
Abstract
The c-Jun N-terminal kinase (JNK) pathway plays essential roles in regulating a variety of physiological processes including cell migration and invasion. To identify critical factors that regulate JNK-dependent cell migration, we carried out a genetic screen in Drosophila based on the loss-of-cell polarity-triggered cell migration in the wing epithelia, and identified MKK3 licorne (lic) as an essential regulator of JNK-mediated cell migration and invasion. We found that loss of lic suppressed ptc > scrib-IR or ptc > Egr triggered cell migration in the wing epithelia, and Rasv12/lgl−/− induced tumor invasion in the eye discs. In addition, ectopic expression of Lic is sufficient to induce JNK-mediated but p38-independent cell migration, and cooperate with oncogenic Ras to promote tumor invasion. Consistently, Lic is able to activate JNK signaling by phosphorylating JNK, which up-regulates the matrix metalloproteinase MMP1 and integrin, characteristics of epithelial–mesenchymal transition (EMT). Moreover, lic is required for physiological JNK-mediate cell migration in thorax development. Finally, expression of human MKK3 in Drosophila is able to initiate JNK-mediated cell migration, cooperates with oncogenic Ras to trigger tumor invasion, and rescue loss-of-lic induced thorax closure defect. As previous studies suggest that MKK3 specifically phosphorylates and activates p38MAPK, our data provide the first in vivo evidence that MKK3 regulates JNK-dependent cell migration and invasion, a process evolutionarily conserved from flies to human.
Similar content being viewed by others

Introduction
About 90% of cancer patients die from tumor metastasis rather than primary tumor growth1. Therefore, finding effective ways to prevent or even reverse tumor cell invasion is of great significance to the treatment of cancer. To investigate the underlying genetic mechanisms, several invasion and metastasis models have been established in Drosophila melanogaster2,3,4. For example, depletion of cell polarity genes such as scrib along the anterior/posterior (A/P) compartment boundary in the wing epithelia produces an invasive cell migration phenotype5, while loss of cell polarity cooperates with oncogenic Ras (RasV12) in the eye discs to promote tumor growth and invasion6. Previous work has identified the c-Jun N–terminal kinase (JNK) signaling as a crucial mediator of both invasive cell migration and tumor invasion in Drosophila7,8,9.
JNK belongs to the mitogen-activated protein kinase superfamily, and the JNK pathway plays crucial roles in many kinds of cellular behaviors, such as cell migration, proliferation, differentiation, apoptosis and stress reaction. The JNK pathway is highly conserved from Drosophila to human, while dysregulation of JNK signaling has been implicated in various human diseases, including cancer and neurodegenerative diseases10,11. Yet, it remains elusive how this pathway is tightly regulated in development, and factors that modulate this pathway have not been fully identified.
In mammalian cells, the p38 mitogen-activated protein kinase (MAPK) pathway is stimulated in response to a variety of environmental stresses and inflammatory stimuli. MKK3 is a protein kinase with dual specificity and belongs to the MAPK kinase family. Previous studies suggest that MKK3 specifically phosphorylates and activates p38 MAPK. In Drosophila, p38 MAPK is activated via dual phosphorylation at the Thr-Gly-Tyr motif by the MKK3 ortholog Licorne (Lic). For example, during oogenesis, Lic-p38 signaling is required in the germ line for correct asymmetric development of the egg12. Lic has also been shown to modulate other signaling pathways. For instance, Lic overexpression affects target gene expression of the Wingless (Wg) or Hippo pathway13,14. Consistently, MKK3 promotes nuclear localization of YAP via the actin cytoskeleton14. Despite all of the above, a role of MKK3 in JNK signaling has not been previously reported.
We have carried out a genetic screen in Drosophila, and found that lic is required for loss-of-cell polarity-triggered JNK-dependent invasive cell migration in the wing epithelia and oncogenic cooperation-induced JNK-mediated tumor invasion from the eye disc to the ventral nerve cord (VNC). In addition, ectopic expression of Lic activates JNK pathway by promoting JNK phosphorylation, triggers JNK-dependent invasive cell migration and cooperates with RasV12 to promote tumor invasion. Moreover, lic is required for physiological JNK-mediated cell migration in thorax development. Furthermore, genetic epistasis analysis suggests that Lic acts in parallel with Hep as a potential JNK kinase. Finally, we found that expression of human MKK3 in Drosophila also activates JNK signaling, triggers JNK-dependent cell migration, cooperates with oncogenic Ras to promote tumor invasion, and rescues loss-of-lic induced JNK-mediated thorax closure defect. Thus, we provide the first in vivo evidence that MKK3 regulates JNK-mediated cell migration and invasion, and this function of MKK3 is likely conserved from flies to human.
Results
lic is required for depletion of scrib-induced invasive cell migration
JNK signaling plays an important role in the regulation of cell migration and tumor invasion. Depletion of cell polarity genes, such as scrib, dlg and lgl, along the A/P compartment boundary of the Drosophila wing disc, triggers JNK-mediated invasive cell migration, a widely accepted in vivo model to study cell migration and invasion15,16,17.
To identify additional factors that regulate JNK-mediated cell migration and invasion, a candidate screen for dominant modifiers of ptc > scrib-IR induced invasive phenotype was carried out, in which the ptc-Gal4 driver was used to knock down scrib along the A/P boundary in the wing disc18. We have screened > 1000 UAS-RNAi lines from the Bloomington, Vienna Drosophila RNAi Center (VDRC) and National Institute of Genetics (NIG) stock centers targeting potential factors upstream of JNK, or factors that interact with JNK pathway components genetically or biochemically as reported in the literature. We have previously identified Rho1, wnd, Src42, ben and dUev1 as modulators of JNK-mediated cell invasion from the screen18,19,20. The screen is still ongoing, as more RNAi lines are being added to the stock centers.
Compared with the ptc > GFP control (Fig. 1a–d), RNAi-mediated depletion of scrib resulted in a large number of cells delaminated from the A/P boundary and migrated to the posterior compartment (Fig. 1e, f), accompanied by the upregulation of MMP1 levels (Fig. 1g)21, which is one of the important molecular features of epithelial–mesenchymal transition (EMT)22,23. puckered (puc) is a transcriptional target of JNK signaling, but also encodes a JNK phosphatase that inhibits JNK activity24,25. As a positive control, Puc expression blocked ptc > scrib-IR induced cell migration and MMP1 activation (Fig. 1h–j), confirmed that both phenotypes depend on JNK signaling. To rule out the possibility of Gal4 titration by another UAS transgene, we used UAS-LacZ as a negative control, and confirmed that LacZ expression did not affect the cell migration phenotype (Supplementary Figure 1). From the screen, expression of two independent lic-RNAi lines were found to significantly inhibit depletion-of-scrib triggered cell migration and MMP1 upregulation (Fig. 1k–q). Furthermore, knockdown of lic significantly suppressed scrib-IR-induced JNK signaling activation (revealed by expression of the TRE-RFP and puc-LacZ reporters), JNK phosphorylation and accumulation of β-integrin (Supplementary Figure 2). The knockdown efficiencies of the lic-RNAi lines were verified by qRT-PCR (Supplementary Figure 3). Taken together, these data suggest that lic is physiologically required for JNK-dependent invasive cell migration triggered by loss of cell polarity.

Fluorescent micrographs of third instar wing discs (a–p) are shown. The dotted line depicts the area where cell migration occurs with ptc > GFP in the imaginal wing disc (a). ptc-Gal4 driven overexpression of scrib-RNAi promoted cells from A/P boundary migrating to posterior and MMP1 activation (e–g), compared to the GFP-expressing control (b–d). As a positive control, Puc blocked the cell migration phenotype and MMP1 staining (h–j). Expression of two independent lic-RNAi lines significantly inhibited cell invasion and MMP1 expression (k–p). Scale bars, 20 μm. The number of migrated cells were quantified and shown in (q), and one-way ANOVA test was used to calculate statistical significance, n ≥ 17, mean + s.d., ***P < 0.001
Lic promotes JNK-dependent cell migration
Next, we sought to test whether Lic promotes JNK-dependent cell migration in vivo. Compared with the ptc > GFP control (Fig. 2a–f), ectopic expression of Lic promotes some of the GFP-positive cells migrated to the posterior part (Fig. 2g, s), along with upregulated staining of MMP1 (Fig. 2h) and integrin (Fig. 2i), another biomarker for EMT26,27. Consistent with previous reports, both MMP1 and integrin were activated autonomously and non-autonomously4. Importantly, ectopic Lic-induced EMT-like invasive phenotypes were fully inhibited by the expression of Puc (Fig. 2m–o), or a dominant negative form of Bsk (BskDN) encoding the Drosophila JNK ortholog (Supplementary Figure 4).

Fluorescent (a–e, g–k, m–q) and light (f, l, r) micrographs of third instar wing discs are shown. Compared with the control (a–f), ptc-Gal4-driven overexpression of Lic induced cell migration (g), elevated expression of MMP1 (h), β-integrin (i), TRE-RFP (j), JNK phosphorylation (k) and puc-LacZ (l), which were inhibited by Puc expression (m–r). Scale bars, 20 μm. Statistics of migrated cell numbers was shown in (s), and One-way ANOVA test was used to calculate statistical significance, n ≥ 14, mean + s.d., ***P < 0.001
These data suggest that JNK signaling is required for Lic-induced EMT-like cell invasion. To investigate whether Lic is sufficient to activate JNK signaling, we checked the expression of a puc-LacZ reporter, a commonly used readout of JNK signaling. Compared with the control (Fig. 2f), puc-LacZ expression was considerably activated along the A/P boundary by ectopic Lic expression driven by ptc-Gal4 (Fig. 2l), which could be significantly suppressed by co-expression of Puc (Fig. 2r). A TRE-RFP reporter containing four copies of an optimal Jun/Fos (AP-1) heterodimer binding site fused with RFP has been used to characterize the activation of JNK signaling28. Compared with the control (Fig. 2d), expression of Lic dramatically enhanced TRE-RFP expression (Fig. 2j), which was abolished by expressing Puc (Fig. 2p). Consistent with previous reports that JNK signaling propagates in the wing disc, Lic is able to activate JNK signaling both autonomously and non-autonomously (Supplementary Figure 5). Lic encodes the Drosophila MKK3 that regulates the p38 signaling in immunity29, oogenesis12, stress response30 and tissue growth14,31, yet its role in JNK signaling has not been previously reported. To investigate whether Lic is a kinase upstream of JNK, we checked JNK phosphorylation by a specific anti-p-JNK antibody32. We found that ectopic Lic was sufficient to promote JNK phosphorylation (Fig. 2k, q; Supplementary Figure 6). Thus, Lic is sufficient to activate JNK-dependent invasive cell migration in the wing disc. In contrast, expressing a kinase dead form of Lic (LicKD) failed to produce any of the above phenotypes (Supplementary Figure 7), indicating that the kinase activity is required to induce invasive cell migration.
p38b has been proposed to play the central role in Drosophila p38 signaling33,34,35,36. However, Lic-induced JNK pathway activation and invasive cell migration were not affected by expressing a p38b-RNAi or a dominant-negative allele of p38b (p38bDN) (Supplementary Figure 8). Together, these data suggest that Lic activates the JNK signaling by promoting JNK phosphorylation in a mechanism independent of p38.
Human MKK3 promotes JNK-dependent cell migration in Drosophila
Given that Lic encodes the Drosophila MKK3 ortholog and triggers JNK-dependent cell migration, it is of interest to know whether MMK3 retains Lic’s ability to promote JNK-mediated cell migration. A few in vitro studies have suggested that MKK3 is possibly involved in cancer cell invasion37, but this function of MKK3 has not been confirmed in vivo, and it remains unclear whether it is JNK dependent. To address this issue, a UAS-MKK3 transgene was generated and ectopically expressed the human MKK3 along the A/P boundary in the wing disc. Compared with the control (Fig. 3a–f), expression of MKK3 phenocopied that of Lic with induced cell migration (Fig. 3g, s), JNK phosphorylation (Fig. 3k), upregulation of MMP1, β-Integrin, TRE-RFP and puc-LacZ (Fig. 3h–j, l). Similar to Lic, these functions of MKK3 depend on JNK (Fig. 3m–r, Supplementary Figure 9), but not p38 (Supplementary Figure 10). Collectively, these data reveal that human MKK3 is functionally conserved with Drosophila Lic to promote JNK-dependent epithelia cell invasion in vivo, which is of great interest for future drug research targeting MKK3.

Fluorescent (a–e, g–k, m–q) and light (f, l, r) micrographs of third instar wing discs are shown. Compared with the control (a–f), ectopic expression of MKK3 induced cell migration (g) accompanied by activation of MMP1 (h), integrin (i), TRE-RFP (j), p-JNK (k) and puc-LacZ (l), which were significantly suppressed by co-expression of Puc (m–r). Scale bars, 20 μm. Statistics of migrated cell numbers was shown in (s), and One-way ANOVA test was used to calculate statistical significance, n ≥ 14, mean + s.d., ***P < 0.001
Lic is required for Ras v12 /lgl −/−-induced tumor invasion
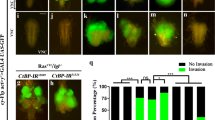
Expression of oncogenic Ras is able to cooperate with loss of cell polarity, e.g. lgl mutation, in eye disc clones to promote tumor-like growth (Fig. 4a) and invasion into the ventral nerve cord (VNC) (Fig. 4e)6,38, a previously established in vivo model to study the mechanism of tumor invasion19,32,39. Consistent with previous data indicating that the JNK signaling plays a crucial role in Rasv12/lgl−/− -induced tumor invasion19,20, expression of Puc completely blocked tumor cell invasion to VNC (Fig. 4f, i). We found that Rasv12/lgl−/− -induced tumor invasion from eye to VNC was significantly inhibited by knocking-down lic or in heterozygous lic mutants (Fig. 4g–i). Intriguingly, the sizes of primary tumors in eye discs appeared to be not affected by JNK inactivation or loss of lic (Fig. 4b–d), implying Lic is specifically required for Rasv12/lgl−/−-triggered JNK-dependent tumor invasion.

Dorsal views of the cephalic complexes (a–d, j–m) and VNCs (e–h, n–q) are shown. RasV12/lgl−/− induced tumor invasion into the VNC (e) was fully blocked by expression of Puc (f), and significantly suppressed by expressing a lic-RNAi (g) or in lic heterozygous mutants (h). A quantification of tumor invasion phenotype in e–h was shown in (i). One-way ANOVA test was used to calculate statistical significance, mean + s.d., n.s., P > 0.05; ***P < 0.001, and more than 30 samples were counted for each group. Eye disc clones expressing Lic (j) or RasV12 (k) never invaded into VNC (n, o), while massive tumor growth and invasion to the VNC were observed by co-expression of RasV12 and Lic (l, p), or RasV12 and MKK3 (m, q). Scale bars, 200 μm in (a–d, j–m) and 100 μm in (e–h, n–q). A quantification of tumor invasion phenotype in n–q was shown in (r). One-way ANOVA test was used to calculate statistical significance, mean + s.d., ***P < 0.001, and more than 40 samples was counted for each group
Lic/MKK3 synergizes with Ras V12 to promote tumor growth and invasion
Loss of cell polarity or activation of cell morphogenetic genes induces JNK-dependent invasive cell migration and synergizes with RasV12 to initiate tumor growth and invasion40,41. Given that Lic triggers JNK-dependent invasive cell migration, we speculated that Lic might be able to cooperate with RasV12 to promote tumor growth and invasion. Overexpression of Lic alone does not induce tumorigenesis or invasion (Fig. 4j, n), which is similar to that of RasV12 (Fig. 4k, o)6. When Lic was simultaneously expressed with RasV12, tumors formed from eye discs and invaded into the VNC region 8 days after egg laying (Fig. 4l, p, r), suggesting an oncogenic cooperation between Lic and RasV12 is sufficient to promote tumor growth and invasion. Importantly, such synergistic effect also occurred between MKK3 and RasV12 (Fig. 4m, q, r), suggesting that this function of Lic/MKK3 has been evolutionary conserved.
Lic modulates JNK activity in parallel with Hep
Next, to investigate how Lic regulates JNK signaling, we performed epistasis analysis between Lic and JNK pathway components. As shown above, ptc > Lic-induced invasive cell migration and JNK activation were significantly inhibited by expressing Puc (Fig. 2m–s) or BskDN (Supplementary Figure 4), suggesting that Lic acts upstream of Bsk. Next, we checked the genetic interaction between Lic and other kinases upstream of Bsk. Hemipterous (Hep) is a serine/threonine protein kinase involved in the JNK pathway by phosphorylating Bsk42,43. Both Lic and Hep belong to the mitogen-activated protein kinase kinase family (MAPKKs), while Wnd and dTAK1 are MAPKKKs that activate JNK via Hep activation. We found that knockdown of hep, dTAK1 or wnd did not inhibit Lic-triggered cell migration (Fig. 5a-d, m and Supplementary Figure 11a–e). On the other hand, ectopic Hep-induced cell migration (Fig. 5e) was not suppressed by depleting lic (Fig. 5f), but was synergistically enhanced by co-expressing Lic or MKK3 (Fig. 5g, h, n). Egr encodes the Drosophila TNF ortholog that activates JNK-mediated cell migration (Fig. 5i), which was suppressed partially in heterozygous hep or lic mutants (Fig. 5j, k), but completely in transheterozygous mutants (Fig. 5l, o). Collectively, these data suggest that Lic likely activates JNK in parallel with Hep (Supplementary Figure 11f).

Fluorescent micrographs of third instar wing discs (a–l) are shown. Compared with the GFP-expressing control (a), RNAi-mediated depletion of hep, dTAK1 or wnd failed to suppress ptc > Lic induced cell invasion (b–d). ptc-Gal4 driven expression of HepWT induced a large number of cells migrating to the posterior of wing disc, which was not suppressed by expressing GFP (e) or lic-RNAi (f), but was synergistically enhanced by co-expressing Lic (g) or MKK3 (h). ptc > Egr induced cell migration (i) was partially inhibited in hep (j) or lic (k) heterozygous mutants, and fully blocked in their transheterozygous mutants (l). Scale bars, 20 μm. Statistics of migrated cell numbers was shown in (m–o), and One-way ANOVA test was used to calculate statistical significance, n ≥ 18, mean + s.d., n.s., P > 0.05; ***P < 0.001
Lic regulates physiological JNK-dependent thorax closure in development
Thorax closure serves as another in vivo model to study cell migration in Drosophila development. JNK signaling is crucial for thorax closure44, as reduced JNK activity results in a dorsal cleft phenotype in the thorax45. Consistent with this, endogenous JNK activation, indicated by puc-LacZ expression, was detected in the dorsal patch of the wing disc (Fig. 6a)46. RNAi mediated downregulation of lic under the control of pannier promoter (pnr-Gal4) abrogated puc-LacZ expression (Fig. 6b), and produced a thorax cleft (Fig. 6d) that phenocopies that of JNK inactivation45. The thorax defect was further enhanced in heterozygous bsk mutants (Fig. 6f), but was suppressed in heterozygous puc mutants (Fig. 6h) in which the endogenous JNK activity is increased47. Therefore, lic is required for physiological JNK-mediated cell migration in thorax closure. Intriguingly, depletion-of-lic-induced thorax defect was suppressed by expressing MKK3 (Fig. 6j), implying this function of Lic has been conserved in evolution as well.

Light micrographs of Drosophila wing discs (a, b) and adult thoraxes (c–j) are shown. The endogenous puc expression pattern in the notum region of wing disc (a) was impeded by knockdown lic (b). Compared with the control thorax (c), loss of Lic-produced a thorax cleft phenotype (d), which was enhanced in heterozygous bsk mutants (f), and restored in heterozygous puc mutants (h), or by overexpression of MKK3 (j), whereas heterozygous mutation in bsk or puc, or expression of MKK3 gave no obvious phenotype (e, g, i, respectively)
Lic-induced JNK-mediated cell invasion is not a consequence of cell death
Previous studies have shown that JNK pathway activation promotes cell invasion in a context-dependent manner, but cell death in a non-tissue-specific manner. For instance, activation of JNK along the A/P compartment boundary in wing discs by ptc > Hep triggers both cell death and invasion (Supplementary Figure 12f, g and j), while GMR > Hep only induces cell death, but not cell invasion in eye discs (Supplementary Figure 12a, b and e). Consistently, we found that Lic promotes JNK-mediated cell invasion in a tissue-specific manner, and cell death non-tissue specifically (Supplementary Figure 12c, d, h and i). These data suggest cell death and cell invasion are independent outcomes of JNK activation. Consistent with this notion, expression of the baculovirus protein p35, which has been reported to inhibit programmed cell death in Drosophila48,49, fully suppressed ptc > Lic-triggered cell death (Fig. 7g, h, j), but had no effect on cell invasion and MMP1 activation (Fig. 7d–f, i). Taken together, the above results suggest that Lic-induced cell invasion is a direct outcome of JNK activation, but not a secondary effect of cell death.

Fluorescent micrographs of third instar wing discs are shown (a–h). ptc > Lic induced cell death (g), but not cell migration (a, b) and MMP1 activation (c), was inhibited by p35 overexpression (d–f, h). Scale bars, 20 μm. The statistical analysis of the number of migrated cells and apoptotic cells are shown in (i, j). One-way ANOVA test was used to calculate statistical significance, n ≥ 15, mean + s.d., n.s., P > 0.05; **P < 0.01
Discussion
lic encodes the Drosophila MKK3 ortholog that has been previously characterized as the MAP kinase kinase regulating p38 signaling in cell growth, stress response, innate immunity and asymmetric egg development in oogenesis12. In this study, we provide the first in vivo evidence that Lic acts as an essential regulator of the JNK signaling crucial for invasive cell migration triggered by the loss-of-cell polarity in the wing epithelia and oncogenic cooperation induced eye tumor invasion to the VNC. Furthermore, Lic regulates physiological JNK-dependent cell migration in thorax development. Finally, these functions of Lic are evolutionarily conserved as human MKK3 appears to work similarly in flies. Interestingly, MKK3 has previously been implicated in tumor cell invasion in vitro37,50, yet this function has not been confirmed in vivo, and its underlying mechanism remains elusive. Thus, our results not only shed light on the mechanism of MKK3-mediated tumor cell invasion, but also provide potential therapeutic strategies for MKK3-related cancer treatment.
Although previous studies have characterized Lic as a MAPKK for the p38 kinase, our data suggest that Lic regulates JNK signaling in cell invasion via a mechanism that is independent of p38. Firstly, loss of lic, but not p38, suppressed loss-of-cell polarity-triggered JNK-dependent cell invasion (Fig. 1l, o, q and data not shown). Secondly, ectopic Lic-triggered JNK activation and cell invasion were not affected by loss of p38 (Supplementary Figure 8a–f). Thirdly, Lic acts in parallel with Hep, as depletion of lic did not suppress ectopic Hep-triggered cell invasion (Fig. 5e, f, n), and vice versa (Fig. 5a, b and Supplementary Figure 11a, b and e). It is plausible that Lic acts as another JNK kinase that performs redundant function with Hep (Supplementary Figure 11f), since co-expression of Lic and Hep displays synergistic effect in promoting cell invasion (Fig. 5g, n), and Egr-triggered cell invasion is only partially suppressed in heterozygous hep or lic mutants, but fully blocked in transheterozygous double mutants (Fig. 5i–l, o).
A close relationship between cell death and migration has been reported51,52,53, and under certain situations dying cells could cause compensatory proliferation and subsequent migration of surrounding cells54,55,56. Consistent with the role of JNK signaling in cell death, ectopic expression of Lic triggers not only EMT-like cell invasion, but also cell death marked by acridine orange (AO) staining (Fig. 7a–c and g). Blocking cell death by expressing the baculovirus protein p35 effectively abolished Lic-induced cell death, but does not affect Lic-initiated cell invasion and MMP1 activation (Fig. 7d–f, h–j), suggesting Lic-triggered cell invasion is a direct outcome of JNK signaling activation, but not a secondary effect caused by cell death.
Materials and methods
Drosophila stocks and genetics
Stocks were raised on standard Drosophila media and crosses were performed at 25 °C, cell migration assay were performed at 29 °C. Following fly stocks have been described previously: w11184; TRE-RFP28; GMR-Gal4, ey-Gal4, pnr-Gal4, ptc-Gal4, UAS-RasV12, lgl4, UAS-HepWT20; UAS-GFP, UAS-p35, UAS-BskDN, UAS-Puc, pucE69, UAS-scrib-RNAi18; GFP-RNAi57; UAS-Lic14 hep158; bsk159; licG025260; UAS-EgrRegg161 and UAS-HepCA62. Strains obtained from the Bloomington Drosophila Stock Center (BDSC) are hs-Gal4 (#1799), UAS-LacZ (#3956), UAS-p38b-RNAi (#29405), UAS-lic-RNAi (#31643), UAS-p38bDN (#59005), UAS-hep-RNAi (#28710), UAS-dTAK1-RNAi (#31045) and UAS-wnd-RNAi (#27525). Strains received from the Vienna Drosophila RNAi Center (VDRC) are UAS-lic-RNAi (#20166); UAS-wnd-RNAi (#13786) and UAS-hep-RNAi (#26929). UAS-dTAK1-RNAi (#1388R-2) is acquired from the National Institute of Genetics (NIG-FLY). Fluorescently labeled invasive tumors were produced by the following strains: y, w, ey-Flp; tub-Gal80, FRT40A; Act > y+ > Gal4, UAS-GFP (40A tester), lgl4 FRT40A UAS-RasV12 (40A tester) and ey-Flp, Act > y+ > Gal4, UAS-GFP63. UAS-LicKD were gifts from Prof. Haiyun Song.
UAS-MKK3 transgenic flies
The MKK3 coding region was amplified by RT-PCR from 293 cell line, and analyzed by standard sequencing. The pUAST-MKK3 construct was introduced into the germ line by injection and UAS-MKK3 transgenic lines were established by standard genetics.
Antibodies
The following primary antibodies were used: mouse anti-MMP1 (1:200), rabbit anti-phospho-JNK (1:200, Calbiochem, San Diego, CA, USA) and mouse-anti-β-Gal (1:500, DSHB). The following secondary antibodies were used: anti-mouse CY3 (1:1000, CST) and anti- rabbit CY3 (1:1000, CST). The following primary antibodies were used for western blot analysis: rabbit anti-p-JNK (9251S, 1:1,000, Cell Signaling Technology), Rabbit anti-JNK (sc-7345, 1:500, Santa Cruz Biotechnology).
X-gal staining
Eye and wing discs were dissected from third instar larvae in PBST (1× PBS pH 7.0, 0.1% Triton X-100) and stained for β-galactosidase activity as described64.
AO staining
Eye and wing discs were dissected from 3rd instar larvae in 1× PBS (phosphate-buffered saline) and incubated in 1 × 10−5 M acridine orange (AO) for 5 min at room temperature65.
Imaging of fly eyes and wings
Three-day-old flies were collected and frozen at −80 °C. When taking pictures, flies were unfrozen at room temperature and placed on 1% agarose plate. Light images of eye was taken by OLYMPUS stereo microscope SZX16. Wings were dissected and placed on slide with alcohol/glycerol (1:1) buffer. Light images of wing were taken by OLYMPUS BX51 microscope.
qRT-PCR
Total RNA was extracted from flies’ cephalosome of indicated genotype using PureLink RNA Mini Kit (Ambion). Total RNA was reverse-transcribed into cDNA with the PrimeScript RT Master Mix (Takara), quantitative PCR was performed with SYBR Premix ExTaq II (Takara) and quantified by the Stratagene MX3000P system (Stratagene). RP49 was used as an internal control. The following primers were used for real-time PCR.
lic, sense primer: 5′-GGCCGCTACCCATACGACAA-3′;
lic, antisense primer: 5′-ACTGTCCTCAACCACCTGA-3′.
p38b, sense primer: 5′-GAAGCGCACCTATCGGGAAC-3′;
p38b, antisense primer: 5′-GACATCCAGCAGACCAATAA-3′.
Rp49, sense primer: 5′-CCACCAGTCGGATCGATATGC-3′;
Rp49, antisense primer: 5′-CTCTTGAGAACGCAGGCGACC-3′.
References
Valastyan, S. & Weinberg, R. A. Tumor metastasis: molecular insights and evolving paradigms. Cell 2, 275–292 (2011).
Gonzalez, C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 3, 172–183 (2013).
Rudrapatna, V. A., Cagan, R. L. & Das, T. K. Drosophila cancer models. Dev. Dyn. 1, 107–118 (2012).
Ma, X. et al. Hippo signaling promotes JNK-dependent cell migration. Proc. Natl Acad. Sci. USA 8, 1934–1939 (2017).
Miles, W. O., Dyson, N. J. & Walker, J. A. Modeling tumor invasion and metastasis in Drosophila. Dis. Model Mech. 6, 753–761 (2011).
Igaki, T., Pagliarini, R. A. & Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 11, 1139–1146 (2006).
Uhlirova, M. & Bohmann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 22, 5294–5304 (2006).
Leong, G. R., Goulding, K. R., Amin, N., Richardson, H. E. & Brumby, A. M. Scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol. 7, 62 (2009).
Wu, M., Pastor-Pareja, J. C. & Xu, T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 7280, 545–U165 (2010).
Nateri, A. S., Spencer-Dene, B. & Behrens, A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 7056, 281–285 (2005).
Morfini, G. et al. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci. 7, 907–916 (2006).
Suzanne, M. et al. The Drosophila p38 MAPK pathway is required during oogenesis for egg asymmetric development. Genes Dev. 11, 1464–1474 (1999).
Schertel, C. et al. Systematic screening of a Drosophila ORF library in vivo uncovers Wnt/Wg pathway components. Dev. Cell. 2, 207–219 (2013).
Huang, D. et al. Regulation of Hippo signalling by p38 signalling. J. Mol. Cell Biol. 4, 328–337 (2016).
Vidal, M., Larson, D. E. & Cagan, R. L. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell. 1, 33–44 (2006).
Hainaut, P. & Plymoth, A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr. Opin. Oncol. 1, 50–51 (2013).
Portela, M. & Richardson, H. E. Death takes a holiday--non-apoptotic role for caspases in cell migration and invasion. EMBO Rep. 2, 107–108 (2013).
Ma, X. et al. Rho1-Wnd signaling regulates loss-of-cell polarity-induced cell invasion in Drosophila. Oncogene 7, 846–855 (2016).
Ma, X. et al. Src42A modulates tumor invasion and cell death via Ben/dUev1a-mediated JNK activation in Drosophila. Cell Death Dis. 4, e864 (2013).
Ma, X. et al. dUev1a modulates TNF-JNK mediated tumor progression and cell death in Drosophila. Dev. Biol. 2, 211–221 (2013).
Elsum, I., Yates, L., Humbert, P. O. & Richardson, H. E. The Scribble-Dlg-Lgl polarity module in development and cancer: from flies to man. Essays Biochem. 53, 141-168 (2012).
Yamben, I. F. et al. Scrib is required for epithelial cell identity and prevents epithelial to mesenchymal transition in the mouse. Dev. Biol. 1, 41–52 (2013).
Elsum, I. A., Martin, C. & Humbert, P. O. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J. Cell. Sci. 17, 3990–3999 (2013).
Martin-Blanco, E. et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 4, 557–570 (1998).
McEwen, D. G. & Peifer, M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 17, 3935–3946 (2005).
Yamakoshi, S. et al. Expression of mesenchymal-related genes by the bovine trophectoderm following conceptus attachment to the endometrial epithelium. Reproduction 3, 377–387 (2012).
Yue, J. N. et al. Epigenetic silencing of miR-483-3p promotes acquired gefitinib resistance and EMT in EGFR-mutant NSCLC by targeting integrin beta 3. Oncogene 31, 4300–4312 (2018).
Chatterjee, N. & Bohmann, D. A versatile PhiC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS ONE 4, e34063 (2012).
Han, Z. S. et al. A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol. Cell. Biol. 6, 3527–3539 (1998).
Inoue, H. et al. A Drosophila MAPKKK, D-MEKK1, mediates stress responses through activation of p38 MAPK. EMBO J. 19, 5421–5430 (2001).
Cully, M. et al. A role for p38 stress-activated protein kinase in regulation of cell growth via TORC1. Mol. Cell. Biol. 2, 481–495 (2010).
Ma, X. et al. Myc suppresses tumor invasion and cell migration by inhibiting JNK signaling. Oncogene 22, 3159–3167 (2017).
Seisenbacher, G., Hafen, E. & Stocker, H. MK2-dependent p38b signalling protects Drosophila hindgut enterocytes against JNK-induced apoptosis under chronic stress. PLoS Genet. 8, e1002168 (2011).
Vrailas-Mortimer, A. et al. A Muscle-Specific p38 MAPK/Mef2/MnSOD Pathway Regulates Stress, Motor Function, and Life Span in Drosophila. Dev. Cell. 4, 783–795 (2011).
Belozerov, V. E., Lin, Z. Y., Gingras, A. C., McDermott, J. C. & Siu, K. W. M. High-Resolution protein interaction map of the drosophila melanogaster p38 mitogen-activated protein kinases reveals limited functional redundancy. Mol. Cell. Biol. 18, 3695–3706 (2012).
Tian, L. L. et al. Thep38 pathway regulates oxidative stress tolerance by phosphorylation of mitochondrial protein IscU. J. Biol. Chem. 46, 31856–31865 (2014).
Shin, I., Kim, S., Song, H., Kim, H. R. & Moon, A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway: its critical role in invasion and migration of breast epithelial cells. J. Biol. Chem. 15, 14675–14683 (2005).
Brumby, A. M. & Richardson, H. E. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 21, 5769–5779 (2003).
Ma, X. et al. PP6 disruption synergizes with oncogenic ras to promote jnk-dependent tumor growth and invasion. Cell Rep. 13, 2657–2664 (2017).
Brumby, A. M. et al. Identification of novel ras-cooperating oncogenes in drosophila melanogaster: a rhogef/rho-family/jnk pathway is a central driver of tumorigenesis. Genetics 1, 105–125 (2011).
Vidal, M., Warner, S., Read, R. & Cagan, R. L. Differing src signaling levels have distinct outcomes in drosophila. Cancer Res. 21, 10278–10285 (2007).
Sluss, H. K. et al. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 21, 2745–2758 (1996).
RiesgoEscovar, J. R., Jenni, M., Fritz, A. & Hafen, E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 21, 2759–2768 (1996).
Stronach, B. & Perrimon, N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes & Dev. 3, 377–387 (2002).
Zeitlinger, J. & Bohmann, D. Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 17, 3947–3956 (1999).
Campuzano, S. & Modolell, J. Patterning of the Drosophila nervous system: the achaete-scute gene complex. Trends Genet. 6, 202–208 (1992).
Taniguchi, K. et al. D-JNK signaling in visceral muscle cells controls the laterality of the Drosophila gut. Dev. Biol. 311, 251-263 (2007).
Ohtsubo, T., Kamada, S. & Tsujimoto, Y. [Inhibition of apoptosis by a baculovirus p35 gene]. Nihon rinsho Jpn. J. Clin. Med. 7, 1907–1911 (1996).
Sahdev, S., Saini, K. S. & Hasnain, S. E. Baculovirus P35 protein: an overview of its applications across multiple therapeutic and biotechnological arenas. Biotechnol. Prog. 2, 301–312 (2010).
Behren, A. et al. Phenotype-assisted transcriptome analysis identifies FOXM1 downstream from Ras-MKK3-p38 to regulate in vitro cellular invasion. Oncogene 10, 1519–1530 (2010).
Yang, L., Cao, Z., Yan, H. & Wood, W. C. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res. 20, 6815–6824 (2003).
Huang, Q. et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 7, 860–866 (2011).
Boehm, D. et al. Caspase-3 is involved in the signalling in erythroid differentiation by targeting late progenitors. PLoS ONE 5, e62303 (2013).
Chen, F. JNK-Induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2, 379–386 (2012).
Yamamoto, M., Ohsawa, S., Kunimasa, K. & Igaki, T. The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition. Nature 7640, 246–250 (2017).
Sun, G. P. & Irvine, K. D. Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev. Biol. 1, 139–151 (2011).
Wang, X. Pontin/Tip49 negatively regulates JNKmediated cell death in Drosophila. Cell Death Discov. 4, 74 (2018).
Fernandez, B. G., Jezowska, B. & Janody, F. Drosophila actin-Capping Protein limits JNK activation by the Src proto-oncogene. Oncogene 16, 2027–2039 (2014).
Li, P. et al. Tankyrase mediates K63-Linked ubiquitination of jnk to confer stress tolerance and influence lifespan in Drosophila. Cell Rep. 2, 437–448 (2018).
Baril, C., Sahmi, M., Ashton-Beaucage, D., Stronach, B. & Therrien, M. The PP2C Alphabet is a negative regulator of stress-activated protein kinase signaling in Drosophila. Genetics 2, 567–579 (2009).
Igaki, T. et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 12, 3009–3018 (2002).
Kanda, H., Igaki, T., Kanuka, H., Yagi, T. & Miura, M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J. Biol. Chem. 32, 28372–28375 (2002).
Pagliarini, R. A. & Xu, T. A genetic screen in Drosophila for metastatic behavior. Science 5648, 1227–1231 (2003).
Xue, L. & Noll, M. Drosophila female sexual behavior induced by sterile males showing copulation complementation. Proc. Natl Acad. Sci. USA 7, 3272–3275 (2000).
Ma, X. et al. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res. 2, 425–431 (2012).
Acknowledgements
We thank Bloomington, VDRC, National Institute of Genetics (NIG-FLY), the Core Facility of Drosophila Resource and Technology at SIBCB, CAS and Dr. Haiyun Song for fly stocks and reagents, Dr. Margaret Ho for comments and suggestion. This work is supported by the National Natural Science Foundation of China (31571516, 31771595) and Shanghai Committee of Science and Technology (09DZ2260100, 18430711600, 18140900400).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Raschellà
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, Y., zhang, D., Guo, X. et al. MKK3 modulates JNK-dependent cell migration and invasion. Cell Death Dis 10, 149 (2019). https://doi.org/10.1038/s41419-019-1350-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1350-6
This article is cited by
-
Comprehensive bioinformatic analysis of the expression and prognostic significance of TSC22D domain family genes in adult acute myeloid leukemia
BMC Medical Genomics (2023)
-
The CRTC-CREB axis functions as a transcriptional sensor to protect against proteotoxic stress in Drosophila
Cell Death & Disease (2022)
-
Epidermal growth factor receptor cascade prioritizes the maximization of signal transduction
Scientific Reports (2022)
-
ANKFN1 plays both protumorigenic and metastatic roles in hepatocellular carcinoma
Oncogene (2022)
-
Usp8 promotes tumor cell migration through activating the JNK pathway
Cell Death & Disease (2022)
