
Abstract
Mechanisms underlying the generation of induced pluripotent stem cells (iPSC) and keeping iPSC stability remain to be further defined. Accumulated evidences showed that iPSC reprogramming may be controlled by the cell-division-rate-dependent model. Here we reported effects of absence of mouse p27 or p18 on iPSC generation efficiency and genomic stability. Expression levels of cyclin-dependent kinases inhibitors (CDKIs), p21, p27, and p18 decreased during iPSC reprogramming. Like p21 loss, p27 or p18 deficiency significantly promoted efficiency of iPSC generation, whereas ectopic expression of p27, p18, or treatment with CDK2 or CDK4 inhibitors repressed the reprogramming rate, suggesting that CDKIs-regulated iPSC reprogramming is directly related with their functions as CDK inhibitors. However, unlike p21 deletion, absence of p27 or p18 did not increase DNA damage or chromosomal aberrations during iPSC reprogramming and at iPSC stage. Our data not only support that cell cycle regulation is critical for iPSC reprogramming, but also reveal the distinction of CDKIs in somatic cell reprogramming.
Similar content being viewed by others


Introduction
The reprogramming of somatic cells into induced pluripotent stem cells (iPSC) by introduction of the four defined transcription factors (Oct4, Sox2, Klf4, and c-Myc) is an intensively investigated area in stem cell research for its enormous potential in regenerative medicine since 20061. The efficiency of iPSC generation however remains low, and the iPSC genomic stability is still concerned.
Accumulated evidences demonstrated that iPSC reprogramming is mainly controlled by the cell-division-rate-dependent model2,3,4,5,6,7. p21, p27, and p18 are three important cell cycle regulators and cyclin-dependent kinase inhibitors (CDKIs)8,9,10,11,12. Limited studies showed that p21, p27, and p18 are important for iPSC reprogramming2,13,14,15,16. We and others demonstrated previously that loss of p21 could promote somatic reprogramming, however, caused markedly genomic instability2,13,14. Deletion of p27 enhances somatic reprogramming in the absence of ectopic Sox215. p18 reduces iPSC reprogramming by targeting CDK4/6-mediated cell cycle regulation16. However, their roles in controlling iPSC quality and genomic stability are still unclear.
Here we examined iPSC generation from murine cells that are deficient in p27 or p18 in comparison with p21 loss. We found that although loss of different CDKIs can improve iPSC colony formation efficiency, but iPSC quality with loss of p21, p27, or p18 was significantly different. In comparison to loss of p21, iPSC with absence of p27 or p18 were associated with fewer chromosomal aberrations. Our results demonstrated that deletion of p27 or p18 may be a better choice to enhance iPSC generation efficiency with a guaranteed quality.
Methods
Mice
Wild type, p21−/−, p18−/−, p27−/−, and NOD/SCID/Gamma (NSG) mice were purchased from The Jackson Laboratory. All the mice strains had been crossed to generate a pure C57BL/6J background and maintained in the pathogen free animal facility, Institute of Hematology and Blood Diseases Hospital. All procedures and animal experiments were approved by the Institutional Animal Care and Use Committee at Institute of Hematology and Blood Diseases Hospital, CAMS/PUMC.
Cells culture conditions
Primary MEFs were obtained from 13.5-day embryos of the indicated genotypes based on the protocol from Wicell and cultured in standard DMEM medium containing 10% FBS (Millipore) in our lab. Murine ES cell was purchased from Wicell and iPS cells were reprogramming from MEF cells with four factors (Oct4, Sox2, Klf4, and c-Myc). Murine ES and iPSC cells were cultured in ‘ES culture medium’ composed of Knock-Out DMEM (Invitrogen) supplemented with ES cell qualified FBS (20%, Millipore), mouse LIF (1000 U/ml), non-essential amino acids, L-Glutamine, and β-mercaptoethanol. Bone marrow c-kit+ cells were harvested from femurs of mice, enriched by CD177-streptavidin kit (Miltenyi), and cultured in standard BIT9500 (Stemcell Technologies) containing 10% FBS supplemented with murine SCF, Flt-3, and TPO before transduction.
Plasmids
Retroviral constructs pMXs-Klf4 (#13370)1, pMXs-Sox2 (#13367)1, pMXs-Oct4 (#13366)1, pMXs-c-Myc (#13375)1, were obtained from Addgene. The cDNA of mouse p27 and p18 were also purchased from Addgene. They were both cloned into pMXs-GFP retroviral vectors.
Generation of mouse iPS cells
Reprogramming of primary (passage 2) MEFs was performed as previously described1. In brief, primary MEFs of indicated genotypes were seeded in 100-mm-diameter dish (5 × 105 cells per dish) pre-coated with 0.1% gelatin (Sigma). They were transduced twice in the next two days at 24-h interval by virus supernatant collected from Plat-E cells transfected with the previously mentioned retroviral plasmids. At the end of transduction, medium was changed to ES culture medium. After cultured for 10–12 days, colonies with ES-cell-like morphology became visible. They were then either scored after SSEA1 staining or picked for further expansion on feeder fibroblasts using standard ES culture methods.
Reprogramming efficiency analysis
For quantification of iPSC generation efficiency, retroviral transduction was measured in parallel infections containing all the retroviruses used for reprogramming plus a GFP retrovirus (pMXs-GFP) (equal volumes of each retrovirus were used during the transduction). The efficiency of transduction was measured by FACS analysis the next day after medium was changed to ES culture medium. Total numbers of iPSC colonies were counted after staining plates for SSEA1 antibody (R&D). Briefly, 5 × 105 cells per 100-mm dish were seeded after retroviral transduction and measured GFP positive rates in different genotypes. The numbers of SSEA1+ colonies were counted on Day 12. The percentage of SSEA1+ colonies over all the transduced MEFs was determined. The efficiency of reprogramming was also calculated as the relative change compared to that of control.
CDK inhibitors and p27 siRNA sequence
CDK4 (CDK4/6) inhibitor Palbociclib (PD-0332991, Cat#: S1116, Selleck), CDK2 (Cdc2, CDK2, and CDK5) inhibitor Roscovitine (Seliciclib, CYC202, Cat: S1153, Selleck), CDK4/6 inhibitor Abemaciclib (Cat: HY-16297, MCE), CDK7 inhibitor THZ1 (Cat: S7549, Selleck), p18 inhibitor NSC23005 sodium (Cat: HY-100791, MCE). p27 siRNA sequence: 5′-GTGGAATTTCGACTTTCAG-3′.
Teratoma formation
Cells (2 × 106) of indicated mouse iPS cell lines were subcutaneously injected into NOD/SCID mice. Teratomas were recovered and surgically removed after 3 weeks. Tissues were snap-frozen, embedded in tissue-tek with O.C.T. compound, and stored at −80 °C. The samples were sectioned at a thickness of 5 mm and stained with haematoxylin and eosin for pathological examination.
Western blot analysis
Cell extracts were prepared using RIPA buffer, resolved on NuPAGE 12% gradient Bis-Tris gels, transferred to nitrocellulose and hybridized using antibodies against p27 (1:500), p18 (1:500), p21 (1:500), RAD51 (1:500), and β-actin (1:500 dilution, Santa Cruz); PARP (1:1000 dilution, cell signaling technology); γH2AX-S139 (1:1000) and, 53BP1 (1:1000, abcam).
Immunofluorescence
Cells of mouse iPS cell lines were cultured in 12-well plates with feeders for 2–3 days until colonies formed. The cells were then fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.3% Triton X-100 in PBS for 15 min and blocked with 5%BSA in PBS for 1 h at room temperature. After incubation with antibodies against mouse Oct-4, Sox-2, Nanog (1:100 diluted in PBS containing 4%BSA, Santa Cruz) overnight at 4 °C, cells were washed with PBS the next day and incubated with secondary antibodies conjugated with Alexa 488 or Alexa 555 (1:1000 diluted in PBS containing 4% BSA, Molecular Probes). For staining of SSEA-1(R&D), the permeabilization step was emitted.
Detection of double-strand breaks of DNA
Reprogramming of MEFs of indicated genotypes with the four factors was performed as we previously described. Reprogramming cells were digested by trypsin and resuspended in PBS. SSEA1+ cells were sorted and then cytospun onto the slides. Or four genotype iPS cell lines were irradiated with 2 Gy and collected at indicated time points. The slides were then fixed in 4% paraformaldehyde for 15 min, permeabilized by 0.3% Triton X-100 in PBS, and blocked by 5% BSA in PBS for 1 h at room temperature. The slides were incubated with mouse monoclonal antibodies against γH2AX (1:150 diluted in PBS containing 2% BSA, Trevigen) overnight at 4 °C and secondary antibodies conjugated with Alex-555 for 1 h at room temperature the next day. DAPI was used as nuclear staining. Images were produced from confocal microscope (Leica) at ×63 magnification and analyzed for foci/nucleus.
Comet assay
At reprogramming Day 12, the cells were collected and digested. Then sorted SSEA1+ positive cells were sorted. Or four genotype iPS cell lines were irradiated with 2 Gy, collected at different time points. The cells were then processed for alkaline comet assay using Comet SCGE Assay kit (Enzo Life Sciences) according to the manufacturer’s protocol. Each slide was photographed under a Zeiss Axio Observer Z1 microscope and the percentage of tail intensity was computed by the Comet Assay IV software (Perceptive Instruments Ltd.).
Quantitative real-time PCR analysis
For the determination of mRNA levels of p18, p21, and p27 during reprogramming and in MEF, ESC, and iPS cells, cells were harvested by treating with trypsin-EDTA solution and washed with PBS three times, SSEA1+ cells were sorted at the different time points during reprogramming. Total RNA was extracted by using RNeasy kit (Qiagen) according to the manufacturer’s instructions. RNA was treated with RNase–free DNase (Invitrogen) for 15 min at room temperature before reverse transcription with superscript II RT (Invitrogen). Real-time PCR was performed on the chromo 4TM detector (M J Research) with SYBR Green PCR master mix (Thermo Scientific). PCR conditions consisted of a 10-min hot start at 95 °C followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min and incubation for 3 s at 77 °C with a final extension for 10 min at 72 °C. The average threshold cycle (Ct) for each gene was determined from triplicate reactions and the levels of gene expression relative to β-actin were determined as we previously described17. p18 primers: 5′-CTCCGGATTTCCAAGTTCA-3′ and 5′-GGGGGACCTAGAGCAACTTAC-3′. p21 primers: 5′-GTGGGTCTGACTCCAGCCC-3′ and 5′-CCTTCTCGTGAGAC GCTTAC-3′. p27 primers: 5′-CGATCGGAATTCATGTCAAACGTGCGAGTG-3′ and 5′-CGATCGAGATCTTTACGTTTGACGTCTTCTGAGGCC-3′. ACTB primers: 5′-ATGGAGGGGAATACAGCCC-3′ and 5′-TTCTTTGCAGCTCCTTCGTT-3′.
Cell cycle and proliferation analysis
Cell cycle and proliferation analysis assays were performed using Click-iT EdU Assay Kits at the indicated time points during reprogramming as we previously described13. We selected pacific blue to show the EDU and 7-AAD to stain the DNA. The flow cytometry data were analyzed by Syan software.
Karyotyping and G-banding assays
G-banding chromosome analysis of the iPS cell lines were performed as we previously described13. Data were interpreted by a certified cytogenetic technologist.
Centrosome number and spindle assays
Cells growing on coverslips were irradiated at the different time point. The cells were fixed with prechilled methanol for 20 min at −20 °C, washed with phosphate buffered saline (PBS), and permeabilized with 1% NP-40 in PBS for 5 min at 25 °C. Cells were blocked with 10% normal goat serum in PBS for 1 h and probed with anti-γ-tubulin monoclonal antibody (1:400, GTU-88, Sigma) for 1 h at 25 °C. The antibody–antigen complexes were detected with secondary antibodies conjugated with Alexa 555 (1:1000 diluted in PBS containing 4% BSA, Molecular Probes) by incubation for 1 h at 25 °C. The samples were counterstained with DAPI. These panels show merge of DNA (blue) and γ-tubulin (red). Scale bars, 10 mm. for showing the spindle, based on centrosome staining, added α-Tubulin antibody (1:400, Sigma) conjugated with FITC to show the spindle fibers (green).
Statistical analyses
Data are expressed as mean ± SD. All analyses were two-tailed and considered statistically significant when P values were less than or equal to 0.05.
Results
Expression levels of p21, p27, and p18 are decreased during iPSC reprogramming
To determine the roles of CDKIs, p21, p27, and p18 in iPSC reprogramming, we first tested cell proliferation in wild type (WT), p21−/−, p27−/−, and p18−/− MEFs. As shown in Fig. 1a, compared to WT, deletion of CDKIs all promoted cell proliferation. Then, we assessed the mRNA expression of p21, p27, and p18 in MEF, iPSC, and ESC derived from the same mice, and found that all CDKIs expressions were lower in iPSC and ESC compared to MEFs (Fig. 1b). We further tested the mRNA expression of p21, p27, and p18 during iPSC reprogramming, and found that all CDKIs expression levels were reduced (Fig. 1c). Moreover, at the early stage (Day 4) and middle stage (Day 8) after transduction, p21 expression was relatively higher than p27 and p18 (Fig. 1c). This suggests that these CDKIs’ expression levels are negatively correlated with cell stemness and pluripotency.

a Cell proliferation analysis of MEFs wild type (WT), p21−/−, p27−/−, or p18−/− mutant. Cell proliferation was determined by counting cell numbers using trypan blue staining. b qRT-PCR analysis of the mRNA expression levels of p18, p27, and p21 in mouse WT ESCs, iPSCs and MEFs. c Expression levels of p18, p27, and p21 mRNA in WT MEFs at indicated time points during reprogramming. d Flow cytometry (FACS) analysis of cell cycle using EDU and 7-AAD staining at the indicated time points during reprogramming. e Proliferation measured by EDU staining in different genotype MEFs at different time points during reprogramming. f Proliferation measured by EDU staining in different genotype iPSCs. Data are representative of two or three independent experiments. Error bars, ±SD. *p < 0.05; **p < 0.01, by two-tailed t test
Next, we examined the effects of deletion of CDKIs on cell cycle progression during iPSC reprogramming. As shown in Fig. 1d, e, the percentages of G1-phase cells of all genotypes were decreased and the cell proliferation rates were increased. However, at the early stage (Day 4) and middle stage (Day 8) after transduction, the percentages of G1 stage cells in the group with p21−/−, p27−/−, and p18−/− genotypes were markedly lower than that with WT genotype (Fig. 1d), which was correlated with increased cell proliferation rates measured by EDU incorporation (Fig. 1e). We further determine effects of p21−/−, p27−/−, or p18−/− KO on iPSC proliferation. As shown in Fig. 1f, p21 KO did not affect iPSC proliferation and p18 or p27 KO significantly inhibited iPSC proliferation compared to WT. These data show that expression levels of CDKIs are decreased during iPSC reprogramming and thereby cell cycle and proliferation of reprogramming cells with absence of CDKIs are changed.
Deletion of CDKIs increases iPSC reprogramming efficiency
To compare iPSC reprogramming efficiency of MEFs with p21−/−, p27−/−, and p18−/− genotypes, we measured the quantities of SSEA1+ iPSC colonies. As shown in Fig. 2a, like p21 loss, absence of p27 or p18 increased iPSC reprogramming efficiency compared to WT. SSEA1+ iPSC colonies with p27−/− or p18−/− all expressed pluripotency markers, Oct4, Nanog and Sox2, (Supplementary Figure 1a) and produced teratomas that could differentiate into all three germ layers (Supplementary Figure 1b). Moreover, absence of either p27 or p18 increased iPSC generation to an extent comparable to that of p21 deficiency not only with four factors (Oct4, Sox2, Klf4, and c-Myc) but also three factors (Oct4, Sox2, and Klf4) (Fig. 2a and Supplementary Figure 2a). Besides, p27 or p18 deletion mediated increase in iPSC generation efficiency was dose dependent (Fig. 2b and Supplementary Figure 2b). Ectopic expression of p27 or p18 in WT, p27−/−or p18−/− genotype MEFs all reduced iPSC reprogramming efficiency (Fig. 2c–e). To further validate these observations, we used p18 small-molecular inhibitor NSC23005 and p27 siRNA to treat during the reprogramming of WT MEFs. As shown in Fig. 2f, g, both NSC23005 treatment and p27 siRNA knockdown significantly promoted iPSC generation. These data showed that all CDKIs are critical for iPSC reprogramming, and reprogramming efficiency has no difference among them.

a SSEA1 positive colonies in p21−/−, p27−/−, and p18−/− iPSCs relative to that of WT iPSCs at day 14 after transduction with four factors. b Upper, SSEA1 positive colonies in p27+/−, p27−/−, p18+/−, and p18−/− iPSCs relative to that of WT iPSCs at day 14 after transduction with four factors. Lower, Western blot analysis of endogenous expression of p27 and p18 in various genotype MEFs. c–e Effects of ectopic expression of p27 or p18 on iPSC generation in WT, p18−/−, and p27−/− MEFs. Upper, numbers of SSEA1 positive colonies. Lower, Western blots analysis of ectopic expression of p18 and p27 in various genotype MEFs at day 4 of reprogramming after transduced with 4F and p27 or p18. f Effects of p18 inhibitor on iPSC generation in WT MEFs. Upper, numbers of SSEA1 positive colonies relative to untreated control. Lower, Western blots analysis of p18 expression under p18 inhibitor treated at day 4 of reprogramming after transduced with 4F. g Effects of p27 siRNA on iPSC generation in WT MEFs. Upper, numbers of SSEA1 positive colonies relative to vector control. Lower, Western blots analysis of p27 expression at day 4 of reprogramming after transduced with 4F and p27 SiRNA. Data are representative of two or three independent experiments. Error bars, ±SD. *p < 0.05; **p < 0.01, by two-tailed t test
Small-molecule inhibitors of CDKs can reduce iPSC generation as CDKIs
p21 and p27 are inhibitors of broad cyclin-CDK complexes, and p18 is a specific inhibitor of CDK4/6 kinases8,11,18,19. To test whether roles of p21, p27, and p18 in iPSC reprogramming are directly related with them as CDK inhibitors, we used small-molecule CDK2 and CDK4 inhibitors to treat MEFs with various genotypes during reprogramming. As shown in Fig. 3a, b, both the treatment of Small-molecule CDK2 and CDK4 inhibitors reduced all genotypes iPSC reprogramming efficiency. We also found that treatment with both CDK4 and CDK2 inhibitors almost blocked iPS generation in WT MEFs (supplemental Fig. 3a). To further validate CDK inhibitors’ function in iPS generation, the small-molecule CDK4/6 (Abemaciclib) and CDK7 inhibitors (THZ1) were used to treat WT MEFs with CDK4/6 small-molecule inhibitor Abemaciclib or CDK7 small-molecule inhibitor THZ1, respectively. As shown in supplemental Fig. 3b and c, both Abemaciclib and THZ1 reduced iPSC reprogramming efficiency. These results suggest that CDKIs-regulated iPSC reprogramming is directly related with their functions as CDK inhibitors.

a, b Effects of CDK2 (a) and CDK4 (b) inhibitor treatment on efficiency of iPSC generation in WT, p21−/−, p27−/−, and p18−/− MEFs. Inhibitors were added in the medium during reprogramming. Data are representative of two or three independent experiments. Error bars, ± SD. *p < 0.05; **p < 0.01, ***p < 0.001 by two-tailed t test
p27 and p18 are dispensable for genomic stability during reprogramming
DNA damage frequently occurs during somatic reprogramming5,20,21,22,23,24. We and others previously showed that absence of p53 or p21 could not only improve the reprogramming efficiency but also increase genomic instability13,14. p27 and p18 were also shown to be related with DNA damage25,26,27,28,29. To compare DNA damage status in various genotype MEFs during reprogramming, we examined the DNA double-strand breaks using anti-γ-H2AX staining in the transduced MEFs at Day 0, Day 4 and the iPSC colonies at Day 12 after transduction (Fig. 4a, b). There was no difference among all genotype cells at reprogramming Day 0 and Day 4, whereas γ-H2AX foci formation in p21−/− iPSC colonies at reprogramming Day 12 significantly increased when compared with other genotypes (Fig. 4a, b). The results at reprogramming Day 12 were validated using comet assays (Fig. 4c, d). These data suggest that unlike p21, p27, and p18 are dispensable for DNA damage repair during iPSC reprogramming.

a Representative images of γ-H2AX staining in various genotypes cells at reprogramming Day 0, Day 4, and Day 12. Bars: 50 μm. b Percentage of cells with γ-H2AX foci in (a). c Representative images of comet assay in various genotype cells at reprogramming Day 12. Bars: 200 μm. d Tail intensity of the cells in (c). e The karyotypic alterations in three clones each from three independent iPSCs at passage 5 derived from three passage 3 MEF cell lines per genotype were quantified. Graphic illustration of the number of chromosomal alterations (numerical gains, losses, and structural alterations) in the iPSCs. f Karyotypes from 20 metaphase cells from three sets of each three cell lines per type of iPSCs were analyzed under the microscope or after digital imaging and karyotyping. Images shown were taken with ×100 oil objective. Data are representative of two or three independent experiments. Error bars, ±SD. **p < 0.01, ***p < 0.001, by two-tailed t test
We further quantified the chromosomal alterations in three clones, each from three independent iPSCs at passage 5 derived from three passage 3 MEF cell lines per genotype. Chromosomal alterations, including chromosomal gains, losses, and structural abnormalities were observed and quantified in the iPSCs of all genotypes and the corresponding MEFs (Fig. 4e, f, and Supplementary Tables S1 to S6) as we previously described13. There was no difference in genomic stability among p18−/−, p27−/−, and WT genotypes, and only p21−/− caused significantly more chromosomal alterations in comparison to the others. These data showed that p27 and p18 were critical for somatic cell reprogramming efficiency but dispensable for genomic stability during iPSC reprogramming.
p27 and p18 are dispensable for genomic stability at iPSC stage
As p27 and p18 do not impair genomic stability during reprogramming, we further determined whether p27 and p18 are also dispensable for genomic stability at iPSC stage. To test this, we selected the best genomic stability cell lines in each genotype and then tested the centrosome number and the spindles after irradiation (IR) treatment. Significantly more abnormal centrosomes and spindles were found only in p21−/− iPS cell lines compared with WT, but there was no difference in p27−/−, p18−/−, and WT genotype iPS cell lines (Fig. 5a, b).

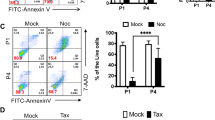
a Representative images of spindle abnormal phenotype. Bars: 1.5 μm. b Abnormal centrosomes in various iPSCs at the indicated time points post 2-Gy IR. c Tail intensity of the cells in various iPSCs at the indicated time points post 2-Gy IR. d Western blot assays of expression of cleaved PARP in various iPSCs at the indicated time points post 2-Gy IR. e FACS analysis of apoptosis at the indicated time points post 2-Gy IR. Data are representative of two or three independent experiments. Error bars, ±SD. **p < 0.01, ***p < 0.001, by two-tailed t test
To confirm the observation, we further tested the percentage of tail intensity in different genotype iPS cell lines after IR treatment. As shown in Fig. 5c–e, loss of p21 induced significantly higher percentage of tail intensity, less cleaved PARP formation, and less late apoptosis compared to WT, whereas loss of p18 or p27 had no effects. These data suggested that p27 and p18 are different from p21 and are dispensable for iPSC genomic stability during reprogramming and at iPSC stage.
Discussion
Overlapping mechanisms between reprogramming and tumorigenesis which were shown in the p53-p21 pathway represent a huge challenge for the therapeutic use of iPS technology2,13,14. The goal of the present study is to gain a better understanding of the roles of CDKIs in iPSC reprogramming and iPSC genomic stability to identify a more suitable molecular target and potentially improve the technology for iPSC generation. We found that compared with loss of p21, absence of p27 or p18 can promote the efficiency of iPSC generation without the induction of genomic instability.
Successful generation of iPS cells from somatic cells need reset of the patterns of cell cycle in those reprogramming cells, suggesting iPSC reprogramming was mainly controlled by a cell-division-rate-dependent model6,30. For example, p53 and its up and downstream regulators such as p16/p19, p21 limit iPSC generation mainly by cell cycle arrest and senescence2,3,4. Here, we found that like p53-p21, absence of p27 and p18 promoted efficiency of iPSC generation by regulating cell cycle. Moreover, using small-molecule CDK inhibitor treatment, we revealed that p21, p27, and p18-mediated iPSC reprogramming are directly associated with them as CDKIs.
Although it is known that loss of CDKIs promotes efficiency of iPSC generation, iPSC quality with absence of CDKI’s genotypes is still unclear. p21 was reported to be directly involved in cellular repair processes by binding to proliferating cell nuclear antigen (PCNA) and inhibiting DNA replication after DNA damage31,32, and p21 also played a role in aneuploidy formation33. p21-deficient mice developed spontaneous tumors at an average age of 16 months, whereas wild-type mice were tumor-free beyond 2 years of age34. Reduced p27 enhanced chromosomal instability in non-small-cell lung carcinomas and other cancers, and ectopic expression of p27 induces a significantly decrease in the accumulation of aneuploidy35,36. The cell cycle inhibitors p21 (Waf1/Cip1) and p27 (Kip1) were frequently downregulated in many human cancers, and correlated with a worse prognosis. Combined deficiency of p21 and p27 proteins in mice was linked to more aggressive spontaneous tumorigenesis, and resulted in a decreased lifespan37,38. p18 was also a known haploinsufficient tumor suppressor39,40, and loss of p18 resulted in widespread hyperplasia and organomegaly after birth of the mice39. We previously showed that loss of p21 during reprogramming can induce DNA damage accumulation13. Consistent with this, here we demonstrated that loss of p21 induces iPSC centrosome abnormal and reduces DNA repair ability. However, absence of p27 or p18 kept not only mitosis but also chromosome well during reprogramming and at iPSC stage, indicating that p27 and p18 are dispensable for iPSC genomic stability.
In summary, here we have reported a distinct role of CDKIs in somatic cell reprogramming. Deletion of p27 or p18 promotes reprogramming efficiency without reduction of genomic stability. Therefore, our results not only support that cell cycle regulation is critical for iPSC reprogramming, but also offer a strong rationale for targeting p27 and p18 in regenerative medicine.
References
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Hong, H. et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460, 1132–1135 (2009).
Kawamura, T. et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460, 1140–1144 (2009).
Li, H. et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136–1139 (2009).
Marion, R. M. et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460, 1149–1153 (2009).
Hanna, J. et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462, 595–601 (2009).
Utikal, J. et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460, 1145–1148 (2009).
Hirai, H., Roussel, M. F., Kato, J. Y., Ashmun, R. A. & Sherr, C. J. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell. Biol. 15, 2672–2681 (1995).
Guan, K. L. et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 8, 2939–2952 (1994).
Cerqueira, A. et al. Genetic characterization of the role of the Cip/Kip family of proteins as cyclin-dependent kinase inhibitors and assembly factors. Mol. Cell. Biol. 34, 1452–1459 (2014).
Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K. & Elledge, S. J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 (1993).
Kiyokawa, H. et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85, 721–732 (1996).
Li, Y. et al. The p53-PUMA axis suppresses iPSC generation. Nat. Commun. 2013, 2174 (2013).
Lake, B. B. et al. Context-dependent enhancement of induced pluripotent stem cell reprogramming by silencing Puma. Stem Cells 30, 888–897 (2012).
Li, H. et al. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell stem Cell 11, 845–852 (2012).
Zhu, S. et al. p18 inhibits reprogramming through inactivation of Cdk4/6. Sci. Rep. 6, 31085 (2016).
Lv, D. et al. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat. Commun. 8, 1454 (2017).
Reynisdottir, I. & Massague, J. The subcellular locations of p15(Ink4b) and p27(Kip1) coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev. 11, 492–503 (1997).
Chu, I. et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 128, 281–294 (2007).
Hussein, S. M. et al. Copy number variation and selection during reprogramming to pluripotency. Nature 471, 58–62 (2011).
Laurent, L. C. et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8, 106–118 (2011).
Blasco, M. A., Serrano, M. & Fernandez-Capetillo, O. Genomic instability in iPS: time for a break. EMBO J. 30, 991–993 (2011).
Kinoshita, T. et al. Ataxia-telangiectasia mutated (ATM) deficiency decreases reprogramming efficiency and leads to genomic instability in iPS cells. Biochem. Biophys. Res. Commun. 407, 321–326 (2011).
Quinlan, A. R. et al. Genome sequencing of mouse induced pluripotent stem cells reveals retroelement stability and infrequent DNA rearrangement during reprogramming. Cell Stem Cell 9, 366–373 (2011).
Liu, B. et al. DICER-dependent biogenesis of let-7 miRNAs affects human cell response to DNA damage via targeting p21/p27. Nucleic Acids Res. 43, 1626–1636 (2015).
Liontos, M. et al. The roles of p27(Kip1) and DNA damage signalling in the chemotherapy-induced delayed cell cycle checkpoint. J. Cell. Mol. Med. 14, 2264–2267 (2010).
Cuadrado, M., Gutierrez-Martinez, P., Swat, A., Nebreda, A. R. & Fernandez-Capetillo, O. p27Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res. 69, 8726–8732 (2009).
Payne, S. R. et al. p27kip1 deficiency impairs G2/M arrest in response to DNA damage, leading to an increase in genetic instability. Mol. Cell. Biol. 28, 258–268 (2008).
Park, B. J. et al. The haploinsufficient tumor suppressor p18 upregulates p53 via interactions with ATM/ATR. Cell 120, 209–221 (2005).
Singh, A. M. & Dalton, S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell 5, 141–149 (2009).
Cazzalini, O., Scovassi, A. I., Savio, M., Stivala, L. A. & Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 704, 12–20 (2010).
Li, R., Hannon, G. J., Beach, D. & Stillman, B. Subcellular distribution of p21 and PCNA in normal and repair-deficient cells following DNA damage. Curr. Biol. 6, 189–199 (1996).
Shen, K. C. et al. ATM and p21 cooperate to suppress aneuploidy and subsequent tumor development. Cancer Res. 65, 8747–8753 (2005).
Martin-Caballero, J., Flores, J. M., Garcia-Palencia, P. & Serrano, M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 61, 6234–6238 (2001).
Matsumoto, Y., Hayashi, K. & Nishida, E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 9, 429–432 (1999).
Meraldi, P., Lukas, J., Fry, A. M., Bartek, J. & Nigg, E. A. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat. Cell Biol. 1, 88–93 (1999).
Garcia-Fernandez, R. A. et al. Combined loss of p21(waf1/cip1) and p27(kip1) enhances tumorigenesis in mice. Lab. Investig. 91, 1634–1642 (2011).
Garcia-Fernandez, R. A. et al. Cooperative role between p21cip1/waf1 and p27kip1 in premature senescence in glandular proliferative lesions in mice. Histol. Histopathol. 29, 397–406 (2014).
Bai, F., Pei, X. H., Godfrey, V. L. & Xiong, Y. Haploinsufficiency of p18 (INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol. Cell. Biol. 23, 1269–1277 (2003).
Hossain, M. G. et al. Expression of p18(INK4C) is down-regulated in human pituitary adenomas. Endocr. Pathol. 20, 114–121 (2009).
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (81470315, 81772663 to Y.L.; 81572467, 81874078 to H.F.; 81421002, 81730006 to T.C.; 81400152 to H.G.); the Ministry of Science and Technology of China (2016YFA0100600 to T.C., 2017YFA0103102 to G.J.); National Key R&D Program of China (2018YFC1313000/2018YFC1313005 Y.L.); CAMS Initiative for Innovative Medicine (2016-I2M-1-017) to T.C.; Shanghai Jiao Tong University Medical Engineering Cross Fund (No. YG2017MS32), Collaborative Innovation Center for Translation Medicine at Shanghai Jiao Tong University School of Medicine (TM201502) to Y.L.; the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (No. 2014024), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (No. 20161310), New Hundred Talent Program (Outstanding Academic Leader) at Shanghai Municipal Health Bureau (2017BR021) to H.F., Local High Level University Construction Project of Shanghai Jiao Tong University School of Medicine to Y.L.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Y. Shi
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhan, Z., Song, L., Zhang, W. et al. Absence of cyclin-dependent kinase inhibitor p27 or p18 increases efficiency of iPSC generation without induction of iPSC genomic instability. Cell Death Dis 10, 271 (2019). https://doi.org/10.1038/s41419-019-1502-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1502-8
This article is cited by
-
Overabundance of Veillonella parvula promotes intestinal inflammation by activating macrophages via LPS-TLR4 pathway
Cell Death Discovery (2022)
-
Molecular study of the proliferation process of beta cells derived from pluripotent stem cells
Molecular Biology Reports (2022)
-
Roles Played by YY1 in Embryonic, Adult and Cancer Stem Cells
Stem Cell Reviews and Reports (2021)
