibitors, such as Trametinib, are currently approved for melanoma treatment, and are being investigated in clinical trials for HNSCC81.
HPV-associated HNSCC
HPV is a risk factor associated with 22% of oropharyngeal (OPSCC) and 47% of tonsillar squamous cell carcinomas (TSCC). The incidence of HPV-positive HNSCC increased 225% from 1984 to 2004, and has now surpassed the incidence of HPV-induced cervical cancer5. This increase is thought to be a consequence of changes in sexual behaviour82. Of the 200 types of HPV viruses, HPV-16 is the most common type found in HNSCCs (90% of HPV-OPSCC), followed by HPV-18 (3%).
Clinically, HPV-positive patients often present with small tumours, but with advanced nodal metastasis. The epidemiological profile for HPV-positive patients is unique, with the majority of patients being young, white and male9. Crucially, HPV-positive HNSCC patients demonstrate favourable prognosis, with a 28% reduced risk of death and almost 50% reduced risk of local recurrence when compared with HPV-negative patients83. HPV-positive patients show improved response to radiotherapy and chemotherapy84. Whether this is due to the molecular pathogenesis, or related to age and better overall health of patients remains unclear.
In contrast to HPV-derived cervical cancers, in HNSCC, dysplastic lesions are rarely found before cancers are diagnosed. The HPV virus is also not commonly found in non-malignant tonsil tissue samples85. Evidence suggests that the tonsillar crypt epithelium on its own might not be permissive of the viral reproductive cycle. Despite this, the microenvironment is such that some 47% of infections within the tonsillar crypts can progress to cancer. Notably, only 3.9% of HPV infections in the oral cavity lead to cancer9.
Genetic alteration in HPV-derived HNSCC is primarily characterised by inactivation of p53 and pRB by viral proteins E6 and E7, respectively (Fig. 3). E5 is also implicated in carcinogenesis through activation of signalling pathways, involving EGFR, immune recognition and regulation of apoptosis86. PI3K mutations are observed in 30% of HPV-associated HNSCC87, based on current genomic data, mutation hotspots of PIK3CA vary between HPV-positive and negative tumours. Mutations predominantly occur in the helical domain of the gene in the HPV-positive subtype, whereas mutations occur throughout the gene in HPV-negative tumours88. These differences may have an impact on clinical outcomes to PI3K/mTOR inhibitors, and even have predictive value48.

HPV oncogenesis occurs mainly in the tonsillar crypt epithelium of the oropharynx. Upon HPV infection, viral DNA is either integrated or exists in the cells in episomes. Regardless, this allows for transcription of viral oncoproteins E5, E6 and E7. The main dysregulation attributed to carcinogenesis in HPV-positive cases stems from the inhibition of p53 by the E6 viral protein, and of Rb by E7. This leads to entry into the cell cycle via release of E2F, and inhibition of p53-mediated cell death. It also results in the accumulation of p16, which subsequently acts as a surrogate marker of HPV infection in HNSCC. Though previously considered a minor player, E5 has also been shown to activate EGFR leading to further oncogenic potential. Amongst the signalling pathways shown to be activated in HPV-HNSCC, the PI3K pathway has been found to be significantly upregulated. This may in part be due to activation by ErbB family members, including both EGFR and Her31,7,23,24,25,26
TCGA data demonstrates the presence of several other genetic differences, including, loss of TRAF (a tumour-suppressor gene implicated in anti-viral immunity response) and aberrant activation of cell-cycle genes89. The most frequently altered genes in HPV-negative HNSCCs are often unaltered in HPV-positive HNSCCs. However, the alteration of p53, p16INK4A and RB as a result E6 and E7 in HPV-positive cases is functionally similar5. A recent study using TCGA data, sought to further classify HNSCC tumours based on HPV status and TP53/CDKN2A mutation status. The results demonstrated that genes involved in DNA mismatch repair were upregulated in HPV-positive tumours, a pathway implicated in the cytotoxicity of chemo/radiotherapy90, thus providing a possible insight into the improved response of these patients to radiotherapy91.
Currently, there are no subtype-specific treatment regimens available9. Several trials have assessed de-intensification of treatment of HPV-positive cancers using various methods, including reducing total dose of radiation, RT as a monotherapy and replacing conventional chemotherapy with targeted therapies. The results of some phase III clinical trials aiming to assess efficacy of replacing platinum-based chemotherapy with Cetuximab and radiotherapy, including RTOG 1016, TROG 12.01 and the De-ESCALaTE trial92,93,94,95, are published. These trials report that EGFR inhibition by Cetuximab conferred a reduction in overall survival and tumour control when compared with cisplatin. The authors of the De-ESCALaTE trial also recommend caution when considering reducing the dose of radiotherapy for HPV-positive patients. Additional efforts have been made towards the development of therapeutic HPV vaccines96.
Database analysis of head and neck squamous cell carcinomas
Following the omics revolution, the amount of data generated with the aim of uncovering biomarkers, network motifs and susceptibility markers has soared. The development of databases to the structure and sharing of these data has been critical. See Sepiashvili et al. for detailed biomarker data with respect to HNSCC97.
Both genomic and proteomic data can be accessed, with databases such as the GEO omnibus98 and Array Express99 containing microarray data, DNA- and RNA-sequencing, and DNA methylation studies (Table 2) and The Human Protein Atlas containing results from tumour tissue staining100,101. These databases have their own merits and pitfalls. Whereas GEO and Array Express comprehensively gather all species and cell types, the Cancer Genome Atlas (TCGA) specifically focuses on patient-derived samples and contains information about genomic changes in different cancer types. For head and neck, this includes 44 healthy and 504 cancer samples102. TCGA comprises mRNA, miRNA and protein expression data as well as DNA methylation status. The Leipzig Health Atlas contains gene expression profiling and targeted sequencing of 50 genes of 290 consecutively recruited HNSCC patients103. For metabolic alterations observed in HNSCC a variety of medium and small-scale data sets have been generated as summarised by Shin et al.104. Furthermore, a genomic information portal for HNSCC cell systems has been set up, detailing clinical and genomic information of 44 cell lines105.
The Danish Head and Neck Cancer group (DAHANCA) established in 1976, was set up with the aim of generating guidelines for the treatment of HNSCCs based on patient cases. To date, they have compiled data from more than 33,000 patients containing variables incorporating symptoms, aetiological factors, diagnostic evaluation and involvement in clinical trials106. This database is a valuable resource for longitudinal studies, which have clear advantages over single case studies. However, the accessibility of DAHANCA to the scientific community is limited by the use of the Danish language in all reports. The National Cancer Database (NCDB), based in the USA, collects information submitted from hospitals detailing the state of cancer care throughout the country. Between 1990 and 2004, information of around 800,000 head and neck tumours were collected107.
In contrast to these large studies, some databases have attempted to group all decentralised information into a single source. One such database for head and neck cancer is HNdb, which was established with the aim to combine genomics, transcriptomics and proteomics data, literature citations, and cross-references from external databases. For this purpose, Henrique et al. established a text mining procedure to identify genes in literature related to HNSCC. Their database provides user-friendly access to information on HNSCC-related genes and their different biological data resources108. Overall, the large amounts of data amassed so far offer a great foundation for the identification of possible biomarkers, to begin working towards personalised medicine for HNSCC.
The microenvironmental landscape
Hypoxia in HNSCC
Hypoxia is a well-established cause of poor response to treatment, metastasis and recurrence. It develops as a result of progressive tumour growth leading to impairment of oxygen supply within tumour tissues. This results in the development of areas starved of oxygen30. Within these regions, central areas (experiencing oxygen levels < 0.1% O2) will likely become necrotic. The cells within regions of acute hypoxia (< 2% O2) that survive these extreme conditions adapt a programme of gene expression leading to treatment resistance and metastasis109,110,111.
The hypoxic response is primarily mediated by the heterodimeric hypoxia-inducible factors (HIF-1-3). Under normal oxygen conditions, the HIFα subunits undergo rapid degradation by the E3-Ubiquitin Ligase Von-Hippel-Lindau protein (VHL). Under hypoxic conditions, HIFα is stabilised and binds to HIFβ in the nucleus, where the activated HIF complex binds to hypoxia response elements (HREs), allowing for adaption of the tumour. HREs are present in genes responsible for altering metabolism (CA9, GLUT1), EMT (Vimentin), extracellular matrix remodelling (LOX, MMPs), angiogenesis (VEGF), immune modulation and inflammation (TNFα, IL1β)112,113,114,115,116.
The state of hypoxia is not clear-cut, with several examples of stabilisation of HIFs in normoxic conditions. mTOR has been shown to induce expression of HIF-1α in tumour areas that are not significantly hypoxic, and has been attributed to various oncogenic mechanisms, such as inactive p53 mutations, excessive accumulation of oxygen radicals, RAS mutations, inactivation of PTEN and ineffective degradation of HIF-1α by mutations in VHL112. Gain-of-function mutations in p53 have shown a synergism with HIF signalling. A recent study in hypoxic NSCLC found p53 mutations that regulate a selective gene signature including extracellular matrix (ECM) components, such as type VIIa1 collagen and laminin-γ2. This study illustrated the impact of p53 mutants on the microenvironment in co-operation with HIF-1 to promote cancer progression117,118. By considering the high frequency of p53 mutations in HPV-negative HNSCC and the hypoxia observed in these tumours, the co-operation between p53 mutants and HIF-1 signalling might represent a meaningful avenue for clinical investigation.
Hypoxic tumours represent a distinct subtype of HNSCCs with poor prognosis and treatment resistance. Attempts have been made to make clinical use of this information. Hypoxic modulation has been suggested to revert the changes that occur in low oxygen and restore sensitivity to treatment. Hyperbaric oxygen therapy is one such method which aims to improve tumour oxygenation119,120. Hypoxia-activated pro-drugs including Tirapazamine and Nimorazole are thought to act as oxygen-mimetic radiosensitizers. The latter is now included in the standard of care for Danish HNSCC patients, following guidelines from DAHANCA110,121. A trial using Nimorazole alongside RT for patients with R/M-HNSCC is currently recruiting in the UK (NIMRAD)122.
Targeting hypoxia therapeutically to overcome treatment resistance needs accurate and reliable detection methods. Several approaches including needle electrodes, endogenous or exogenous hypoxia tissue markers and hypoxia imaging have been tested, but have limitations. Needle electrodes are an invasive method limited to accessible tumours, albeit with demonstrable prognostic value in HNSCC tumours123. To overcome these limitations, endogenous hypoxia biomarkers have been identified in patient tissues and serum, the most commonly used being GLUT-1, CA9, VEGF and serological biomarker OPN124. Several hypoxia-specific gene signatures (HGS) have been developed125,126,127, with evidence of HGS having prognostic and predictive values. Notably, a 15-gene HGS from human HNSCC xenografts has been identified and further validated in a cohort of 302 HNSCC patients with successful discrimination between low and high hypoxic tumours127.
Positron emission tomography (PET)/computer tomography (CT) using hypoxia-specific radiotracers, combined with assessment of endogenous or exogenous hypoxia markers could provide a reliable clinical method for both detection and measurement of tumour hypoxia during treatment126,128. In HNSCC, PET imaging using the hypoxia radiotracer 18F-MISO has been investigated, demonstrating successful identification of HNSCC patients who benefited from addition of Tirapazamine with prediction of recurrence after radiotherapy129. Likewise, a recent study by Suh et al. combined hypoxia imaging with PET/CT using 64Cu-ATSM and gene expression of patient tumours to validate a HGS, linked to prognosis126.
The immune-microenvironment in HNSCC
Immune evasion is considered a key hallmark of cancer, generating an environment permissive of survival and progression. HNSCCs are immune-suppressive, with the ability to avoid recognition and clearance by immune cells. This evasion of immunosurveillance is achieved by alterations, including loss or downregulation of human leucocyte antigens (HLAs) expression, impaired recognition of cancer cells by T cells and activation of MAPK, STAT3 and β-catenin/Wnt signalling pathways130. The HNSCC tumour microenvironment has also been shown to have impaired function of tumour-infiltrating lymphocytes (TILs)131.
Both HPV-negative and HPV-positive tumours show high levels CD8 + cytotoxic T cells and activated NK cells132. However, these tumours still evade their cytotoxic mechanisms. For clinical purposes, research is ongoing to investigate efficacy of enhancing immune cell targeting of tumours. A potential method for this could be to exploit the presence of tumour-associated neoantigens expressed on cancer cells resulting from genetic reconfigurations133.
Adoptive immunotherapy, which involves administration of genetically modified T cells targeting specific antigens expressed on the surface of tumour cells, is a recent revolutionary advance in cancer immunotherapy. The synthetic chimeric antigen receptor (CAR)-T cell therapy has shown remarkable success in haematological cancers134. An FDA-approved CD19-targeting CAR-T cell therapy has demonstrated success in leukaemias and lymphomas, encouraging the development of similar therapies for solid tumours135. However, these therapies face multiple challenges such as a need to identify specific tumour-associated antigens that are overexpressed in tumours, but not in normal tissues136. A recent ongoing trial developed a pan-ErbB-targeted CAR-T cell therapy for HNSCC (NCT01818323)137.
The momentous discovery that certain proteins act as “immune checkpoints” by regulating the T-cell response has spurred efforts to develop treatments to reverse this effect and restore anti-tumour immune responses138,139. In normal circumstances proteins including programmed death protein (PD-1), its ligand (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), function to prevent autoimmunity140,141. PD-L1 has been detected in most human cancers and leads to anergy and apoptosis of activated T cells and subsequent immune evasion of the tumour cells142,143,144,145. These proteins are often overexpressed and co-opt this protective mechanism to inhibit immune activation. Immune checkpoint blockade therapies have shown remarkable clinical success for some cancers, including metastatic melanoma140,146,147. In 2016, anti-PD1 mAb Nivolumab was FDA-approved for recurrent/metastatic HNSCC patients with progressive disease or failure of platinum-based therapy. The approval followed the promising results of the CheckMate 14 trial, demonstrating a statistically significant improvement in OS and quality of life in patients treated with nivolumab148. Pembrolizumab, another anti-PD1 mAb, was in the same year approved in R/M-HNSCC treatment after the positive results of the KEYNOTE-12 trial149. Studies to approve these drugs as mono or combination therapies for different cancer types is ongoing.
Cancer-associated fibroblasts
Cancer-associated fibroblasts (CAFs) are fibroblast-like cells associated with the tumour that develop an activated phenotype, expressing markers such as α-smooth muscle actin (α-SMA) and fibroblast activation protein (FAP)150. These fibroblasts, alongside infiltrating immune cells, can resemble the architecture of a wounded tissue. However, unlike wound fibroblasts, CAFs are often resistant to cell death mechanisms and remain activated151,152,153. Communication, through direct contact and secretion of signalling molecules between CAFs and tumour cells is vital for their function154. With perpetual activation of CAFs, the tumour can benefit from CAF secretion of proteins for ECM remodelling155, growth factors152, cytokines, chemokines and recruitment of immune-suppressive cells156. This environment supports tumour growth, migration, invasion, angiogenesis, colonisation of distant tissues by way of the CAF-supported “pre-metastatic niche” and evasion of the immune system157,158,159,160,161. An additional danger engendered by this stroma–tumour communication has been the reported changes in responsiveness to cancer therapy161,162,163. This is particularly relevant in HNSCC, as CAFs have been shown to regulate response to Cetuximab164 and radiotherapy165,166. Research has shown that these treatments in turn impact CAF activation167,168.
A number of potential targeting strategies exist to block CAF-mediated tumour support, including inhibition of CAF cell-surface proteins (such as the anti-FAP antibody Sibrotuzumab169), blocking CAF activation or by targeting CAF-tumour signalling159. Tumour cells are also thought to contribute to activation of CAFs via a transformation process involving exosomes containing nucleic acids like microRNAs170,171, chemokines and cytokines like TGFβ, CXCL12 (CXC-Chemokine ligand 12 also known as SDF1) and IL-6, as well as local stimuli like hypoxia and oxidative stress154,156,172. Though research within this field with respect to HNSCC is somewhat underdeveloped, many of these factors have independently been implicated in its oncogenesis173,174,175,176,177. Recent work by Hersi et al. showed that low expression of the tumour-suppressive miR-9, which targets the CXCL12 receptor CXCR4, was linked to aggressive behaviour of HNSCC cells, counteracted by targeting of this interaction with Plerixafor (a CXCR4 inhibitor)178. Likewise, IL-6, its receptor and downstream JAK/STAT signalling have all been strongly implicated in HNSCC prognosis71,179. A recent study showed that IL-6 was responsible for both promoting oncogenesis when secreted from CAFs and CAF activation when secreted from tumour cells180.
Importantly, targeting some of these mechanisms, such as the CXCL12-CXCR4 and IL-6 pathways, could result in inhibition of both the tumour stroma and the tumour itself. One challenge associated with targeting CAFs in the HNSCC microenvironment relates to their heterogeneity. Their biology is complex, and CAFs likely originate from a number of different cell types181. In practice, this non-uniform pool of cells has been difficult to characterise completely. One benefit of CAFs is that they are genetically stable, unlike tumour cells, which may make long-term treatment plans more viable159. As with much of what has been reviewed herein, holistic approaches involving combinations of immune therapy, conventional therapy and CAF-targeted therapy may prove most useful for future treatment.
The oral microbiome
Changes in the oral microbiome have been proposed to contribute to oncogenesis in the 7–15% of oral cancer cases, which cannot be explained by known risk factors182,183,184. The microbiome denotes the collective genome of complex communities of bacteria, archaea, viruses, fungi and protists, each with crucial roles to play in stabilising microbial diversity185,186. The clinical relevance of the oral microbiome lies in the statistical association between dysbiosis (often a result of poor oral health) and the prevalence of many types of cancer187,188,189. Within the multifarious environment of the human mouth, with its mucosal surfaces and deep-tissue crevices, both healthy and malignant sites contain distinct microbial populations190,191.
Several bacterial species have been associated with oral cancer. A potential biomarker signature has been suggested for oral cancer and consists of the three bacterial species Capnocytophaga gingivalis, Prevotella melaninogenica and Streptococcus mitis. These bacteria are found in 80% of OSCC cases, and the signature has demonstrated a diagnostic sensitivity of 80% and a specificity of 82%192. Another three-fungal signature of Rhodotorula, Geotrichum and Pneumocystis, as well as the microsporidia Phialophora and Cladophialophora are specifically seen in OCSCC183.
Risk factors for HNSCCs including tobacco and alcohol consumption and HPV infection have been shown to affect the oral microbiome193,194,195,196. A shift towards different genera of bacteria has been associated with exposure to these risk factors. Oral microbes have also been shown to contribute to acetaldehyde (the carcinogenic metabollite of ethanol) production, which can induce mutagenesis, and hyperproliferation of the epithelium197. Bacterial strains show significant differences in their ability to produce acetaldehyde, for instance, S. mitis produces high amounts of acetaldehyde and has significant alcohol dehydrogenase activity198. Concurrently, elevated levels of S. mitis have been detected in OSCC192. Acetaldehyde and malondialdehyde production from Streptococcus species such as Streptococcus gordonii V2016 significantly increased bacterial attachment to keratinocytes, facilitated HPV infection by enhanced expression of furin and resulted in malignant transformation of infected keratinocytes199. Oral Streptococcus species and HPV seem to co-operate in order to infect and transform oral keratinocytes after exposure to alcohol. Cleavage of the minor capsid protein L2 by furin is required for HPV infection200,201. Since only a subset of bacteria contains enzymes with furin-like activity, this might contribute to HPV tissue tropism202.
The carcinogenic potential of several bacteria has been demonstrated in vitro and in animal models184. Chronic infection of P. gingivalis or F. nucleatum have been shown to augment the IL-6-STAT3 inflammatory cascade and promote HNSCC development203. Interactions between periodontopathogenic bacteria and single-nucleotide polymorphisms (SNPs) of Toll-like receptors (TLRs) such as TLR2 and TLR4 have also been shown to influence OSCC risk204,205,206. Virulent P. gingivalis strains can induce expression of PD-L1 and PD-L2 receptors in squamous carcinoma cells, mediated by the membrane fraction of P. gingivalis207,208.
Particularly relevant for HNSCC and its treatment options, some microbiota such as C. albicans and E. faecalis can activate EGFR signalling209,210,211. In addition, E. faecalis can drive tumorigenesis by hydrogen peroxide-mediated DNA damage, chromosomal instability and mutagenesis211. The compositional and functional variations of oral microbiota have been associated with the mutational changes in oral cancer. Yang et al. investigated the oral microbiome composition of OSCC patients in association with their mutational profile and identified three patient clusters, which varied in their bacterial species richness and their relative abundance of Firmicutes and Bacteroidetes212.
The clinical relevance of the microbiome is important as treatments such as radiotherapy cause alterations in healthy oral microflora, partially due to treatment-induced xerostomia213,214,215. Ultimately these alterations can result in exacerbation of mucositis and systemic infections. Patients who have good oral care during cancer therapy have better outcomes213. Different treatments in oral cancer patients have distinct effects on microflora216. Opportunistic pathogens such as staphylococci, enteric rods and Candida sp. tend to increase in prevalence after radiotherapy (IMRT) with or without chemotherapy217. C. albicans was found in one or more sites in 54% of patients who received radiotherapy in comparison with 15% of controls188.
A new avenue for personalised treatment could involve targeting the microbiome for therapeutic purposes with microbial supplements, such as synbiotics (probiotics and prebiotics), diet or microbial suppression strategies using antibiotics218,219,220,221,222,223. Probiotics have the potential to protect against cancer development in animal models, and some probiotic strains diminish the incidence of postoperative inflammation in cancer patients224,225,226,227,228,229. Importantly, most commercial probiotic products are generally safe and can improve the health of the host by modulating the intestinal microbiota and immune response230. Therefore, probiotic strains might be useful adjuvants for cancer prevention and/or treatment231. Lactobacillus brevis CD2 lozenges have been shown to reduce the severity and incidence of radio/chemotherapy-induced mucositis in HNSCC patients, thereby increasing the rate of anticancer treatment completion232,233.
In brief, the emerging fields of microbiomics and metagenomics will help to identify the presence of HNSCC-specific microbes and help us understand234 the development of accurate and cost-effective diagnostic and therapeutic strategies.
Conclusion
The heterogeneous nature of HNSCCs has hindered the identification of specific targets for the development of targeted therapies. Over the past 30 years we have developed a better understanding of the genomic, proteomic, microbiomic and metabolomic alterations in HNSCCs. This knowledge is helping to move us closer to personalised therapy, where each subtype can be treated as a separate disease. The significant problems associated with high toxicities as well as resistance to current treatments, and low quality of life for patients, make these efforts particularly crucial. Since our last clinical update in 20144, our understanding of these diseases has broadened to not only consider the endogenous alterations as key contributors to oncogenesis but also to consider the microenvironmental factors that build an environment permissive of these oncogenic mechanisms. How the integration of microenvironmental factors and genetic backgrounds reshapes the HNSCC epigenome is largely unclear and represents one of the new priorities in the field. It is likely that interdisciplinary approaches seeking to link the genetic and microenvironmental biology of HNSCC will allow us to better treat HNSCC in the future.
References
Shaw, R. & Beasley, N. Aetiology and risk factors for head and neck cancer: United Kingdom National Multidisciplinary Guidelines. J. Laryngol. Otol. 130, S9–S12 (2016).
Carbone, M. et al. Consensus report of the 8 and 9th Weinman Symposia on gene x environment interaction in carcinogenesis: novel opportunities for precision medicine. Cell Death Differ. 25, 1885–1904 (2018).
Ha, P. K., Chang, S. S., Glazer, C. A., Califano, J. A. & Sidransky, D. Molecular techniques and genetic alterations in head and neck cancer. Oral. Oncol. 45, 335–339 (2009).
Suh, Y., Amelio, I., Guerrero Urbano, T. & Tavassoli, M. Clinical update on cancer: molecular oncology of head and neck cancer. Cell death Dis. 5, e1018 (2014).
Leemans, C. R., Snijders, P. J. F. & Brakenhoff, R. H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18, 269–282 (2018).
Moskovitz, J., Moy, J. & Ferris, R. L. Immunotherapy for head and neck squamous cell carcinoma. Curr. Oncol. Rep. 20, 7 (2018).
Sacco, A. G. & Worden, F. P. Molecularly targeted therapy for the treatment of head and neck cancer: a review of the ErbB family inhibitors. Onco Targets Ther. 9, 1927–1943 (2016).
Dietz, A. et al. Induction chemotherapy with paclitaxel and cisplatin followed by radiotherapy for larynx organ preservation in advanced laryngeal and hypopharyngeal cancer offers moderate late toxicity outcome (DeLOS-I-trial). Eur. Arch. Oto-Rhino-Laryngol. 266, 1291–1300 (2009).
Maxwell, J. H., Grandis, J. R. & Ferris, R. L. HPV-associated head and neck cancer: unique features of epidemiology and clinical management. Annu Rev. Med 67, 91–101 (2016).
Göttgens, E. L., Ostheimer, C., Span, P. N., Bussink, J. & Hammond, E. M. HPV, hypoxia and radiation response in head and neck cancer. Br. J. Radiol. 92, 20180047 (2018).
Sullivan, K. D., Galbraith, M. D., Andrysik, Z. & Espinosa, J. M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25, 133–143 (2018).
Bykov, V. J. N., Eriksson, S. E., Bianchi, J. & Wiman, K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89 (2017).
Kaiser, A. M. & Attardi, L. D. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 25, 93–103 (2018).
Aubrey, B. J., Kelly, G. L., Janic, A., Herold, M. J. & Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25, 104–113 (2018).
Parrales, A., Thoenen, E. & Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 25, 460–470 (2018).
Levine, A. J. Reviewing the future of the P53 field. Cell Death Differ. 25, 1–2 (2018).
Lawrence, M. S. et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
Kim, M. P. & Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 25, 161–168 (2018).
Castellsagué, X. et al. HPV involvement in head and neck cancers: comprehensive assessment of biomarkers in 3680 patients. J. Natl. Cancer Inst. 108, djv403 (2016).
Wu, D. & Prives, C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 25, 169–179 (2018).
Si, H. et al. TNF-alpha modulates genome-wide redistribution of DeltaNp63alpha/TAp73 and NF-kappaB cREL interactive binding on TP53 and AP-1 motifs to promote an oncogenic gene program in squamous cancer. Oncogene 35, 5781–5794 (2016).
Nemajerova, A. et al. Non-oncogenic roles of TAp73: from multiciliogenesis to metabolism. Cell Death Differ. 25, 144–153 (2018).
Rothenberg, S. M. & Ellisen, L. W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Invest 122, 1951–1957 (2012).
Rocco, J. W. & Ellisen, L. W. p63 and p73: life and death in squamous cell carcinoma. Cell Cycle 5, 936–940 (2006).
Rocco, J. W., Leong, C. O., Kuperwasser, N., DeYoung, M. P. & Ellisen, L. W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9, 45–56 (2006).
Danilov, A. V. et al. DeltaNp63alpha-mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PLoS One 6, e26815 (2011).
Compagnone, M. et al. DeltaNp63-mediated regulation of hyaluronic acid metabolism and signaling supports HNSCC tumorigenesis. Proc. Natl Acad. Sci. USA 114, 13254–13259 (2017).
Klanrit, P. et al. PML involvement in the p73-mediated E1A-induced suppression of EGFR and induction of apoptosis in head and neck cancers. Oncogene 28, 3499–3512 (2009).
Lu, H. et al. TNF-alpha promotes c-REL/Delta Np63 alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res. 71, 6867–6877 (2011).
Kang, H., Kiess, A. & Chung, C. H. Emerging biomarkers in head and neck cancer in the era of genomics. Nat. Rev. Clin. Oncol. 12, 11–26 (2015).
Gipson, B. J., Robbins, H. A., Fakhry, C. & D’Souza, G. Sensitivity and specificity of oral HPV detection for HPV-positive head and neck cancer. Oral. Oncol. 77, 52–56 (2018).
Castellanos, M. R. & Pan, Q. Novel p53 therapies for head and neck cancer. World J. Otorhinolaryngol. - head. neck Surg. 2, 68–75 (2016).
Study of COTI-2 for the treatment of gynecologic malignancies and head and neck squamous cell carcinoma. ClinicalTrials.gov (2018). Critical Outcome Technologies Inc.
Nowell, C. S. & Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 17, 145 (2017).
Sun, W. Y. et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 74, 1091–1104 (2014).
Zhao, Z. L. et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 6, 12 (2016).
Lee, S. H. et al. Notch1 signaling contributes to sternness in head and neck squamous cell carcinoma. Lab. Investig. 96, 508–516 (2016).
Terwilliger, T. & Abdul-Hay, M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 7, 12 (2017).
Kwon, C. et al. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat. Cell Biol. 13, 1244–U1169 (2011).
Agrawal, N. et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333, 1154–1157 (2011).
Ishida, T., Hijioka, H., Kume, K., Miyawaki, A. & Nakamura, N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol. Lett. 6, 1201–1206 (2013).
Song, X. M. et al. Common and complex Notch1 mutations in chinese oral squamous cell carcinoma. Clin. Cancer Res. 20, 701–710 (2014).
Nyman, P. E., Buehler, D. & Lambert, P. F. Loss of function of canonical Notch signaling drives head and neck carcinogenesis. Clin. Cancer Res. 24, 6308–6318 (2018).
Kozakiewicz, P. & Grzybowska-Szatkowska, L. Application of molecular targeted therapies in the treatment of head and neck squamous cell carcinoma. Oncol. Lett. 15, 7497–7505 (2018).
Zhao, Y. Y., Yu, G. T., Xiao, T. & Hu, J. The Notch signaling pathway in head and neck squamous cell carcinoma: a meta-analysis. Adv. Clin. Exp. Med. 26, 881–887 (2017).
Vander Broek, R., Mohan, S., Eytan, D. F., Chen, Z. & Van Waes, C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral. Dis. 21, 815–825 (2015).
Wang, Z., Valera, J. C., Zhao, X., Chen, Q. & Silvio Gutkind, J. mTOR co-targeting strategies for head and neck cancer therapy. Cancer Metastas-. Rev. 36, 491–502 (2017).
Pfisterer, K. et al. PI3K/PTEN/AKT/mTOR polymorphisms: association with clinical outcome in patients with head and neck squamous cell carcinoma receiving cetuximab-docetaxel. Head Neck 37, 471–478 (2015).
Swick, A. D. et al. Cotargeting mTORC and EGFR signaling as a therapeutic strategy in HNSCC. Mol. Cancer Ther. 16, 1257–1268 (2017).
Solomon, B., Young, R. J. & Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2018.01.008 (2018).
Chung, C. H. et al. Genomic alterations in human epidermal growth factor receptor 2 (HER2/ERBB2) in head and neck squamous cell carcinoma. Head Neck 39, E15–E19 (2017).
Sigismund, S., Avanzato, D. & Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 12, 3–20 (2018).
Brand, T. M. et al. Nuclear EGFR as a molecular target in cancer. Radio. Oncol. 108, 370–377 (2013).
Vouri, M. et al. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis 5, 10 (2016).
Azoury, S. C., Gilmore, R. C. & Shukla, V. Molecularly targeted agents and immunotherapy for the treatment of head and neck squamous cell cancer (HNSCC). Discov. Med. 21, 507–516 (2016).
Redlich, N. et al. Anti-Trop2 blockade enhances the therapeutic efficacy of ErbB3 inhibition in head and neck squamous cell carcinoma. Cell Death Dis. 9, 5 (2018).
Yamaoka, T., Ohba, M. & Ohmori, T. Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. Int. J. Mol. Sci. 18, 2420–2442 (2017).
Brand, T. M. et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin. Cancer Res. 21, 2601–2612 (2015).
Leonard, B. et al. BET inhibition overcomes receptor tyrosine kinase-mediated cetuximab resistance in HNSCC. Cancer Res. https://doi.org/10.1158/0008-5472.can-18-0459 (2018).
Hartmann, S. et al. Erlotinib and gefitinib responsiveness in head and neck cancer cell lines–a comparing analysis with cetuximab. Clin. Oral. Invest. 20, 759–769 (2016).
Specenier, P. & Vermorken, J. Afatinib in squamous cell carcinoma of the head and neck. Expert Opin. Pharmacother. 17, 1295–1301 (2016).
De Pauw, I. et al. Simultaneous targeting of EGFR, HER2, and HER4 by afatinib overcomes intrinsic and acquired cetuximab resistance in head and neck squamous cell carcinoma cell lines. Mol. Oncol. 12, 830–854 (2018).
Cohen, E. E. W. et al. Biomarkers predict enhanced clinical outcomes with afatinib versus methotrexate in patients with second-line recurrent and/or metastatic head and neck cancer. Ann. Oncol. 28, 2526–2532 (2017).
Madoz-Gúrpide, J. et al. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J. Transl. Med. 13, 282 (2015).
Cho, Y. A. et al. Alteration status and prognostic value of MET in head and neck squamous cell carcinoma. J. Cancer 7, 2197–2206 (2016).
Szturz, P. et al. Understanding c-MET signalling in squamous cell carcinoma of the head & neck. Crit. Rev. Oncol. Hematol. 111, 39–51 (2017).
Rothenberger, N. J. & Stabile, L. P. Hepatocyte growth factor/c-Met signaling in head and neck cancer and implications for treatment. Cancers 9, https://doi.org/10.3390/cancers9040039 (2017).
Arnold, L., Enders, J. & Thomas, S. M. Activated HGF-c-Met axis in head and neck cancer. Cancers 9, https://doi.org/10.3390/cancers9120169 (2017).
Study of INC280 in patients with c-MET dependent advanced solid tumors. ClinicalTrials.gov (2018). Novartis Pharmaceuticals.
Ficlatuzumab and cetuximab in recurrent/metastatic head and neck squamous cell carcinoma (HNSCC). ClinicalTrials.gov (2018). J. J. Lee, University of Pittsburgh.
Geiger, J. L., Grandis, J. R. & Bauman, J. E. The STAT3 pathway as a therapeutic target in head and neck cancer: barriers and innovations. Oral. Oncol. 56, 84–92 (2016).
Wang, Y. et al. Suppression of the growth and invasion of human head and neck squamous cell carcinomas via regulating STAT3 signaling and the miR-21/beta-catenin axis with HJC0152. Mol. Cancer Ther. 16, 578–590 (2017).
Avalle, L. et al. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2 + ) fluxes and apoptotic responses. Cell Death Differ, https://doi.org/10.1038/s41418-018-0171-y (2018).
Seo, S. U. et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression. Cell Death Dis. 8, e2599 (2017).
Bu, L. L. et al. STAT3 induces immunosuppression by upregulating PD-1/PD-L1 in HNSCC. J. Dent. Res. 96, 1027–1034, https://doi.org/10.1177/0022034517712435 (2017).
Yu, H., Lee, H., Herrmann, A., Buettner, R. & Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746 (2014).
Study to assess MEDI4736 with either AZD9150 or AZD5069 in advanced solid tumors & relapsed metastatic squamous cell carcinoma of head & neck. ClinicalTrials.gov (2018). AstraZeneca.
Chiba, T. et al. MALT1 inhibition of oral carcinoma cell invasion and ERK/MAPK activation. J. Dent. Res. 95, 446–452 (2016).
Peng, Q. et al. Mitogen-activated protein kinase signaling pathway in oral cancer. Oncol. Lett. 15, 1379–1388 (2018).
De Carvalho, T. G. et al. Search for mutations in signaling pathways in head and neck squamous cell carcinoma. Oncol. Rep. 30, 334–340 (2013).
A study to determine the relative bioavailability of the MEK inhibitor, trametinib, in subjects with solid tumor malignancies. ClinicalTrials.gov (2018). GlaxoSmithKline.
Ngan, H. L., Wang, L., Lo, K. W. & Lui, V. W. Y. Genomic landscapes of EBV-associated nasopharyngeal carcinoma vs. HPV-associated head and neck cancer. Cancers 10, https://doi.org/10.3390/cancers10070210 (2018).
Zhang, W., Edwards, A., Fang, Z., Flemington, E. K. & Zhang, K. Integrative genomics and transcriptomics analysis reveals potential mechanisms for favorable prognosis of patients with HPV-positive head and neck carcinomas. Sci. Rep. 6, 24927 (2016).
Coordes, A. et al. Meta-analysis of survival in patients with HNSCC discriminates risk depending on combined HPV and p16 status. Eur. Arch. Oto-Rhino-Laryngol. 273, 2157–2169 (2016).
Faraji, F., Zaidi, M., Fakhry, C. & Gaykalova, D. A. Molecular mechanisms of human papillomavirus-related carcinogenesis in head and neck cancer. Microbes Infect. 19, 464–475 (2017).
Um, S. H. et al. Variable expression of the forgotten oncogene E5 in HPV-positive oropharyngeal cancer. J. Clin. Virol. 61, 94–100 (2014).
Yan, L., Zhan, C., Wu, J. & Wang, S. Expression profile analysis of head and neck squamous cell carcinomas using data from The Cancer Genome Atlas. Mol. Med Rep. 13, 4259–4265 (2016).
Hayes, D. N., Van Waes, C. & Seiwert, T. Y. Genetic landscape of human papillomavirus-associated head and neck cancer and comparison to tobacco-related tumors. J. Clin. Oncol. 33, 3227–3234 (2015).
De Cecco, L. et al. Head and neck cancer subtypes with biological and clinical relevance: meta-analysis of gene-expression data. Oncotarget 6, 9627–9642 (2015).
Martin, L. M. et al. DNA mismatch repair and the DNA damage response to ionizing radiation: making sense of apparently conflicting data. Cancer Treat. Rev. 36, 518–527 (2010).
Tonella, L., Giannoccaro, M., Alfieri, S., Canevari, S. & De Cecco, L. Gene expression signatures for head and neck cancer patient stratification: are results ready for clinical application? Curr. Treat. options Oncol. 18, 32 (2017).
Weekly Cetuximab/RT versus weekly cisplatin/RT in HPV-associated oropharyngeal squamous cell carcinoma. ClinicalTrials.gov (2018). Trans-Tasman Radiation Oncology Group (TROG).
Gillison, M. L. et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet 393, 40–50 (2019).
Mehanna, H. et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): an open-label randomised controlled phase 3 trial. Lancet, https://doi.org/10.1016/S0140-6736(18)32752-1 (2018).
Determination of cetuximab versus cisplatin early and late toxicity events in HPV + OPSCC. ClinicalTrials.gov (2018). J. Dunn, University of Warwick.
Mirghani, H. & Blanchard, P. Treatment de-escalation for HPV-driven oropharyngeal cancer: where do we stand? Clin. Transl. Radiat. Oncol. 8, 4–11, https://doi.org/10.1016/j.ctro.2017.10.005 (2018).
Sepiashvili, L. et al. Novel insights into head and neck cancer using next-generation “omic” technologies. Cancer Res. 75, 480–486, https://doi.org/10.1158/0008-5472.can-14-3124 (2015).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Kolesnikov, N. et al. ArrayExpress update–simplifying data submissions. Nucleic Acids Res. 43, D1113–D1116, https://doi.org/10.1093/nar/gku1057 (2015).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419, https://doi.org/10.1126/science.1260419 (2015).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357, https://doi.org/10.1126/science.aan2507 (2017).
Weinstein, J. N. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Wichmann, G. et al. The role of HPV RNA transcription, immune response-related gene expression and disruptive TP53 mutations in diagnostic and prognostic profiling of head and neck cancer. Int. J. Cancer 137, 2846–2857 (2015).
Shin, J. M., Kamarajan, P., Fenno, J. C., Rickard, A. H. & Kapila, Y. L. Metabolomics of Head and Neck. Cancer.: A Mini-Rev. Front. Physiol. 7, 526 (2016).
Lee, B. K. B. et al. GENIPAC: a genomic information portal for head and neck cancer cell systems. J. Dent. Res. 97, 909–916 (2018).
Overgaard, J., Jovanovic, A., Godballe, C. & Grau Eriksen, J. The Danish head and neck cancer database. Clin. Epidemiol. 8, 491–496 (2016).
Cooper, J. S. et al. National cancer database report on cancer of the head and neck: 10-year update. Head Neck 31, 748–758 (2009).
Henrique, T. et al. HNdb: an integrated database of gene and protein information on head and neck squamous cell carcinoma. Database (Oxford) 2016, 1–11 (2016).
Yeom, C. J., Goto, Y., Zhu, Y. X., Hiraoka, M. & Harada, H. Microenvironments and cellular characteristics in the micro tumor cords of malignant solid tumors. Int. J. Mol. Sci. 13, 13949–13965 (2012).
Mistry, I. N., Thomas, M., Calder, E. D. D., Conway, S. J. & Hammond, E. M. Clinical advances of hypoxia-activated prodrugs in combination with radiation therapy. Int. J. Radiat. Oncol.*Biol. *Phys. 98, 1183–1196 (2017).
Petrova, V., Annicchiarico-Petruzzelli, M., Melino, G. & Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 7, 10 (2018).
Gammon, L. & Mackenzie, I. C. Roles of hypoxia, stem cells and epithelial-mesenchymal transition in the spread and treatment resistance of head and neck cancer. J. Oral. Pathol. Med. 45, 77–82 (2016).
Koukourakis, M. I. et al. Hypoxia-regulated carbonic anhydrase-9 (CA9) relates to poor vascularization and resistance of squamous cell head and neck cancer to chemoradiotherapy. Clin. Cancer Res. 7, 3399–3403 (2001).
Chen, C. H., Pore, N., Behrooz, A., Ismail-Beigi, F. & Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1-interaction between H-ras and hypoxia. J. Biol. Chem. 276, 9519–9525 (2001).
Tsai, Y. P. & Wu, K. J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 19, 7 (2012).
Eltzschig, H. K. & Carmeliet, P. Mechanisms of disease: hypoxia and inflammation. New Engl. J. Med. 364, 656–665 (2011).
Amelio, I. et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl Acad. Sci. USA 115, E10869–E10878 (2018).
Amelio, I. & Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem Sci. 40, 425–434 (2015).
Swartz, J. E., Pothen, A. J., Stegeman, I., Willems, S. M. & Grolman, W. Clinical implications of hypoxia biomarker expression in head and neck squamous cell carcinoma: a systematic review. Cancer Med. 4, 1101–1116 (2015).
Graham, K. & Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 13, 6049–6058 (2018).
Overgaard, J. et al. Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol. 6, 757–764 (2005).
NIMRAD (A Randomised Placebo-controlled Trial of Synchronous NIMorazole Versus RADiotherapy Alone in Patients With Locally Advanced Head and Neck Squamous Cell Carcinoma Not Suitable for Synchronous Chemotherapy or Cetuximab) (NIMRAD), https://clinicaltrials.gov/ct2/show/NCT01950689 (2018).
Bredell, M. G. et al. In: Andrei B. Gudkov, Mikhail V Blagosklonny (eds) Current relevance of hypoxia in head and neck cancer. Oncotarget, 7, 50781–50804 (2016), Impact Journals, Orchard Park, New York, USA.
Sethi, A., Mao, W., Wordinger, R. J. & Clark, A. F. Transforming growth factor–β induces extracellular matrix protein cross-linking lysyl oxidase (LOX) genes in human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 52, 5240–5250 (2011).
Tawk, B. et al. Comparative analysis of transcriptomics based hypoxia signatures in head- and neck squamous cell carcinoma. Radio. Oncol. 118, 350–358 (2016).
Suh, Y.-E. et al. Association between hypoxic volume and underlying hypoxia-induced gene expression in oropharyngeal squamous cell carcinoma (OPSCC). Br. J. Cancer 116, 1057–1064 (2017).
Toustrup, K. et al. Validation of a 15-gene hypoxia classifier in head and neck cancer for prospective use in clinical trials. Acta Oncol. 55, 1091–1098 (2016).
van Dijk, L. K., Boerman, O. C., Kaanders, J. & Bussink, J. PET imaging in head and neck cancer patients to monitor treatment response: a future role for EGFR-targeted imaging. Clin. Cancer Res. 21, 3602–3609 (2015).
Rischin, D. et al. Prognostic significance of F-18 -misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group study 98.02. J. Clin. Oncol. 24, 2098–2104 (2006).
Ling, D. C., Bakkenist, C. J., Ferris, R. L. & Clump, D. A. Role of immunotherapy in head and neck cancer. Semin. Radiat. Oncol. 28, 12–16 (2018).
de Ruiter, E. J., Ooft, M. L., Devriese, L. A. & Willems, S. M. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: a systematic review and meta-analysis. Oncoimmunology 6, e1356148 (2017).
Outh-Gauer, S. et al. Immunotherapy in head and neck cancers: a new challenge for immunologists, pathologists and clinicians. Cancer Treat. Rev. 65, 54–64 (2018).
Begg, K. & Tavassoli, M. Review: Biomarkers towards personalised therapy in cancer, https://www.drugtargetreview.com/article/23631/biomarkers-personalised-therapy-cancer/ (2017).
Fesnak, A. D., June, C. H. & Levine, B. L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581 (2016).
Brentjens, R. J. et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra138 (2013).
Yong, C. S. M. et al. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 95, 356–363 (2017).
van Schalkwyk, M. C. et al. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum. Gene Ther. Clin. Dev. 24, 134–142 (2013).
Kareemaghay, S. & Tavassoli, M. Clinical immunotherapeutic approaches for the treatment of head and neck cancer. Int. J. Oral and Maxillofac. Surg., https://doi.org/10.1016/j.ijom.2018.10.012 (2018).
Dong, M. et al. Diosgenin promotes antitumor immunity and PD-1 antibody efficacy against melanoma by regulating intestinal microbiota. Cell Death Dis. 9, 1039 (2018).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Patel, R. et al. Culling of APCs by inflammatory cell death pathways restricts TIM3 and PD-1 expression and promotes the survival of primed CD8 T cells. Cell Death Differ. 24, 1900–1911 (2017).
Dyck, L. & Mills, K. H. G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 47, 765–779 (2017).
Ott, P. A. & Hodi, F. S. The B7-H1/PD-1 pathway in cancers associated with infections and inflammation: opportunities for therapeutic intervention. Chin. Clin. Oncol. 2, 7 (2013).
McDermott, D. F. & Atkins, M. B. PD-1 as a potential target in cancer therapy. Cancer Med. 2, 662–673 (2013).
Wang, Y., Wu, L., Tian, C. & Zhang, Y. Z. PD-1-PD-L1 immune-checkpoint blockade in malignant lymphomas. Ann. Hematol. 97, 229–237 (2018).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 363, 711–723 (2010).
Guan, X. et al. Blockade of Na/H exchanger stimulates glioma tumor immunogenicity and enhances combinatorial TMZ and anti-PD-1 therapy. Cell Death Dis. 9, 1010 (2018).
Ferris, R. L. et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. New Engl. J. Med. 375, 1856–1867 (2016).
Seiwert, T. Y. et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 17, 956–965 (2016).
Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 4, 62–62 (2014).
Dvorak, H. F., Flier, J. & Frank, H. Tumors—wounds that do not heal—similarities between tumor stroma generation and wound-healing. New Engl. J. Med. 315, 1650–1659 (1986).
Barker, H. E., Paget, J. T. E., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015).
Shiga, K. et al. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers 7, 2443–2458 (2015).
Utispan, K. & Koontongkaew, S. Fibroblasts and macrophages: key players in the head and neck cancer microenvironment. J. Oral. Biosci. 59, 23–30 (2017).
Peltanova, B., Raudenska, M. & Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol. Cancer 18, 24 (2019).
Takahashi, H. et al. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol. Immunother. 64, 1407–1417 (2015).
Psaila, B. & Lyden, D. The metastatic niche: adapting the foreign soil. Nat. Rev. Cancer 9, 285–293 (2009).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423 (2013).
Chen, X. M. & Song, E. W. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 18, 99–115 (2019).
Jung, D. W. et al. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int. J. Cancer 127, 332–344 (2010).
Ji, X. Q., Zhu, X. X. & Lu, X. G. Effect of cancer-associated fibroblasts on radiosensitivity of cancer cells. Future Oncol. 13, 1537–1550 (2017).
Strom, T. et al. Tumour radiosensitivity is associated with immune activation in solid tumours. Eur. J. Cancer 84, 304–314 (2017).
Huelsken, J. & Hanahan, D. A subset of cancer-associated fibroblasts determines therapy resistance. Cell 172, 643–644 (2018).
Johansson, A. C. et al. Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol. Cancer Res. 10, 1158–1168 (2012).
Zhang, H. F. et al. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 8, 13 (2017).
Zhang, H. et al. Cancer-associated Fibroblast–promoted LncRNA < em > DNM3OS < /em > Confers Radioresistance by Regulating DNA Damage Response in Esophageal Squamous Cell Carcinoma. Clin. Cancer Res., https://doi.org/10.1158/1078-0432.CCR-18-0773 (2018).
Schmitz, S., Bindea, G., Albu, R. I., Mlecnik, B. & Machiels, J. P. Cetuximab promotes epithelial to mesenchymal transition and cancer associated fibroblasts in patients with head and neck cancer. Oncotarget 6, 34288–34299 (2015).
Berndt, A. et al. Effects of activated fibroblasts on phenotype modulation, EGFR signalling and cell cycle regulation in OSCC cells. Exp. Cell Res. 322, 402–414 (2014).
Scott, A. M. et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 9, 1639 (2003).
Kahlert, C. & Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med.-JMM 91, 431–437 (2013).
Qin, X. et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 20, 12 (2019).
Cirillo, N. et al. Cancer-associated fibroblasts regulate keratinocyte cell–cell adhesion via TGF-β-dependent pathways in genotype-specific oral cancer. Carcinogenesis 38, 76–85 (2016).
Fitzgerald, A. L. et al. Reactive oxygen species and p21(Waf1/Cip1) are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 6, 10 (2015).
White, R. A., Malkoski, S. P. & Wang, X. J. TGFβ signaling in head and neck squamous cell carcinoma. Oncogene 29, 5437–5446 (2010).
Marullo, R. et al. HPV16 E6 and E7 proteins induce a chronic oxidative stress response via NOX2 that causes genomic instability and increased susceptibility to DNA damage in head and neck cancer cells. Carcinogenesis 36, 1397–1406 (2015).
Zhu, G. et al. Hypoxia promotes migration/invasion and glycolysis in head and neck squamous cell carcinoma via an HIF-1alpha-MTDH loop. Oncol. Rep. 38, 2893–2900 (2017).
Hess, A. K. et al. MiR-200b and miR-155 as predictive biomarkers for the efficacy of chemoradiation in locally advanced head and neck squamous cell carcinoma. Eur. J. Cancer 77, 3–12 (2017).
Hersi, H. M., Raulf, N., Gaken, J., Folarin, N. & Tavassoli, M. MicroRNA-9 inhibits growth and invasion of head and neck cancer cells and is a predictive biomarker of response to plerixafor, an inhibitor of its target CXCR4. Mol. Oncol. 12, 2023–2041 (2018).
Tsai, M. S., Chen, W. C., Lu, C. H. & Chen, M. F. The prognosis of head and neck squamous cell carcinoma related to immunosuppressive tumor microenvironment regulated by IL-6 signaling. Oral. Oncol. 91, 47–55 (2019).
Karakasheva, T. A. et al. IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 78, 4957–4970 (2018).
LeBleu, V. S. & Kalluri, R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Models Mech. 11, dmm029447 (2018).
Vargas-Ferreira, F. et al. Etiologic factors associated with oral squamous cell carcinoma in non-smokers and non-alcoholic drinkers: a brief approach. Braz. Dent. J. 23, 586–590 (2012).
Banerjee, S. et al. Microbial signatures associated with oropharyngeal and oral squamous cell carcinomas. Sci. Rep. 7, 20 (2017).
Perera, M., Al-Hebshi, N. N., Speicher, D. J., Perera, I. & Johnson, N. W. Emerging role of bacteria in oral carcinogenesis: a review with special reference to perio-pathogenic bacteria. J. Oral. Microbiol. 8, 10 (2016).
Rizzetto, L., De Filippo, C. & Cavalieri, D. Richness and diversity of mammalian fungal communities shape innate and adaptive immunity in health and disease. Eur. J. Immunol. 44, 3166–3181 (2014).
Smith, A. J., Jackson, M. S. & Bagg, J. The ecology of Staphylococcus species in the oral cavity. J. Med. Microbiol. 50, 940–946 (2001).
Rajeev, R., Choudhary, K., Panda, S. & Gandhi, N. Role of bacteria in oral carcinogenesis. South Asian J. cancer 1, 78–83 (2012).
Meurman, J. H. Oral microbiota and cancer. Journal of oral microbiology. 2, 10.3402/jom.v3402i3400.5195, https://doi.org/10.3402/jom.v2i0.5195 (2010).
Mao, W. M., Zheng, W. H. & Ling, Z. Q. Epidemiologic risk factors for esophageal cancer development. Asian Pac. J. Cancer Prev. 12, 2461–2466 (2011).
Le Bars, P. et al. The oral cavity microbiota: between health, oral disease, and cancers of the aerodigestive tract. Can. J. Microbiol. 63, 475–492 (2017).
Wang, H. N. et al. Microbiomic differences in tumor and paired-normal tissue in head and neck squamous cell carcinomas. Genome Med. 9, 10 (2017).
Mager, D. L. et al. The salivary microbiota as a diagnostic indicator of oral cancer: a descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J. Transl. Med. 3, 8 (2005).
Ling, L. J. et al. Association between betel quid chewing, periodontal status and periodontal pathogens. Oral. Microbiol. Immunol. 16, 364–369 (2001).
Amaral, C. D. F., da Silva-Boghossian, C. M., Leao, A. T. T. & Colombo, A. P. V. Evaluation of the subgingival microbiota of alcoholic and non-alcoholic individuals. J. Dent. 39, 729–738 (2011).
Gomes, S. C. et al. Periodontal status in smokers and never-smokers: clinical findings and real-time polymerase chain reaction quantification of putative periodontal pathogens. J. Periodontol. 77, 1483–1490 (2006).
Bornigen, D. et al. Alterations in oral bacterial communities are associated with risk factors for oral and oropharyngeal cancer. Sci. Rep. 7, 13 (2017).
Marttila, E. et al. Acetaldehyde production and microbial colonization in oral squamous cell carcinoma and oral lichenoid disease. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 116, 61–68 (2013).
Kurkivuori, J. et al. Acetaldehyde production from ethanol by oral streptococci. Oral. Oncol. 43, 181–186 (2007).
Tao, L., Pavlova, S. I., Gasparovich, S. R., Jin, L. & Schwartz, J. Advances in experimental medicine and biology. in Biological Basis of Alcohol-Induced Cancer, Vol. 815 (eds Vasiliou, V., Zakhari, S., Seitz, H. K. & Hoek, J. B.) 239–264 (Springer-Verlag, Berlin, 2015).
Cruz, L., Biryukov, J., Conway, M. J. & Meyers, C. Cleavage of the HPV16 minor capsid protein L2 during virion morphogenesis ablates the requirement for cellular furin during de novo. Infect. Virus.-Basel 7, 5813–5830 (2015).
Bronnimann, M. P. et al. Furin cleavage of L2 during Papillomavirus infection: minimal dependence on Cyclophilins. J. Virol. 90, 6224–6234 (2016).
Pavlova, S. I. et al. Streptococcus endopeptidases promote HPV infection in vitro. Microbiologyopen 8, 14 (2019).
Gallimidi, A. B. et al. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 6, 22613–22623 (2015).
Gallo, C. D. et al. Toll-like receptor 2rs4696480 polymorphism and risk of oral cancer and oral potentially malignant disorder. Arch. Oral. Biol. 82, 109–114 (2017).
Imamura, Y. et al. Polymorphism of genes encoding toll-like receptors and inflammatory cytokines in periodontal disease in the Japanese population. J Int Acad Periodontol. 10, 95–102 (2008).
Kinane, D. F. et al. Gingival epithelial cells heterozygous for Toll-like receptor 4 polymorphisms Asp299Gly and Thr399lle are hypo-responsive to Porphyromonas gingivalis. Genes Immun. 7, 190–200 (2006).
Groeger, S., Domann, E., Gonzales, J. R., Chakraborty, T. & Meyle, J. B7-H1 and B7-DC receptors of oral squamous carcinoma cells are upregulated by Porphyromonas gingivalis. Immunobiology 216, 1302–1310 (2011).
Groeger, S., Jarzina, F., Mamat, U. & Meyle, J. Induction of B7-H1 receptor by bacterial cells fractions of Porphyromonas gingivalis on human oral epithelial cells: B7-H1 induction by Porphyromonas gingivalis fractions. Immunobiology 222, 137–147 (2017).
Zhu, W. D. et al. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc. Natl Acad. Sci. USA 109, 14194–14199 (2012).
Solis, N. V., Swidergall, M., Bruno, V. M., Gaffen, S. L. & Filler, S. G. The Aryl hydrocarbon receptor governs epithelial cell invasion during oropharyngeal candidiasis. Mbio 8, 16 (2017).
Boonanantanasarn, K. et al. Enterococcus faecalis enhances cell proliferation through hydrogen peroxide-mediated epidermal growth factor receptor activation. Infect. Immun. 80, 3545–3558 (2012).
Yang, S. F. et al. Compositional and functional variations of oral microbiota associated with the mutational changes in oral cancer. Oral. Oncol. 77, 1–8 (2018).
Vasconcelos, R. M. et al. Host-microbiome cross-talk in oral mucositis. J. Dent. Res. 95, 725–733 (2016).
Almstahl, A., Wikstrom, M., Stenberg, I., Jakobsson, A. & Fagerberg-Mohlin, B. Oral microbiota associated with hyposalivation of different origins. Oral. Microbiol. Immunol. 18, 1–8 (2003).
Kuten, A. et al. Oral side-effects of head and neck irradiation - correlation between clinical manifestations and laboratory data. Int. J. Radiat. Oncol. Biol. Phys. 12, 401–405 (1986).
Shao, Z. Y. et al. Effects of intensity-modulated radiotherapy on human oral microflora. J. Radiat. Res. 52, 834–839 (2011).
Schuurhuis, J. M. et al. Head and neck intensity modulated radiation therapy leads to an increase of opportunistic oral pathogens. Oral. Oncol. 58, 32–40 (2016).
Jia, G. et al. The oral microbiota - a mechanistic role for systemic diseases. Br. Dent. J. 224, 447–455 (2018).
Nagpal, R. et al. Probiotics, their health benefits and applications for developing healthier foods: a review. Fems Microbiol. Lett. 334, 1–15 (2012).
Dzidic, M. et al. Oral microbiome development during childhood: an ecological succession influenced by postnatal factors and associated with tooth decay. Isme J. 12, 2292–2306 (2018).
Zhang, Y. H. et al. Human oral microbiota and its modulation for oral health. Biomed. Pharmacother. 99, 883–893 (2018).
Mahasneh, S. A. & Mahasneh, A. M. Probiotics: a promising role in dental health. Dent. J. 5, 26 (2017).
Rajpoot, M., Sharma, A. K., Sharma, A. & Gupta, G. K. Understanding the microbiome: emerging biomarkers for exploiting the microbiota for personalized medicine against cancer. Semin. Cancer Biol. 52, 1–8 (2018).
Liu, Z. H. et al. The effects of perioperative probiotic treatment on serum zonulin concentration and subsequent postoperative infectious complications after colorectal cancer surgery: a double-center and double-blind randomized clinical trial. Am. J. Clin. Nutr. 97, 117–126 (2013).
Calaca, P. R. D., Bezerra, R. P., Albuquerque, W. W. C., Porto, A. L. F. & Cavalcanti, M. T. H. Probiotics as a preventive strategy for surgical infection in colorectal cancer patients: a systematic review and meta-analysis of randomized trials. Transl. Gastroenterol. Hepatol. 2, 10 (2017).
Li, J. et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc. Natl Acad. Sci. USA 113, E1306–E1315 (2016).
Kuugbee, E. D. et al. Structural change in microbiota by a probiotic cocktail enhances the gut barrier and reduces cancer via TLR2 signaling in a rat model of colon cancer. Dig. Dis. Sci. 61, 2908–2920 (2016).
Kumar, R. & Dhanda, S. Mechanistic insight of probiotics derived anticancer pharmaceuticals: a road forward for cancer therapeutics. Nutr. Cancer- Int. J. 69, 375–380 (2017).
Motevaseli, E., Dianatpour, A. & Ghafouri-Fard, S. The role of probiotics in cancer treatment: emphasis on their in vivo and in vitro anti-metastatic effects. Int. J. Mol. Cell. Med. 6, 66–76 (2017).
Yu, A. Q. & Li, L. Q. The potential role of probiotics in cancer prevention and treatment. Nutr. Cancer- Int. J. 68, 535–544 (2016).
Patel, S. & Goyal, A. Evolving roles of probiotics in cancer prophylaxis and therapy. Probiotics Antimicrob. Proteins 5, 59–67 (2013).
Sharma, A. et al. Lactobacillus brevis CD2 lozenges prevent oral mucositis in patients undergoing high dose chemotherapy followed by haematopoietic stem cell transplantation. Esmo Open 1, 7 (2016).
Sharma, A. et al. Lactobacillus brevis CD2 lozenges reduce radiation- and chemotherapy-induced mucositis in patients with head and neck cancer: a randomized double-blind placebo-controlled study. Eur. J. Cancer 48, 875–881 (2012).
Zarco, M. F., Vess, T. J. & Ginsburg, G. S. The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral. Dis. 18, 109–120 (2012).
Acknowledgements
We would like to thank the following funding organisations, E Alsahafi funded by Umm Al-Qura University, Faculty of Dentistry, Mekkah City, Saudi Arabia. Katheryn Begg funded by the NIHR Biomedical Research Centre and King’s College London and Nina Raulf by a grant from the Rosetrees Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Peschiaroli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alsahafi, E., Begg, K., Amelio, I. et al. Clinical update on head and neck cancer: molecular biology and ongoing challenges. Cell Death Dis 10, 540 (2019). https://doi.org/10.1038/s41419-019-1769-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1769-9
This article is cited by
-
Analysis of the effect of CCR7 on the microenvironment of mouse oral squamous cell carcinoma by single-cell RNA sequencing technology
Journal of Experimental & Clinical Cancer Research (2024)
-
IFIT3 accelerates the progression of head and neck squamous cell carcinoma by targeting PD-L1 to activate PI3K/AKT signaling pathway
World Journal of Surgical Oncology (2024)
-
Comparative response to PDT with methyl-aminolevulinate and temoporfin in cutaneous and oral squamous cell carcinoma cells
Scientific Reports (2024)
-
Single-cell RNA sequencing explores the evolution of the ecosystem from leukoplakia to head and neck squamous cell carcinoma
Scientific Reports (2024)
-
Effect of radiotherapy on head and neck cancer tissues in patients receiving radiotherapy: a bioinformatics analysis-based study
Scientific Reports (2024)
