
Abstract
SE translocation (SET), an inhibitor of protein phosphatase 2A (PP2A), plays important roles in mitosis and possesses oncogenic activity in several types of cancer. However, little is known regarding its regulation. Here we reveal a novel phosphorylation site of SET isoform 1, and we have determined its biological significance in tumorigenesis. We found that the mitotic kinase cyclin-dependent kinase 1 (CDK1) phosphorylates SET isoform 1 in vitro and in vivo at serine 7 during antitubulin drug-induced mitotic arrest and normal mitosis. SET deletion resulted in massive multipolar spindles, chromosome misalignment and missegregation, and centrosome amplification during mitosis. Moreover, mitotic phosphorylation of SET isoform 1 is required for cell migration, invasion, and anchorage-independent growth in vitro and tumorigenesis in xenograft animal models. We further documented that SET phosphorylation affects Akt activity. Collectively, our findings suggest that SET isoform 1 promotes oncogenesis in a mitotic phosphorylation-dependent manner.
Similar content being viewed by others


Introduction
SET, also known as inhibitor 2 of protein phosphatase 2A (I2PP2A), directly binds to PP2A and inhibits its phosphatase activity1,2,3. PP2A plays a critical role in neoplastic transformation by negatively regulating many oncogenic signaling pathways, and it serves as a therapeutic target4,5. Accumulated evidence demonstrated that SET functions as an oncogene in several neoplasms and promotes tumorigenesis6,7. Overexpression of SET has been shown in breast cancer8, ovarian cancer9, leukemia10,11, lymphoma12, hepatocellular carcinoma13, prostate cancer14, colon cancer15, non-small-cell lung carcinoma16, and pancreatic cancer17. Furthermore, upregulation of SET correlates with poor clinical outcomes in hepatocellular carcinoma, ovarian cancer, and colorectal cancer13,15,18. In line with these clinical observations, SET silencing or pharmacological inhibition of SET significantly impaired tumor growth in various human malignancies6. Interestingly, ectopic expression of SET also promoted resistance to chemotherapeutics in colon cancer, nonsmall cell lung carcinoma, and hematologic malignancies10,11,13,15,19. Promisingly, several antagonists (e.g., FTY720, OP449, and EMQA) that target the SET–PP2A interface have been shown to inhibit tumor growth and overcome therapeutic resistance in various preclinical models10,11,20. In addition to its oncogenic roles, SET is also involved in various other molecular processes, including histone modification, chromatin remodeling, DNA repair, gene transcription, and androgen synthesis6,7,9. Mechanistically, SET has been shown to interact with several PP2A-regulated oncogenic pathways, including the Akt, mitogen-activated protein kinase, and BCR-ABL pathways6,10. However, SET may regulate cellular physiology through PP2A-independent pathways.
Although extensive studies have demonstrated the important roles for SET-PP2A signaling in tumorigenesis, the underlying mechanisms are less clear. Mitotic aberrations cause aneuploidy or genomic instability, which is a hallmark of human malignancy21,22. Therefore, mitosis has been a long-standing anticancer drug target23,24,25,26. Interestingly, SET has been linked to the mitotic machinery. For example, SET associates with cyclin B and inhibits cyclin B-CDK1 activity27,28. Knockdown (KD) of SET delayed mitotic progression and inhibited G2/M transition29. Moreover, primarily located in the nucleus, SET has been demonstrated to protect histones from acetylation, modulation of chromosome condensation, and cohesion30. Overexpression of SET resulted in precocious separation of chromatids in mouse oocytes31. SET is centromere localized29,31 and forms a complex with linker histones and shugoshins during mitosis29. Several studies demonstrated that SET mediates timely resolution of sister chromatids during mitosis and functions as a mitotic chaperone29,32. These studies suggest that SET might exert its oncogenic function through dysregulation of mitosis.
The human SET gene is located on chromosome 9q34 and contains 11 exons and 10 introns. It was originally identified as part of a fusion gene with nucleoporin Nup214 (CAN) in a patient with acute undifferentiated leukemia in 199233,34. There are four protein isoforms of SET and the only difference is in the first exon among them17,35. Isoforms 1 and 2 are the most thoroughly characterized ones17. SET is a phosphoprotein, and two sites (S9 and S24 in isoform 2) have been identified as phosphorylated by protein kinase C in vivo33. Furthermore, casein kinase 2-mediated phosphorylation at S9 causes cytoplasmic retention of SET and induces hyperphosphorylation of tau in Alzheimer disease36,37. However, the regulation of SET in mitosis and its possible role in cancer have remained unknown. Given the critical roles of SET in mitosis and oncogenesis, we sought to elucidate the regulatory mechanisms of SET in mitosis. We found that the mitotic kinase cyclin-dependent kinase 1 (CDK1) phosphorylates SET at S7 (isoform 1) during mitosis. Moreover, we showed that mitotic phosphorylation of SET is required for precise mitosis and oncogenic activity.
Materials and methods
Expression constructs
The full-length human SET cDNA clone (isoform 1, NM_001122821.1) was purchased from GeneCopoeia (EX-Z5816-M02-B, Rockville, MD, USA). A point mutation (Serine 7 to Alanine) was generated by the QuikChange Site-Directed PCR Mutagenesis Kit (Stratagene, La Jolla, CA, USA) and verified by sequencing. To make the lentivirus-mediated SET expression construct, the above cDNA was cloned into the pSIN4-Flag-IRES-neo vector. The pSIN4-Flag-IRES-neo vector was made by replacing the puromycin-coding sequence of the pSIN4-Flag-IRES-puro vector38 with a neomycin-coding sequence.
Cell culture
HEK293T, HeLa, and RKO cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines were authenticated at ATCC and were used at low (<20) passages. All other cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. HPNE (an immortalized human pancreatic nestin-expressing cell line) was kindly provided by Dr Michel Ouellette (University of Nebraska Medical Center), who originally established the cell line and deposited it at ATCC39. Attractene (Qiagen, Germantown, MD, USA) was used for transient overexpression transfections following the manufacturer’s instructions. Nocodazole (100 ng/ml for 16–20 h) and Taxol (100 nM for 16–20 h) were used to arrest cells at late G2 and prometaphase (G2/M), unless otherwise indicated. VX680 (Aurora-A, -B, -C inhibitor), BI2536 (Plk1 inhibitor), and MK2206 (Akt inhibitor) were purchased from Selleck Chemicals (Houston, TX, USA). U0126 (MEK1/2 inhibitor), SB203580 (p38 inhibitor), LY294002 (PI-3K inhibitor), rapamycin (mTOR inhibitor), and SP600125 (JNK1/2 inhibitor) were from LC Laboratories (Woburn, MA, USA). RO3306 (CDK1 inhibitor) and Purvalanol A (CDK1/2/5 inhibitor) were from ENZO Life Sciences (Farmingdale, NY, USA). MK5108 (Aurora-A inhibitor) and SB216763 (GSK3β inhibitor) were from Merck (Kenilworth, NJ, USA) and Sigma-Aldrich (Burlington, MA, USA), respectively.
Cell line establishment
Stable overexpression and re-expression of SET (wild-type and S7A mutant) in SET-KD cells were achieved by lentivirus-mediated infection and selection40. Gene KD was achieved by an shRNA-mediated method. The MISSION shRNA plasmids targeting human SET were purchased from Sigma-Aldrich (TRCN0000063717). To make the shRNA-resistant (Res) SET cDNA, the target sequence (5′-CCACCGAAATCAAATGGAAATCT-3′) was changed into 5′-CaACtGAgATCAAATGGAAATCT-3′ by PCR mutagenesis. The mutated SET cDNA was then cloned into the pSIN4-Flag-IRES-Neo vector to generate a Flag-tagged shRNA-resistant SET construct.
Recombinant protein purification and in vitro kinase assay
The glutathione S-transferase (GST)-tagged proteins were bacterially expressed and purified on GSTrap FF affinity columns (GE Healthcare, Chicago, IL, USA) following the manufacturer’s instructions. About 0.5 µg of GST-SET proteins were incubated with 10 U recombinant CDK1/cyclin B1 complex (New England Biolabs, Ipswich, MA, USA) in the presence of 5 µCi γ-32P-ATP (3000 Ci/mmol, PerkinElmer, Waltham, MA, USA). Purified CDK1/cyclin B1 complex from SignalChem (Richmond, BC, Canada) was also used for in vitro kinase assays using phospho-specific antibodies. The samples were resolved by SDS-PAGE, transferred onto PVDF (Millipore, Burlington, MA, USA), and visualized by autoradiography or detected by phospho-specific antibodies.
Antibodies
The anti-SET monoclonal antibody from Santa Cruz Biotechnology (SC133138, Dallas, TX, USA) was used throughout the study. Rabbit polyclonal phospho-specific antibody against human SET S7 (isoform 1) was generated and purified by AbMart (Shanghai, China). The peptide for generating the phospho antibody is: APKRQ-pS-PLPPQ. The corresponding non-phospho-peptide was also synthesized for antibody purification. Anti-Flag antibodies were from Sigma-Aldrich. Anti-β-actin, anti-ERK1/2, anti-phospho-S380 RSK, anti-MAD1, anti-MAD2, anti-TTK, anti-CDC27, anti-CDC20, and anti-cyclin B1 antibodies were from Santa Cruz Biotechnology. Anti-Aurora-A, anti-Bub1, anti-BubR1, anti-Cyclin C, and anti-GST antibodies were from Bethyl Laboratories (Montgomery, TX, USA). The following antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA): Phospho-T288/T232/T198 Aurora-A/B/C, phospho-S10 H3, phospho-T202/Y204 ERK1/2, phospho-S127 YAP, phospho-S397 YAP, Yes-associated protein (YAP), ribosomal S6 kinase 1 (RSK1), phospho-S807/S811 Rb, Rb, phospho-S536 NF-kB p65, NF-kB p65, phospho-S176/S180 IKKα/β, IKKα/β, phospho-T308 Akt, phospho-S473 Akt, Akt, phospho-Y705 STAT3, signal transducers and activators of transcription 3 (STAT3), phospho-S727 STAT1, phospho-Y1034/Y1035 JAK1, Janus kinase 1 (JAK1), phospho-S33/S37/T41 β-catenin, β-catenin, CDK1, CDK3, CDK4, CDK5, Cyclin A2, Cyclin D1, Cyclin E1, p21, and CDC25C. Anti-β-tubulin (Sigma-Aldrich) and anti-γ-tubulin (Biolegend, San Diego, CA, USA) antibodies were used for immunofluorescence staining.
Phos-tag and western blot analysis
Phos-tagTM SDS-acrylamide gels were used as we described previously41. Western blotting, immunoprecipitation, and lambda phosphatase treatment assays were done as described40.
Immunohistochemistry (IHC) staining, immunofluorescence staining, and confocal microscopy
Fluorescence staining and confocal microscopy were done, as previously described42. IHC staining (of Ki-67, cleaved caspase 3, and p-T308 Akt) in tumor tissues was performed according to protocols described43,44. Anticleaved caspase 3 (1:100) and p-T308 Akt (1:100) antibodies were from Cell Signaling Technology. Anti-Ki-67 antibody was from Thermo Scientific (Waltham, MA, USA) and was used at 1:100 dilutions.
Cell proliferation, colony formation, migration, and invasion assays
Cell proliferation and colony formation assays in soft agar (anchorage-independent growth) were performed as described44,45. In vitro analysis of invasion and migration (1.0 × 105 cells) was assessed using the BioCoat invasion system (BD Biosciences, Franklin Lakes, NJ, USA) and Transwell system (Corning, Corning, NY, USA), respectively, according to the manufacturer’s instructions. The invasive and migratory cells were stained with 0.1% crystal violet and counted manually.
Animal studies
For in vivo xenograft studies, RKO cells (1.0 × 106 cells each line) were subcutaneously injected into both flanks of 6-week-old male athymic nude mice (Ncr-nu/nu, Harlan, Indianapolis, IN, USA). Cells were suspended in phosphate-buffered saline (PBS) and mixed with Matrigel in 1:1 ratio (volume). Five animals were used per group. Tumor sizes were measured twice a week using an electronic caliper 10 days post injection. Tumor volume (V) was calculated by the formula: V = 0.5 × length × width2,45. Mice were euthanized by CO2 inhalation at the end of the experiment and the tumors were excised for subsequent analysis. The animals were housed in pathogen-free facilities. All animal experiments were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee.
Statistical analysis
Statistical significance was performed using a two-tailed, unpaired Student’s t test.
Results
SET is phosphorylated during antitubulin drug-induced mitotic arrest
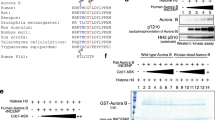
To explore the phospho status of SET during mitosis, we treated HeLa cells with taxol or nocodazole (both agents arrest cells in prometaphase after an overnight treatment) and examined the response of SET on a Phos-tag gel. SET proteins were shown as a doublet (isoform 1 and isoform 2) on an SDS-PAGE gel (Fig. 1a). Interestingly, a significant portion of SET protein was upshifted/retarded on a Phos-tag gel during mitotic arrest, suggesting that SET is phosphorylated under these conditions. Lambda phosphatase treatment eliminated the slow-migrating band (the top band on the gel), indicating that the mobility shift of SET during mitotic arrest is caused by phosphorylation (Fig. 1b). The middle and bottom bands remained unchanged during phosphatase treatment (Fig. 1b).

a HeLa cells were treated with DMSO (control), taxol (100 nM for 16 h), or nocodazole (Noco, 100 ng/ml for 16 h). Total cell lysates were electrophoresed on regular and Phos-tag SDS polyacrylamide gels and probed with the indicated antibodies. Increased cyclin B1 levels marks cells in mitosis. An asterisk (*) marks the phosphorylated/shifted band. b HeLa cells were treated with nocodazole as indicated and cell lysates were further treated with (+) or without (−) λ phosphatase (ppase). Total cell lysates were probed with the indicated antibodies. Increased cyclin B1 levels marks cells in mitosis. An asterisk marks the phosphorylated/shifted band. c HeLa cells were treated with nocodazole, with or without various kinase inhibitors as indicated. Inhibitors were added 1.5 h before harvesting the cells (with MG132 to prevent cyclin B degradation and subsequent mitotic exit). The concentrations used for each inhibitor were as follows: VX680 2 μM, RO3306 5 μM, Purvalanol A 10 μM, BI-2536 100 nM, SB216763 10 μM, MK-2206 10 μM, SB203580 10 μM, U0126 20 μM, SP600125 20 μM, LY294002 30 μM, and rapamycin 100 nM. Total cell lysates were electrophoresed on regular and Phos-tag SDS polyacrylamide gels and probed with the indicated antibodies. Increased cyclin B1 levels mark cells in mitosis. An asterisk marks the phosphorylated/shifted band. d In vitro kinase assays with purified CDK1/cyclin B1 complex using GST-tagged SET isoform 1 proteins as substrates. RO3306 (5 µM) was used to inhibit CDK1/cyclin B1 kinase activity. e GST-SET and GST-SET-S7A proteins were used for in vitro kinase assays with purified CDK1/cyclin B1 complex. f In vitro kinase assays were done as in e except anti-phospho-SET S7 antibody was used
Identification of the corresponding kinase for SET isoform 1 phosphorylation
In order to determine which upstream kinase(s) could be responsible for SET phosphorylation, we treated cells with various kinase inhibitors together with MG132 (stabilizes cyclin B1 and prevent cells from exiting mitosis). Interestingly, the most significant inhibition of phosphorylation of SET was observed in cells treated with RO3306 (a CDK1 inhibitor) and Purvalanol A (inhibits CDK1/2/5) (Fig. 1c), suggesting that CDK1, a well-known mitotic kinase, is the candidate kinase for SET phosphorylation. Taken together, these data suggest that mitotic arrest-induced SET phosphorylation is CDK1 dependent.
CDK1 phosphorylates SET isoform 1 in vitro
Next, we performed in vitro kinase assays with GST-tagged SET proteins as substrates to determine whether CDK1 kinase can directly phosphorylate SET. Figure 1d shows that purified CDK1/cyclin B1 complex phosphorylated GST-SET in vitro (Fig. 1d). As expected, addition of RO3306 abolished the 32P incorporation into SET (Fig. 1d).
CDK1 phosphorylates an S/TP consensus sequence46. Database analysis (www.phosphosite.org) identified serine 7 (followed by a proline) as a possible phosphorylation site in SET during mitosis47. Of interest, mutating S7 to alanine largely eliminated the phosphorylation (32P incorporation) of SET (Fig. 1e), suggesting that S7 is the main phosphorylation site of SET in vitro. Next, we generated a phospho-specific antibody against SET S7. Using this antibody, we confirmed that GST-SET proteins were robustly phosphorylated at S7 by CDK1/cyclin B1 kinase complex in vitro (Fig. 1f).
SET isoform 1 is phosphorylated at S7 in cells in a CDK1-dependent manner
After confirming SET phosphorylation at S7 by CDK1 in vitro, we next examined this phosphorylation in cells. Nocodazole or taxol treatment significantly increased phosphorylation of S7 of endogenous SET (Fig. 2a). The shRNA-mediated depletion of SET (both isoform 1 and isoform 2) largely blocked the phospho signal, confirming the specificity of the phospho antibody (Fig. 2b). Furthermore, nocodazole treatment significantly increased the phosphorylation of S7 of transfected SET isoform 1, and the signal was abolished by mutating S7 to alanine (Fig. 2c). Treatment with kinase inhibitors RO3306 or Purvalanol A greatly decreased phosphorylation of S7 induced by nocodazole, suggesting that phosphorylation of SET S7 is CDK1 kinase dependent (Fig. 2d). Consistent with these observations, an enhanced expression of constitutive active CDK1 or cyclin B1 was sufficient to stimulate SET phosphorylation at S7 (Fig. 2e). Taken together, these results indicate that SET isoform 1 is phosphorylated at S7 in cells during mitotic arrest in a CDK1-dependent manner.

a HeLa cells were treated with DMSO, nocodazole (Noco), or taxol and total cell lysates were probed with the indicated antibodies. Increased cyclin B1 levels mark cells in mitosis. b Control and SET-knockdown (both isoforms 1 and 2) HeLa cells were subjected to the indicated treatment. Total cell lysates were analyzed by western blotting with the indicated antibodies. Increased cyclin B1 levels mark cells in mitosis. c HEK293T cells were transfected with isoform 1 SET or SET-S7A plasmid. At 30 h posttransfection, the cells were treated with nocodazole for 16 h. Total cell lysates were probed with the indicated antibodies. Increased p-Aurora A/B/C levels mark cells in mitosis. d HeLa cells were treated with nocodazole for 16 h and RO3306 (5 µM) or Purvalanol A (10 µM) was added to cells 1.5 h before harvesting as indicated. Proteasome inhibitor MG132 was also added (together with inhibitors) to prevent cyclin B from degradation and cells from exiting from mitosis. Increased cyclin B1 levels mark cells in mitosis. e HEK293T cells were transfected with expression constructs as indicated and total cell lysates were analyzed by western blotting. GFP-Cyc B1-CA: GFP-Cyclin B1-R42A (a nondegradable/constitutive active mutant). Flag-CDK1-CA: Flag-CDK1-T14A/Y15A (nonphosphorylatable/constitutive active CDK1). f HeLa cells were synchronized by a double thymidine (DT) block and release method. Total cell lysates were harvested at the indicated time points and subjected to western blotting analysis. Increased p-Aurora-A and cyclin B1 levels mark cells in mitosis
To determine whether mitotic phosphorylation at SET isoform 1 S7 occurs during normal mitosis, we determined the phospho status of cells collected from a double thymidine block and release42. After being released from double thymidine block, cells enter into mitosis at 10–12 h, as revealed by increased p-S10 H3 levels41,48. We found that the p-SET S7 signal was coincidently increased in these cells (Fig. 2f). These observations suggest that SET isoform 1 is phosphorylated at S7 during mitosis.
Mitotic phosphorylation of SET isoform 1 is required for precise mitosis
SET has been shown to be involved in mitotic progression and its alteration resulted in mitotic defects27,29. To determine the functional significance of S7 phosphorylation, we established cell lines expressing shRNA-resistant SET or SET-S7A in SET-KD cells (Fig. 3a). Consistent with previous studies, we found that KD of SET (both isoform 1 and isoform 2) in HeLa cells resulted in a significant higher percentage of cells with multiple nuclei, suggesting a role of SET in cytokinesis (Fig. 3b, c). This phenotype was largely rescued by re-expressing wild-type SET in SET-KD cells, and the phospho-deficient mutant (SET-S7A) failed to restore this defect, suggesting that mitotic phosphorylation at S7 is essential for mitotic cell division (Fig. 3c).

a Western blotting analysis of HeLa SET-knockdown cells stably expressing Flag-tagged shRNA-resistant (Res) isoform 1 SET-WT or SET-S7A. b, c Knockdown of SET resulted in multinucleated cells. Representative photos of control (shControl) and SET-knockdown (shSET) cells were shown in b. Quantification of multinucleated cells (c) in cell lines established in a. Arrows in b indicate some of the multinucleated cells. Data in c were expressed as the mean ± SD of three independent experiments. ***p < 0.001 (unpaired Student’s t test). A total of 250 cells in each group were counted. d, e Knockdown of SET resulted in massive mitotic defects. Representative photos of normal mitosis (shControl) and mitotic abnormalities (shSET) in HeLa cells were shown. Cells were stained with β-tubulin, γ-tubulin antibodies, and DAPI to visualize microtubules (green), centrosomes (red), and chromosomes (blue), respectively. Data in e were collected from 160 mitotic cells for each group (mean ± SD of three independent experiments). ***p < 0.001 (unpaired Student’s t test)
We observed largely normal chromosomal alignment/segregation (DAPI staining), microtubule/spindle formation (immunofluorescence staining with β-tubulin), and centrosome number (γ-tubulin staining) during mitosis in control cells (Fig. 3d). In contrast, massive mitotic defects (e.g., multipolar spindles, supernumerary centrosomes, and chromosome misalignment/missegregation) were detected in SET-KD cells (Fig. 3d, e). Again, re-expression of wild-type SET, but not the SET-S7A mutant, completely rescued the mitotic defects in SET-KD cells (Fig. 3e). These data suggest that mitotic phosphorylation of SET isoform 1 is required for precise mitosis in HeLa cells.
Mitotic phosphorylation of SET isoform 1 is required for cell migration and invasion
Next, we further explored the biological significance of mitotic phosphorylation of SET in cancer cell growth. First, wound scratch assays were performed in control, SET-KD, and SET-KD cells expressing SET or SET-S7A. As expected, KD of SET (both isoforms 1 and 2) significantly decreased the rate of wound closure for HeLa cells (Fig. 4a, b). Similar results were obtained with colon cancer RKO cells (relatively high expression of SET) (Fig. 4c–e). Exogenous expression of wild-type SET, but not SET-S7A, restored migration of both HeLa and RKO cells (Fig. 4a–e). In line with loss-of-function phenotypes, ectopic expression of SET isoform 1 substantially increased the rate of wound closure in immortalized human pancreatic cells (HPNE, express relatively low SET) (Fig. 4f, g). The ability to increase wound closure for the isoform 1 SET-S7A mutant was greatly reduced when compared with wild-type SET isoform 1 (Fig. 4g).

a, b Cell migration (wound healing) assays with cell lines established in Fig. 3a. Data were expressed as the mean ± SD of three independent experiments. ***p < 0.001 (unpaired Student’s t test). c–e Cell migration (wound healing) assays in RKO cells. Data were expressed as the mean ± SD of three independent experiments. ***p < 0.001; **p < 0.01 (unpaired Student’s t test). f, g Cell migration (wound healing) assays in immortalized human pancreatic nestin-expressing (HPNE) cells. Data were expressed as the mean ± SD of four independent experiments. ***p < 0.001 (unpaired Student’s t test)
Second, we used Transwell and Matrigel systems to examine the migratory and invasive properties of these cells. Consistent with the data from Fig. 4, re-expression of wild-type SET isoform 1 completely rescued migration and invasion in HeLa and RKO cells (Fig. 5a–e). However, expression of the nonphosphorylatable mutant SET-S7A only modestly restored these characteristics (Fig. 5a–e). Enhanced expression of wild-type SET, and SET-S7A to a significantly lesser extent, promoted migratory and invasive abilities of HPNE cells (Fig. 5f–i). These observations suggest that mitotic phosphorylation of SET isoform 1 at S7 is essential for cell motility.

a–c Cell migration (Transwell) and invasion (Matrigel) assays with cell lines established in Fig. 3a (HeLa). Data were expressed as the mean ± SD of three independent experiments. Migratory and invasive cells were stained with crystal violet and representative fields were shown in c. ***p < 0.001 (unpaired Student’s t test). d, e Cell migration (Transwell) and invasion (Matrigel) assays in RKO cells established in Fig. 4c. Data were expressed as the mean ± SD of three independent experiments. ***p < 0.001 (unpaired Student’s t test). f–i Cell migration (Transwell) and invasion (Matrigel) assays in HPNE cells established in Fig. 4f. Data were expressed as the mean ± SD of four independent experiments. ***p < 0.001 (unpaired Student’s t test)
Mitotic phosphorylation of SET isoform 1 is required for cell proliferation and anchorage-independent growth
Consistent with the results in migration and invasion, KD of SET significantly decreased cell proliferation and importantly, expression of wild-type SET isoform 1 completely rescued the cell proliferation defects (Fig. 6a, b). However, cells expressing SET-S7A proliferated at a rate similar to that of SET-KD cells, suggesting that mitotic phosphorylation of SET isoform 1 is required for proper cell proliferation (Fig. 6a, b). Furthermore, SET KD also greatly decreased anchorage-independent growth (colony formation) in soft agar, and again, re-expression of SET-S7A failed to rescue the defects while expression of wild-type SET did (Fig. 6c–e). These data suggest that mitotic phosphorylation is essential for SET isoform 1 to promote cell proliferation and anchorage-independent growth.

Mitotic phosphorylation of SET isoform 1 is required for cell proliferation and anchorage-independent growth. a, b Cell proliferation curves of cell lines established in Figs. 3a (HeLa) and 4c (RKO). Data were expressed as the mean ± SD of three independent experiments. ***p < 0.001; **p < 0.01 (unpaired Student’s t test). Green asterisks mark comparisons between shControl and shSET groups. Red asterisks mark comparisons between isoform 1 SET-WT and SET-S7A groups. c–e Anchorage-independent growth (colony formation assays in soft agar) of cell lines established in Figs. 3a (HeLa) and 4c (RKO). Data were expressed as the mean ± SD of three independent experiments. ***p < 0.001 (unpaired Student’s t test)
Mitotic phosphorylation of SET isoform 1 is required for its oncogenic activity in vivo
Having demonstrated the role of mitotic phosphorylation of SET in cell culture models, we next evaluated the influence of SET and its mitotic phosphorylation on tumor growth in animals. Various RKO cell lines (Fig. 4c) were subcutaneously inoculated into immunodeficient mice. Interestingly, SET-KD cells formed significantly smaller tumors (if any) when compared with those from control cells (Fig. 7a, b). No significant difference was observed in the sizes of tumors from control and SET-KD cells with exogenous wild-type SET isoform 1. However, cells expressing the isoform 1 SET-S7A mutant formed tumors in size similar to that of SET-KD cells (Fig. 7a, b). Consistent with our observations in Fig. 6, Ki-67 positivity (a proliferation marker) was significantly higher in wild-type SET-expressing tumors than SET-S7A tumors (Fig. 7c). In contrast, massive apoptosis (cleaved caspase-3 staining) was detected and Akt activity (p-AKT T308) was strongly suppressed in SET-S7A tumors (Fig. 7c, d). These results support the hypothesis that mitotic phosphorylation is essential for SET isoform 1-promoted tumor growth in vivo.

a Representative tumors in each group were excised and photographed at the endpoint. b Tumor growth curve with various RKO cell lines. Cells were subcutaneously inoculated into athymic nude mice on both flanks (five mice each group). The tumor volume shown at each point was the average from 6 to 10 tumors (two animals in shSET and shSET+SET-S7A groups did not form any palpable tumors, mean ± SD). ***p < 0.001 (unpaired Student’s t test). c Ki-67 (proliferation) and cleaved caspase-3 (apoptosis) staining in tumors shown in a. Tumors from shSET+ isoform 1 SET-WT and shSET+ isoform 1 SET-S7A groups were analyzed. d IHC staining with p-AKT T308 antibody in tumors shown in a. e Western blotting analysis of SET (isoforms 1 and 2) expression in immortalized colon epithelial (HCEC) and colon cancer cell lines. f–j High expression of SET (all isoforms) correlates with poor clinical outcome. Kaplan–Meier curves of overall survival in various cancers. Data were generated from an online analysis tool (http://kmplot.com) using RNA-seq data49
Many reports showed that SET (isoforms 1 and 2) is upregulated in cancer cell lines and clinical tumor samples6. We found that SET (isoforms 1 and 2) expression is upregulated in colon cancer cells compared with immortalized colon epithelial cells (Fig. 7e). We further analyzed the correlation between SET expression and clinical outcome in published data49 and confirmed that high mRNA levels of SET (all isoforms) were significantly correlated with poor survival in various malignancies (Fig. 7f–j).
Mitotic phosphorylation of SET isoform 1 affects Akt signaling
SET regulates cell cycle progression and we hypothesized that one of the underlying mechanisms is through binding to cell cycle regulators27,28,50. We surveyed the expression of a panel of cell cycle regulators to determine which one(s) is affected by SET and its mitotic phosphorylation in our system. While most of them were not altered upon SET KD, we found p-S642 WEE1 levels were increased in SET-KD cells (Fig. 8a).

a Expression of cell cycle regulators in shControl (Ctrl) and shSET (KD) HeLa cells. Total cell lysates were probed with the indicated antibodies. b Examination of major signaling pathways in control (Ctrl) and SET-knockdown (KD) RKO cells. c Western blotting analysis with the indicated antibodies in RKO cells. Data from a to c are representative of at least two independent experiments
SET has been shown to regulate many signaling pathways, including Wnt/β-catenin, the JAK/STAT, Akt, mitogen-activated protein kinase, etc6. To understand the possible involvement of these signaling pathways, we examined their activities in SET KD RKO cells. We found JAK/STAT signaling, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), extracellular-signal-regulated kinase/RSK (ERK/RSK), as well as β-catenin and YAP activities were not affected in SET-KD cells when compared to control cells (Fig. 8b). Consistent with previous studies51, Akt activity (revealed by phosphorylation at T308 and S473) was greatly decreased upon SET inhibition in RKO cells (Fig. 8b). Again, re-expression of wild-type SET, but not the SET-S7A mutant, rescued the phenotype (Fig. 8c), suggesting that SET phosphorylation is required for SET-mediated Akt activation (at least via T308 phosphorylation).
Discussion
Among the two major isoforms of SET, isoform 2 remains at a relatively constant expression level among different tissues and cell lines, and isoform 1 expression varies in a cell- or tissue-specific manner17,52. For instance, isoform 1 is absent in some early-stage hematopoietic cell lines and pancreatic cancer cell lines17,52. Our current study identifies S7 as a mitotic phosphorylation site, and this modification is essential for its oncogenic activity (Fig. 7). We noted that serine 7 is unique to isoform 1, which raises the question of why some cells/tissues need an additional layer of regulation for SET. Future studies are also needed to determine the biological significance of expression patterns of SET isoforms. Moreover, it will be equally important to elucidate the clinical relevance of CDK1 phosphorylation of SET in future studies. Addressing these questions is anticipated not only to strengthen the comprehension of the biological relevance of CDK1 phosphorylation of SET, but also to be useful in determining potential overlapping and distinguishing functions of the various isoforms of SET during oncogenesis.
SET is also called I2PP2A and interacts with PP2A and another closely related protein I1PP2A/PP321,2,3. Database analysis (www.phosphosite.org) and phospho-proteomic studies revealed potential mitotic phosphorylation sites at the N-terminus of I1PP2A (T15/S17)53,54. The role and regulation of I1PP2A in mitosis have not been defined. We are also interested in investigating how I1PP2A is regulated and whether it also plays a role in mitosis similar to SET/I2PP2A. As an inhibitor of PP2A, SET promotes tumorigenesis mainly through forming an inhibitory protein complex with PP2A6. The activity of PP2A is tightly regulated by the C-terminal region of SET that is responsible for the binding to PP2A catalytic subunit PP2A-C1,2. Since mitotic phosphorylation occurs on the N-terminus of SET isoform 1, we do not expect that loss of oncogenic activity of SET-S7A is due to alterations in binding with PP2A. Indeed, our coimmunoprecipitation experiments revealed no significant difference between SET and SET-S7A in association with PP2A-C (data not shown). Therefore, it is currently unclear how phosphorylation of SET promotes its oncogenic activity and Akt signaling (Fig. 8). In addition to PP2A, SET has been shown to interact with many other proteins (including CK2, eIF2α, glycogen phosphorylase, TCP1-β, Cyclin B, and p21),28,50,55 several of which are involved in mitotic cell cycle progression and tumorigenesis. Does SET phosphorylation affect these binding partners? The future elucidation of these mechanisms will provide novel insights underlying SET-driven tumorigenesis.
References
Li, M., Guo, H. & Damuni, Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 34, 1988–1996 (1995).
Li, M., Makkinje, A. & Damuni, Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 271, 11059–11062 (1996).
Li, M. & Damuni, Z. I1PP2A and I2PP2A. Two potent protein phosphatase 2A-specific inhibitor proteins. Methods Mol. Biol. 93, 59–66 (1998).
Mumby, M. PP2A: unveiling a reluctant tumor suppressor. Cell 130, 21–24 (2007).
O’Connor, C. M., Perl, A., Leonard, D., Sangodkar, J. & Narla, G. Therapeutic targeting of PP2A. Int. J. Biochem. Cell Biol. 96, 182–193 (2018).
Hung, M. H. & Chen, K. F. Reprogramming the oncogenic response: SET protein as a potential therapeutic target in cancer. Expert Opin. Ther. Targets 21, 685–694 (2017).
Switzer, C. H. et al. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 30, 2504–2513 (2011).
Janghorban, M. et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl Acad. Sci. USA 111, 9157–9162 (2014).
Jiang, S. W., Xu, S., Chen, H., Liu, J. & Duan, P. Oncogenic role of SET/I2PP2A for gynecologic cancers. Curr. Drug Targets 18, 1152–1157 (2017).
Neviani, P. et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 8, 355–368 (2005).
Neviani, P. et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 123, 4144–4157 (2013).
Christensen, D. J. et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: a predictor of aggressive disease and a new treatment target. Blood 118, 4150–4158 (2011).
Hung, M. H. et al. Upregulation of the oncoprotein SET determines poor clinical outcomes in hepatocellular carcinoma and shows therapeutic potential. Oncogene 35, 4891–4902 (2016).
Hu, X. et al. Inhibition of Pten deficient castration resistant prostate cancer by targeting of the SET—PP2A signaling axis. Sci. Rep. 5, 15182 (2015).
Cristobal, I. et al. Deregulation of the PP2A inhibitor SET shows promising therapeutic implications and determines poor clinical outcome in patients with metastatic colorectal cancer. Clin. Cancer Res. 21, 347–356 (2015).
Hung, M. H. et al. SET antagonist enhances the chemosensitivity of non-small cell lung cancer cells by reactivating protein phosphatase 2A. Oncotarget 7, 638–655 (2016).
Mody, H. R. et al. SET contributes to the epithelial-mesenchymal transition of pancreatic cancer. Oncotarget 8, 67966–67979 (2017).
Kandilci, A., Mientjes, E. & Grosveld, G. Effects of SET and SET-CAN on the differentiation of the human promonocytic cell line U937. Leukemia 18, 337–340 (2004).
Oaks, J. J. et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood 122, 1923–1934 (2013).
Agarwal, A. et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 20, 2092–2103 (2014).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Holland, A. J. & Cleveland, D. W. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487 (2009).
Janssen, A. & Medema, R. H. Mitosis as an anti-cancer target. Oncogene 30, 2799–2809 (2011).
Manchado, E., Guillamot, M. & Malumbres, M. Killing cells by targeting mitosis. Cell Death Differ. 19, 369–377 (2012).
Henriques, A. C. et al. Mitosis inhibitors in anticancer therapy: when blocking the exit becomes a solution. Cancer Lett. 440–441, 64–81 (2019).
Dominguez-Brauer, C. et al. Targeting mitosis in cancer: emerging strategies. Mol. Cell 60, 524–536 (2015).
Canela, N. et al. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J. Biol. Chem. 278, 1158–1164 (2003).
Kellogg, D. R., Kikuchi, A., Fujii-Nakata, T., Turck, C. W. & Murray, A. W. Members of the NAP/SET family of proteins interact specifically with B-type cyclins. J. Cell Biol. 130, 661–673 (1995).
Krishnan, S., Smits, A. H., Vermeulen, M. & Reinberg, D. Phospho-H1 decorates the inter-chromatid axis and is evicted along with shugoshin by SET during mitosis. Mol. Cell 67, 579–593.e6 (2017).
Seo, S. B. et al. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 104, 119–130 (2001).
Qi, S. T. et al. Overexpression of SETbeta, a protein localizing to centromeres, causes precocious separation of chromatids during the first meiosis of mouse oocytes. J. Cell Sci. 126, 1595–1603 (2013).
Kitajima, T. S. et al. Shugoshin collaborates with protein phosphatase 2A to protect cohesin. Nature 441, 46–52 (2006).
Adachi, Y., Pavlakis, G. N. & Copeland, T. D. Identification of in vivo phosphorylation sites of SET, a nuclear phosphoprotein encoded by the translocation breakpoint in acute undifferentiated leukemia. FEBS Lett. 340, 231–235 (1994).
von Lindern, M. et al. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene. Mol. Cell Biol. 12, 3346–3355 (1992).
Feng, Y. et al. Regulation of SET gene expression by NFkB. Mol. Neurobiol. 54, 4477–4485 (2017).
Yu, G. et al. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol. Aging 34, 1748–1758 (2013).
Zhang, Q. et al. CK2 phosphorylating I2(PP2A)/SET mediates tau pathology and cognitive impairment. Front. Mol. Neurosci. 11, 146 (2018).
Stauffer, S. et al. CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity. Cell Signal. 39, 74–83 (2017).
Lee, K. M., Yasuda, H., Hollingsworth, M. A. & Ouellette, M. M. Notch 2-positive progenitors with the intrinsic ability to give rise to pancreatic ductal cells. Lab. Investig. 85, 1003–1012 (2005).
Xiao, L., Chen, Y., Ji, M. & Dong, J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J. Biol. Chem. 286, 7788–7796 (2011).
Chen, X., Stauffer, S., Chen, Y. & Dong, J. Ajuba phosphorylation by CDK1 promotes cell proliferation and tumorigenesis. J. Biol. Chem. 291, 14761–14772 (2016).
Zhang, L. et al. KIBRA regulates Aurora kinase activity and is required for precise chromosome alignment during mitosis. J. Biol. Chem. 287, 34069–34077 (2012).
Zhang, L. et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell Biol. 35, 1350–1362 (2015).
Zhou, J. et al. Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation. Proc. Natl Acad. Sci. USA 115, E6760–E6769 (2018).
Dong, J. et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 (2007).
Nigg, E. A. Cellular substrates of p34(cdc2) and its companion cyclin-dependent kinases. Trends Cell Biol. 3, 296–301 (1993).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
Yang, S. et al. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 73, 6722–6733 (2013).
Gyorffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010).
Estanyol, J. M. et al. The protein SET regulates the inhibitory effect ofp21(Cip1) on cyclin E-cyclin-dependent kinase 2 activity. J. Biol. Chem. 274, 33161–33165 (1999).
Huang, C. Y. et al. Antagonizing SET augments the effects of radiation therapy in hepatocellular carcinoma through reactivation of PP2A-mediated Akt downregulation. J. Pharmacol. Exp. Ther. 366, 410–421 (2018).
Nagata, K. et al. Cellular localization and expression of template-activating factor I in different cell types. Exp. Cell Res. 240, 274–281 (1998).
Olsen, J. V. et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3 (2010).
Dephoure, N. et al. A quantitative atlas of mitotic phosphorylation. Proc. Natl Acad. Sci. USA 105, 10762–10767 (2008).
Vera, J. et al. Proteomic analysis of SET-binding proteins. Proteomics 7, 578–587 (2007).
Acknowledgements
All fluorescence images were acquired at the Advanced Microscopy Core at the University of Nebraska Medical Center. The core is supported in part by the grant P30 GM106397 from the National Institutes of Health (NIH). Research in the Dong laboratory is supported by Fred & Pamela Buffett Cancer Center support grant (P30 CA036727), and grants P30 GM106397 and R01 GM109066 from the NIH. L.Y. and Y.Z. are supported by fellowships from Chinese Scholarship Council, China. We also thank Dr Joyce Solheim for critical reading and comments on the manuscript.
Authors’ contributions
J.D., L.Y., and H.S. designed the experiments and wrote the manuscript. L.Y., Y.X., Y.Z. and Y.C. performed the experiments, analyzed the data, and interpreted the results. Y.C. provided technical support. All authors reviewed and approved the manuscript prior to submission. In Fig. 1, Y.X. generated and prepared panels a–c, and Y.C. and L.Y. generated and prepared panels d–f. In Fig. 2, Y.X. generated and prepared panel a, and L.Y. generated and prepared panels c–f. L.Y. generated and prepared Figs. 3–6 and 8. In Fig. 7, Y.Z. and L.Y. generated and prepared panels a and b, L.Y. generated and prepared panels c–e and Y.Z. prepared f–j.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by S. Inoue
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yin, L., Zeng, Y., Xiao, Y. et al. Cyclin-dependent kinase 1-mediated phosphorylation of SET at serine 7 is essential for its oncogenic activity. Cell Death Dis 10, 385 (2019). https://doi.org/10.1038/s41419-019-1621-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1621-2
This article is cited by
-
Mitotic phosphorylation of Tau/MAPT modulates cell cycle progression in prostate cancer cells
Journal of Cancer Research and Clinical Oncology (2023)
