
Abstract
Protein acetylation has a crucial role in energy metabolism. Here we performed the first large-scale profiling of acetylome in rat islets, showing that almost all enzymes in core metabolic pathways related to insulin secretion were acetylated. Label-free quantitative acetylome of islets in response to high glucose revealed hyperacetylation of enzymes involved in fatty acid β-oxidation (FAO), including trifunctional enzyme subunit alpha (ECHA). Acetylation decreased the protein stability of ECHA and its ability to promote FAO. The overexpression of SIRT3, a major mitochondrial deacetylase, prevented the degradation of ECHA via decreasing its acetylation level in β-cells. SIRT3 expression was upregulated in rat islets upon exposure to low glucose or fasting. SIRT3 overexpression in islets markedly decreased palmitate-potentiated insulin secretion, whereas islets from SIRT3 knockout mice secreted more insulin, with an opposite action on FAO. ECHA overexpression partially reversed SIRT3 deficiency-elicited insulin hypersecretion. Our study highlights the potential role of protein acetylation in insulin secretion.
Similar content being viewed by others


Introduction
Pancreatic β-cells display a great degree of plasticity to secrete insulin in response to nutrient availability1,2. Although many metabolic coupling factors have been proposed to modulate metabolic networks involved in fuel-induced insulin secretion, the enormous complexity of metabolism-triggered signaling processes is beyond our understanding3. The growing landscape of protein posttranslational modification (PTM) has highlighted its regulatory roles in cellular metabolism4. Therefore, application of large-scale proteomics should help us comprehensively understand the mechanism for islet β-cells to adapt to metabolic changes and provide insights into the pathogenesis of type 2 diabetes.
Protein lysine acetylation (Kac) is a conserved PTM that is emerging as a crucial regulator of protein function5,6. Recent advances in mass spectrometry have led to the identification of thousands of acetylated proteins7,8,9,10,11, highlighting the regulatory potential of acetylation in many biological processes. Acetylation level is tightly governed by lysine acetyltransferases (KATs) and deacetylases (KDACs)12. All KATs require acetyl-CoA as substrate for acetylation reactions. Another intermediary metabolite NAD+ directly alters KDAC activities to link energy status to cellular homeostasis, making acetylation especially favorable in regulating metabolic enzymes.
As fuel sensors, β-cells are extremely sensitive to nutrients alterations. The primary stimulus for insulin secretion is glucose, whose metabolism is achieved by tightly linking glycolysis with mitochondrial metabolism13. Fatty acids also have enormous capacity to amplify glucose-stimulated insulin secretion (GSIS), in part via their metabolism into lipid signaling molecules14. Given that β-cell function is closely coupled to fuel metabolism and protein acetylation may be at the nexus of coordinating metabolic flux, it is reasonable to hypothesize that protein acetylation may provide a link between fuel metabolism and insulin secretion. It has been shown that inhibition of class I histone deacetylases (HDACs) prevents cytokine-induced toxicity in β-cells15,16. The class III HDACs, sirtuins, also have important roles in insulin secretion17,18,19,20. These studies implicate the involvement of acetylation in regulating islet function. The substantial differences of acetylation patterns across tissues underly the importance of tissue-specific acetylome mapping9. However, the scope and targets of protein acetylation in islets remain largely unknown.
Here we report the first proteomic analysis of lysine acetylation in rat islets using affinity enrichment and high-resolution liquid chromatography tandem mass spectrometer (LC-MS/MS). Further quantitative acetylome of islets in response to high glucose revealed a critical role of acetylation in fatty acid oxidation (FAO) enzymes, among which trifunctional enzyme subunit alpha (ECHA, coding gene HADHA) and its deacetylase SIRT3 inhibited palmitate-stimulated insulin secretion via increasing FAO. This current study establishes protein acetylation as a link of glucose and fatty acid metabolism to adaptive islet β-cell function.
Materials and methods
Islet preparation, MS, and data analysis
Pancreatic islets were isolated from 8- to 12-week-old wild-type Sprague–Dawley (SD) male rats (Shanghai Laboratory Animal Company, Shanghai, China) by collagenase digestion and density-gradient centrifugation. For label-free sample preparation, isolated islets were incubated with either 3.3 mM (low) or 16.7 mM (high) glucose for 6 h in KRB buffer. Freshly isolated or incubated islets were washed twice with glucose-free phosphate buffer and were snap-frozen in liquid nitrogen before proteomic analyses. Islets from 25 rats were pooled to create a uniform sample of 20,000 total islets. Label-free samples were collected from three independent biological experiments. Mass spectrometry and data analysis were performed as described21 (see Supplementary Materials data for details). The quantitative proteomic method was label-free quantification (LFQ) and minimum score for modified peptides score was set ≥ 40. The acetylome datasets generated and analysed during this current study are available in the PRIDE repository, https://www.ebi.ac.uk/pride/archive/login. Username: reviewer56234@ebi.ac.uk Password: LiDVJTfM
Insulin secretion assay
Isolated islets were cultured with indicated reagents in RPMI 1640 medium (0.25% bovine serum albumin (BSA)). To stimulate insulin secretion, islets were preincubated in Krebs-Ringer Buffer (KRB) containing 3.3 mM glucose for 30 min. Then, ten islets per assay in triplicate were incubated with KRB buffer containing either 3.3 mM glucose, 16.7 mM glucose, or other reagents as indicated for 1 h at 37 °C. Supernatants containing insulin were removed and stored at − 20 °C until analysis. Insulin content was extracted with acid-ethanol. Insulin levels of all samples were measured by ELISA kit (Mercodia).
Animal studies
Breeding pairs of SIRT3 knockout mice (original 129/Sv background, purchased from Jackson Laboratories) were a generous gift from Dr. Weili Shen (Shanghai Key Laboratory of Hypertension, Department of Hypertension, Ruijin Hospital, Shanghai Jiaotong University School of Medicine) and have been backcrossed for at least five generations onto the C57BL/6 background (Shanghai Laboratory Animal Company, Shanghai, China). Ten- to 12-week-old male SIRT3KO mice and wild-type (C57BL/6) littermate controls on a standard chow diet were used. For fasting studies, SD rats were transferred to a new cage without food for 24 h.
Cell culture and treatment
INS-1 cells were cultured in RPMI 1640 medium with 11.1 mM glucose that contained 10% fetal bovine serum (FBS). HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS. Cells were treated with 200 nM trichostatin A (TSA; Cell Signaling Technology) for 20 h, 5 mM nicotinamide (NAM; Sigma) for 6 h, or 10 μM MG132 (Sigma) for 6 h in the presence of 5.6 mM glucose. Plasmid transfection was carried out by Lipofectamine 2000 (Invitrogen).
Western blotting and immunoprecipitation
Islets or INS-1 cells were collected in lysis buffer (Cell Signaling Technology). Protein lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (BioRad). Primary antibodies were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies. Anti-ECHA and anti-SIRT4 was from Abcam. Anti-SIRT3, anti-SIRT5, and anti-rabbit IgG conjugated with HRP were from Cell Signaling Technology. Anti-α-tubulin was from Proteintech. Anti-Flag was from Sigma-Aldrich. Anti-acetyllysine was from PTM Biolab. Immunoprecipitation was performed by incubating protein lysates with FLAG M2 Affinity Gel (Sigma) or ECHA antibody for 2 h and then with Protein A/G PLUS-Agarose (Santa-Cruz) overnight at 4 °C. The binding complexes were washed and then eluted with loading buffer. Standard western blotting was followed using anti-acetyllysine antibody for acetylation analysis.
RNA isolation and qRT-PCR
Islet RNAs were extracted using RNeasy Plus Mini kit (Qiagen). Quantitative reverse transcription PCR (qRT-PCR) was performed with SYBR Premix Ex Taq (Takara) on a Light-Cycler 480 instrument (Roche Applied Science). Primer sequences are as follows: 18S (Forward: 5′-CACGGGTGACGGGGAATCAG-3′ and Reverse: 5′-CGGGTCGGGAGTGGGTAATTTG-3′); SIRT3 (Forward: 5′-GGCACTACAGGCCCAATGTC-3′ and Reverse: 5′-TCTCTCAAGCCCGTCGATGT-3′); SIRT4 (Forward: 5′-TTACAGCGCTTCATTAGCCTTTC-3′ and Reverse: 5′-CCCACCTTTTCTGACCTGTAGTCT-3′); SIRT5 (Forward: 5′-AGAGCAAGATCTGCCTCACCAT-3′ and Reverse: 5′-AGCCCCCGAGATGATGACTAT-3′);
Adenovirus infection
For SIRT3 and ECHA overexpression, adenoviruses expressing rat target protein were constructed with a full-length target gene coding sequence. Islets or INS-1 cells were infected with target protein or vector adenovirus according to the manufacturer’s instructions (GeneChem).
Oxygen consumption rate measurement
For FAO assay, except for the indicated treatment, islets were incubated in substrate-limited medium (RPMI 1640 medium with 0.5 mM glucose). Before the assay, islets were washed two times and then transferred to the Islet Capture Microplate (Seahorse Bioscience) with FAO assay medium containing 2 mM glucose. Oxygen consumption rate (OCR) was measured after XF Palmitate–BSA substrate was injected into the wells using a Seahorse XF24 flux analyzer (Seahorse Bioscience). For glucose oxidation assay, 20 mM glucose was preloaded into cartridges and injected into XF wells.
For FAO mitochondrial stress test, HEK293T cells were seeded in XF cell culture microplates and incubated in substrate-limited medium for 24 h after plasmid transfection. Cells were washed with FAO assay medium (supplemented with 2 mM glucose) and XF palmitate–BSA substrate or BSA was added to the wells just before starting the assay. The chemicals (1 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone/antimycin A) were preloaded into cartridges and injected into XF wells in succession. OCR was measured using Seahorse XF24 flux analyzer. Basal/maximal respiration due to utilization of exogenous palmitate was calculated as basal/maximal palmitate rate minus basal/maximal BSA rate.
Statistical analysis
Unless otherwise indicated, all values are expressed as mean ± SEM and statistical significance was assessed by Student’s t-test. Differences were considered to be statistically significant when p < 0.05.
Results
Impact of HDAC inhibition on insulin secretion in rat islets
To directly evaluate the role of protein acetylation in islet function, we investigated the combined effect of two HDAC inhibitors, TSA (inhibitor of HDAC I and II), and NAM (inhibitor of sirtuins), on insulin secretion. TSA and NAM co-treatment acutely enhanced GSIS in rat islets (Fig. 1a). In addition, insulin secretion in the presence of 3.3 mM glucose was markedly elicited with prolonged treatment of TSA and NAM (Fig. 1b). These data suggest that overall hyperacetylation promotes insulin secretion.

a Rat islets were stimulated with 3.3 or 16.7 mM glucose in the presence or absence of 200 nM TSA and 5 mM NAM for 1 h, and then insulin secretion was measured. b Rat islets were treated with 200 nM TSA and 5 mM NAM at 3.3 mM glucose for the indicated time and then insulin secretion was measured. c Proteomic workflow for the identification of acetylated sites and proteins in rat islets. d Comparison of the acetylated proteins identified in the present study (red circle) with that in CPLM database (blue circle). e Comparison of the acetylated lysine sites identified in the present study (red circle) with that in CPLM database (blue circle). f Distribution of the number of acetylated sites per protein. *p < 0.05, **p < 0.01 vs. control (CON)
Identification of acetylated sites and proteins in rat islets
To identify the acetylome in islets, we employed an integrated acetyl-proteomic approach for large-scale profiling. Primary islets from SD rats were isolated and pooled to account for biological variation. Protein extracts were enriched for lysine-acetylated peptides by immunoprecipitation and analyzed by LC-MS/MS. After searching against UniProt_rat database using MaxQuant, we identified 3067 lysine-acetylated sites in 1365 islet proteins (Fig. 1c and supplemental Table S1), among which 328 proteins and 1740 sites were never retrieved (Fig. 1d, e) in the CPLM database for lysine acetylation of rat proteins22. Evaluation of the high-quality MS data was shown in Figure S1A and S1B. We further calculated the number of acetylation sites per protein in this islet acetylome. Fifty-four percent of the identified proteins contained only one acetylation site, whereas 46% were multiacetylated (Fig. 1f).
Characterization of the islet acetylation proteome
Subcellular distribution of islet acetylated proteins showed that the most abundant localization was cytosol. Proteins with exclusively mitochondrial annotation were also highly represented (Fig. 2a). Further analysis of molecular functions showed that the acetylated proteins were significantly enriched in binding and catalytic activities (Fig. 2b).

a Subcellular distribution of acetylated proteins identified in islets. b Representative GO molecular function term enrichment analysis of acetylated proteins. c Representative KEGG pathways enriched with islet acetylome. d Representative GO biological processes enriched with islet acetylome. Logarithmized corrected p-values for significant overrepresentation are shown
To understand islet-specific functions of the acetylated proteins, we performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. In agreement with acetylation-modulated cellular metabolism in the liver11, our data demonstrated that islet acetylated proteins were significantly enriched in metabolic pathways (Fig. 2c). Moreover, there was a significant enrichment for endoplasmic reticulum, ribosome, and vesicle transport pathways (Fig. 2c), implicating a regulatory role of acetylation in insulin biosynthesis and exocytosis machinery. We further performed Gene Ontology (GO) analysis of the biological processes. Consistent with KEGG pathway analysis, citrate metabolism, tricarboxylic acid (TCA) cycle, ɑ-amino acid metabolism, and FAO were among the top ten enriched processes (Fig. 2d), which are essential for β-cell function. Processes related to protein folding and vesicle-mediated transport were also enriched (Fig. 2d). This pattern suggests that islet acetylated proteins are involved in diverse insulin-secreting related functions.
Identification of acetylated proteins in the context of metabolic signaling pathways for insulin secretion
To gain an in-depth and integrated view of the correlation between acetylated proteins and β-cell function, we explored the coverage of our islet acetylome in the context of metabolic signalings for fuel-induced stimulus-secretion coupling3. As the main driver for insulin secretion, glucose undergoes glycolysis and is converted to pyruvate, which enters into mitochondria to participate in TCA cycle and oxidative phosphorylation, resulting in increased insulin exocytosis23. Almost every enzyme involved in the above processes was acetylated (Fig. 3).

Proteins identified in the islet acetylome are labeled in bluish green and those not identified in orange. Arrows are color coded according to their metabolic processes
This classical triggering process is complemented by amplification pathways and activation of auxiliary metabolic pathways3,24. Pyruvate in islets is converted to oxaloacetate via pyruvate carboxylase and further forms into citrate to enter TCA cycle or participate in pyruvate/citrate cycling, which is critical to modulate insulin secretion25,26. The involved enzymes (Pc, Slc25a1, Acly, and Mdh1) were all acetylated. Another pyruvate cycling is the pyruvate/malate cycle that exchanges malate to produce NADPH by Me127, which could be acetylated. There also exists an isocitrate/α-KG shuttle in the control of GSIS28. The enzymes catalyzing this process (Idh1, Idh2, Idh3) have been identified in our acetylome (Fig. 3).
As to amino acid metabolism, glutamate gives rise to α-KG via Glud1 and GABA via Gad2, which further metabolized to succinate and enters TCA cycle. The involved enzymes in this GABA shunt are preeminent for anaplerosis to promote insulin secretion3,29, all of which were acetylated (Fig. 3).
Glucose metabolism is tightly linked to the production of lipid signaling molecules. Cellular free fatty acids (FFAs) could be synthesized by Fasn and further activated by Acsl, which were acetylated (Fig. 3). The generated FA-CoA either joins the glycerolipid/FFA (GL/FFA) cycle to promote insulin secretion, or diverts into mitochondria for β-oxidation30. Interestingly, as an off signal for insulin secretion, every enzyme involved in β-oxidation was found to be acetylated in islet (Fig. 3).
Quantitative acetylome of rat islets in response to glucose
Islets pretreated with high glucose for 24 h secreted more insulin in response to 3.3, 16.7 mM glucose and KCl (Fig. S2A). Pretreatment with TSA and NAM showed a similar result (Fig. S2B). We further pretreated islets with TSA and NAM in the presence of either 1.4 or 5.6 mM glucose. Although 5.6 mM glucose alone potentiated GSIS compared with 1.4 mM glucose, GSIS was dramatically amplified by TSA and NAM pretreatment at 1.4 mM glucose (Fig. S2C), suggesting high glucose shared a similar pathway with acetylation to amplify insulin secretion.
To explore whether protein acetylation is linked to glucose-elicited metabolic flux involved in insulin secretion, we quantified the acetylomes of rat islets exposed to 3.3 and 16.7 mM glucose for 6 h. Using label-free quantitative proteomic approach, we identified 1270 lysine-acetylated sites in 749 proteins, among which 1244 acetylated sites in 739 proteins were accurately quantified in three parallel LC-MS/MS analyses (Fig. 4a). The quality of MS data is evaluated (Figure S1C and S1D). The fold-change cutoff was set to be 1.5. In high glucose-treated islets, the acetylation levels of 14 lysine sites in 14 proteins were significantly upregulated and those of 17 lysine sites in 16 proteins were significantly downregulated (Supplementary Table S2).

a Proteomic workflow for the quantification of Kac sites and proteins in rat islets in response to low and high glucose. b GO-based enrichment analysis of glucose-regulated hyperacetylated proteins. c Subcellular distribution of glucose-regulated hyperacetylated proteins. d KEGG pathway-based enrichment analysis of glucose-regulated hyperacetylated proteins. e Mitochondrial fatty acid β-oxidation and TCA cycle enzymes (gene symbols) are highlighted next to the reaction they catalyze, with hyperacetylated enzymes targeted by high glucose colored in yellow. Logarithmized corrected p-values for significant overrepresentation are shown
To understand the functions of glucose-controlled acetylated proteins, we divided the quantifiable acetylated proteins into four categories according to their fold-change ratios (Fig. S2D) and then plotted for KEGG enrichment-based cluster analysis. We found that the great majority of metabolic pathways were exclusively enriched with hyperacetylated proteins (Fig. S2E). Glucose-upregulated Kac proteins were more frequently present in mitochondria and exhibited 3-hydroxy-CoA dehydrogenase activity (Fig. 4b). The GO biological process analysis demonstrated that these proteins were involved in TCA cycle, aerobic respiration, and fatty acid metabolism (Fig. 4b). Among 14 upregulated Kac proteins, 9 were located in mitochondria, highlighting the importance of mitochondrial metabolism in glucose-modulated islet function (Fig. 4c).
Further KEGG pathway analysis found that many metabolic pathways were strongly influenced by glucose. The top pathways most significantly enriched with glucose-upregulated Kac targets were TCA cycle and fatty acid metabolism (Fig. 4d). It has been demonstrated that malonyl-CoA derived from glucose metabolism inhibits mitochondrial FAO in β-cells31,32. As multiple enzymes of FAO and TCA cycle were highly acetylated by glucose (Fig. 4e), it is possible that reversible protein acetylation is involved in glucose-modulated FAO.
Glucose increases ECHA acetylation and decreases fatty acid β-oxidation of islets
Among glucose-upregulated acetylated enzymes in FAO, ECHA containing enoly-CoA hydratase and long-chain 3-hydroxyacyl-CoA dehydrogenase functions catalyzes the second and third step of long-chain fatty acid β-oxidation33. Glucose increased the acetylation levels of lysine residue K644 and K505 in ECHA (Fig. S2F). Rat islets treated with 16.7 mM glucose showed markedly induced ECHA acetylation, confirming the acetylome results (Fig. 5a). To investigate whether FAO in islets could be functionally impacted by glucose, we directly assessed OCR of palmitate under low glucose condition using Seahorse assay as a parameter of FAO. As previously reported34, 16.7 mM glucose pretreatment significantly decreased palmitate oxidation in rat islets compared with 3.3 mM glucose (Fig. 5b).

a Acetylation levels of endogenous ECHA in rat islets treated with 3.3 mM (LG) and 16.7 mM glucose (HG). b Oxidation consumption rate (OCR) of palmitate was measured after rat islets were treated with 3.3 or 16.7 mM glucose for 6 h. c Acetylation levels of endogenous ECHA in rat islets treated with or without deacetylase inhibitors TSA plus NAM. d Detection of acetylation levels of ectopically expressed ECHA in HEK293T cells treated with or without NAM. e Flag-tagged wild type, K644K505Q (2 K/Q), and K644K505R (2 K/R) ECHA were overexpressed in HEK293T cells and acetylation levels were detected. f OCR of palmitate was measured after wild type, 2 K/Q, and 2 K/R ECHA were overexpressed in HEK293T cells. g Wild type and 2 K/Q ECHA were ectopically expressed in HEK293T cells and OCR was measured in the presence of palmitate–BSA or BSA control. Arrows indicate the time of addition for oligomycin, FCCP and rotenone/antimycin A. h Determination of basal and maximal respiration due to the utilization of exogenous palmitate in g. Data are expressed as mean ± SEM of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (CON or WT)
Similar to high glucose, TSA and NAM treatment also resulted in ECHA hyperacetylation of islets (Fig. 5c). Exogenously expressed ECHA in HEK293T cells was highly acetylated by NAM treatment (Fig. 5d). To determine whether the acetylation of ECHA K644 and K505 changes its ability to promote FAO, we further generated double glutamine (Q) or arginine (R) mutants of K644 and K505 (2 K/Q and 2 K/R). Both 2 K/Q and 2 K/R mutants resulted in significant decreases in ECHA acetylation (Fig. 5e). Moreover, the acetylation mimetic 2 K/Q mutant significantly decreased palmitate oxidation compared with wild-type ECHA, whereas the non-acetylable 2 K/R mutant hardly showed any difference (Fig. 5f). Then we explored the ability of 2 K/Q mutant to utilize exogenous fatty acids under mitochondrial stress (Fig. 5g). Both basal and maximal respiration rates due to utilization of exogenous palmitate were significantly decreased in cells expressed with 2 K/Q mutant (Fig. 5h), confirming that ECHA acetylation decreases FAO.
SIRT3 deacetylates ECHA and increases fatty acid β-oxidation in β-cells
To confirm that ECHA was indeed acetylated in β-cells, we explored ECHA acetylation in a commonly used β-cell line INS-1 cells. ECHA was hyperacetylated in INS-1 cells treated with TSA plus NAM (Fig. S3A) or NAM alone (Fig. 6a). Similar to high glucose, palmitate oxidation was also decreased by NAM pretreatment (Fig. 6b). Furthermore, TSA and NAM co-treatment significantly reduced ECHA protein abundance in INS-1 cells (Fig. S3B), without changing its mRNA expression (Fig. S3C), suggesting that acetylation decreases protein stability of ECHA. Neither the acetylation (Fig. S3D) nor protein expression (Fig. S3E) levels of ECHA were changed by TSA. However, NAM treatment alone significantly decreased ECHA protein level (Fig. 6c), indicating ECHA is regulated only by sirtuins. After cycloheximide treatment, the protein level of ECHA was decreased over time (Fig. 6d). Treatment of MG132, a proteasome inhibitor, increased ECHA protein level and canceled the destabilization effect of NAM on ECHA (Fig. 6e), suggesting that acetylation-promoted degradation of ECHA is likely mediated by the proteasome pathway.

a Acetylation level of endogenous ECHA in INS-1 cells treated with or without 5 mM NAM for 6 h. b OCR of palmitate was measured after rat islets were treated with or without 5 mM NAM for 6 h. c ECHA protein level in INS-1 cells treated with or without 5 mM NAM for 6 h. d ECHA protein level in INS-1 cells treated with cycloheximide (CHX) for the indicated time. e After INS-1 cells were treated with or without NAM in the absence or presence of MG132, ECHA protein level was detected. f After rat islets were transfected with control vector (Ad-Vector) or SIRT3-overexpressing adenovirus (Ad-SIRT3), protein levels of SIRT3, SIRT4, and SIRT5 were detected. g After INS-1 cells were transfected with control vector or SIRT3-overexpressing adenovirus, cell lysates were immunoprecipitated with ECHA antibody and subjected to Western blot with anti-acetyllysine antibody. h INS-1 cells transfected with vector or SIRT3-overexpressing adenovirus were treated with cycloheximide for the indicated time and then ECHA protein level was detected. i Palmitate oxidation rate was measured after rat islets were transfected with control vector or SIRT3-overexpressing adenovirus. Data are expressed as mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, **p < 0.001 vs. control. #p < 0.05 vs. NAM
Of all the sirtuin members, SIRT3 is the only one with robust deacetylation activity35. ECHA has been reported to be a putative SIRT3 target in mice liver36. We wondered whether it was also regulated by SIRT3 in islet β-cells. After adenovirus-mediated SIRT3 overexpression, the acetylation abundance of several proteins was markedly reduced (Fig. S4A), without changing SIRT4 and SIRT5 mRNA and protein expressions (Figs. S4B and 6f). SIRT3 overexpression significantly decreased ECHA acetylation level (Fig. 6g) and prevented the degradation of ECHA protein in INS-1 cells (Fig. 6h). As SIRT3 promotes FAO in the liver37,38, we detected the impact of SIRT3 on FAO in islets. As expected, SIRT3 overexpression led to a substantial increase in palmitate oxidation rate of islets (Fig. 6i). Therefore, it is possible that SIRT3-controlled FAO flux is involved in fuel-mediated insulin secretion.
Roles of SIRT3 and ECHA in regulating islet function
As illustrated in Fig. 7a, insulin secretory response at basal glucose was significantly augmented by a 24 h palmitate pretreatment as shown previously14. SIRT3 overexpression decreased basal insulin hypersecretion in rat islets pretreated with palmitate (Fig. 7a). This was also the case in SIRT3-overexpressing mouse islets (Fig. 7b). We further used SIRT3 knockout mice to evaluate the ex vivo impact of SIRT3 on insulin secretion. Genotypes showed deletion of SIRT3 gene (Fig. S4C) and western blotting confirmed knockout of SIRT3 protein in these mice (Fig. 7c). SIRT3 knockout mice showed no significant alterations in SIRT4 and SIRT5 expressions in both islets (Fig. 7c and S4D) and the liver (Fig. S4E). Palmitate-stimulated insulin secretion was markedly increased in SIRT3KO islets (Fig. 7d), which was reversed by SIRT3 overexpression (Fig. 7f). Consistent with the effect of NAM, SIRT3 knockout islets showed a significant decrease in palmitate oxidation rate (Fig. 7e). Taken together, these data highlight an important role of SIRT3 in regulating palmitate-stimulated insulin secretion.

Rat islets (a) and mouse islets (b) transfected with control vector or SIRT3-overexpressing adenovirus were pretreated with 0.25 mM palmitate for 24 h and then stimulated with 3.3 mM glucose for insulin secretion assay. c Western blot analysis of SIRT3, SIRT4, and SIRT5 protein levels in islets isolated from wild type or SIRT3 knockout mice. d Islets isolated from wild type or SIRT3 knockout mice were stimulated with or without 0.25 mM palmitate at 3.3 mM glucose for 1 h and insulin secretion was assayed (n = 8). e Palmitate oxidation rate was measured in islets isolated from wild type or SIRT3 knockout mice (n = 5). f Islets from wild type or SIRT3 knockout mice were transfected with control vector and SIRT3 or ECHA-overexpressing adenovirus, and then stimulated with 0.25 mM palmitate for 1 h for insulin secretion assay. g Islets isolated from wild type or SIRT3 knockout mice were stimulated with 3.3 or 16.7 mM glucose for 1 h, and insulin secretion was assayed (n = 8). h OCR in islets isolated from wild type or SIRT3 knockout mice was measured at baseline or following glucose addition (n = 5). SIRT3 mRNA (i) and protein (j) expressions were detected after rat islets were treated with 3.3 or 16.7 mM glucose for 24 h. k Western blot analysis of SIRT3 protein levels in islets isolated from rats fed or fasted for 24 h. Data are expressed as mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control (CON), WT, or 3.3 G. #p < 0.05, ##p < 0.01 vs. vector
To determine whether SIRT3 inhibited insulin secretion via deacetylating ECHA, rat islets were transfected with ECHA-overexpressing adenovirus (Fig. S5A). Both basal and high glucose-potentiated insulin release were significantly decreased in ECHA-overexpressing islets (Fig. S5B). ECHA overexpression markedly suppressed basal insulin hypersecretion induced by 16.7 mM glucose pretreatment (Fig. S5C). However, ECHA overexpression only partially reversed SIRT3 deficiency-stimulated insulin secretion under palmitate treatment (Fig. 7f).
Moreover, both basal and GSIS were significantly increased in SIRT3KO islets (Fig. 7g). We further explored mitochondrial glucose oxidation of SIRT3KO islets. Neither basal nor glucose oxidation showed any difference, indicating that increased insulin secretion of SIRT3KO islets is not attributed to glucose oxidation (Fig. 7h).
To determine SIRT3 expression in islets in response to nutrient change, rat islets were incubated for 24 h with 3.3 and 16.7 mM glucose. Both SIRT3 mRNA (Fig. 7i) and protein (Fig. 7j) expressions displayed significant decreases after high glucose treatment, in support for an increased mitochondrial acetylation status under this condition (Fig. 4c). We then observed SIRT3 expression in islets under fasting condition, which promotes FAO to suppress insulin secretion39. Consistent with the result of low glucose incubation, SIRT3 protein level was increased in islets of fasted rats (Fig. 7k).
Discussion
Protein lysine acetylation represents an important mechanism to regulate overall energy metabolism4. In pancreatic islets, studies addressing the biological role of protein acetylation have been hampered by lacking knowledge of the modified targets. The present study performed the first comprehensive acetylation analysis in rat islets. Further islet quantitative acetylome revealed hyperacetylation of fatty acid β-oxidation enzymes under high glucose condition, which resulted in decreased FAO and enhanced β-cell function. Finally, we proposed a mechanism by which SIRT3 negatively regulates insulin secretion through stabilizing ECHA by deacetylation. These findings identify protein acetylation as a novel regulatory mechanism for insulin secretion.
The lysine acetylation profiles of many metabolic tissues highlighted the importance of protein acetylation in maintaining glucose homeostasis7,9,11. One study has provided an organ-wide map of lysine acetylation from 16 rat tissues and identified 2036 acetylated sites on 878 proteins in pancreas9. However, islets were not extracted from pancreas for this experiment. Interestingly, only partial acetylated sites and proteins we identified overlap with those in other tissues, suggesting a distinct pattern of acetylation in islets. We found of interest the broad coverage and highly abundance of the islet acetylome in metabolic pathways of β-cells related to nutrient sensing for insulin secretion (Fig. 3), emphasizing the role of acetylation in modulating tissue-specific function. As acetylation has the potential to affect enzyme activity, it is reasonable to suppose that acetylation-driven cooperativity of these metabolic pathways may be crucial to enhance β-cell function in the face of metabolic demand.
Pancreatic β-cells decrease insulin secretion during fasting to prevent hypoglycemia. It is widely accepted that elevated glucose inhibits FAO by decreasing AMPK activity and increasing malonyl-CoA production in β-cells, thus diverts FA-CoA into the GL/FFA cycle, providing “on” signals for insulin secretion40. On the contrary, fasting activates AMPK, enhances FAO, and “turns off” the switch for insulin secretion. The flux of FAO tightly links glucose and fatty acid metabolism to signaling for insulin secretion14,30. The present study revealed significantly increased SIRT3 expression in rat islets under low glucose and fasting status. It is reasonable to suppose that SIRT3 links the low energy status of islets to insulin hyposecretion. However, the physiological function of SIRT3 in β-cells remains largely obscure. It has been demonstrated that short hairpin RNA-mediated SIRT3 knockdown in MIN6 cells aggravated palmitate-induced dysfunction41, whereas SIRT3 overexpression partially recovered palmitate-impaired GSIS42. These in vitro studies only partially suppressed SIRT3 expression and assessed its function in the context of glucolipotoxicity. A recent study found that SIRT3 knockout in β-cell line had no effect on fuel-stimulated insulin secretion. In their islet perifusion experiments, islets of chow diet SIRT3KO mice showed an increased trend for insulin secretion in response to glucose and fatty acids, without statistical significance43. In our static incubation assay, primary islets of SIRT3KO mice secreted significantly more insulin compared with wild-type controls, in consistent with the results of Hirschey et al.44 that SIRT3KO mice revealed higher fasting and glucose-stimulated insulin levels. Therefore, the hyperinsulinemia of these mice may be attributed at least in part to SIRT3 deficiency caused insulin hypersecretion from β-cells, which accelerates the development of insulin resistance and metabolic syndrome. Moreover, we observed a marked decrease of palmitate-potentiated insulin secretion in SIRT3-overexpressing islets. In basal state, elevated plasma FFAs were responsible for some of the hyperinsulinemia in normoglycemic obese subjects45. Basal insulin levels are such an important determinant of insulin sensitivity that hyperinsulinemia initiates and contributes to insulin resistance in patients with obesity or type 2 diabetes46. Therefore, interfering this process by elevating SIRT3 expression or activity will protect β-cells against FFA-driven excess insulin secretion in these patients.
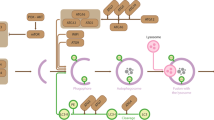
As a metabolic sensor, SIRT3 has been noted for its numerous roles in regulating mitochondrial biology36,47, especially fatty acid β-oxidation38. However, there is still no consensus as to its functional contribution to FAO, with both stimulatory and inhibitory effects observed48. Our study revealed that SIRT3 overexpression increased FAO rate, whereas SIRT3 knockout exhibited an opposite result in islets. The FAO enzymes ECHA and short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD, coding gene HADH) were hyperacetylated by high glucose, and acetylation mimetic mutant of ECHA decreased FAO. SCHAD deficiency is associated with islet cell-autonomous hyperinsulinemic hypoglycemia49. Patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by ECHA mutation also develop hypoglycemia50. To our knowledge, the effect of ECHA on islet function has never been reported. The present study showed that ECHA overexpression reduced basal and fuel-potentiated insulin secretion. Moreover, SIRT3 deacetylated ECHA and prevented its degradation. Apparently, SIRT3 couples the energy status of islets with insulin secretion via protein deacetylation. Under low fuel condition, AMPK-modulated activities of acetyl-CoA carboxylase (ACC) and carnitine palmitoyltransferase 1 (CPT-1) by phosphorylation determine the entry of FA-CoA into mitochondria for β-oxidation3 and SIRT3-mediated deacetylation of ECHA directly promotes FAO, in concert leading to decreased lipid signals for insulin secretion (Fig. 8). Therefore, SIRT3 exerts a new negative metabolic regulation of insulin secretion through a mechanism distinct from AMPK. Glucose decreases FAO flux and provides more lipid signals for insulin secretion, which may at least partially be attributed to the hyperacetylation of FAO enzymes.

a Under fed condition, high glucose reduces AMPK activity, resulting in decreased phosphorylation of ACC and increased production of malonyl-CoA (Mal-CoA), which blocks fatty acyl-CoA (FA-CoA) entrance into mitochondria for β-oxidation via inhibiting CPT-1. On the other hand, high glucose decreases the expression or activity of SIRT3, leading to increased acetylation of ECHA and reduced mitochondrial β-oxidation. Thus, phosphorylation and acetylation programs cooperatively increase cytosolic lipid signaling molecules to promote insulin secretion in islet β-cell. b During fasting, activation of AMPK phosphorylates and inhibits ACC, resulting in reduced Mal-CoA and increased entry of substrates into mitochondria for β-oxidation. Low glucose also increases the expression or activity of SIRT3, which deacetylates ECHA and promotes fatty acid β-oxidation. The two signaling pathways cooperatively decrease lipid signaling molecules for insulin secretion
In summary, this study provides a comprehensive picture of protein acetylation in rat islets and expands the inventory of known acetylated sites and proteins. The acetylation status of islet mitochondrial enzymes in response to nutrient change is crucial for insulin secretion via regulating FAO flux. As the mitochondrial deacetylase, SIRT3 reduces islet acetylation level of FAO enzymes via sensing energy status and promotes FAO, resulting in decreased lipid signaling molecules for insulin secretion. Therefore, modulating SIRT3-mediated acetylation status could be a new and promising approach to prevent the onset of hyperinsulinemia and insulin resistance.
References
Hinke, S. A., Hellemans, K. & Schuit, F. C. Plasticity of the beta cell insulin secretory competence: preparing the pancreatic beta cell for the next meal. J. Physiol. 558, 369–380 (2004).
Ashcroft, F. M. & Rorsman, P. Diabetes mellitus and the beta cell: the last ten years. Cell 148, 1160–1171 (2012).
Prentki, M., Matschinsky, F. M. & Madiraju, S. R. Metabolic signaling in fuel-induced insulin secretion. Cell. Metab. 18, 162–185 (2013).
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E. & Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 (2014).
Menzies, K. J., Zhang, H., Katsyuba, E. & Auwerx, J. Protein acetylation in metabolism - metabolites and cofactors. Nat. Rev. Endocrinol. 12, 43–60 (2016).
Verdin, E. & Ott, M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264 (2015).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Henriksen, P. et al. Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol. Cell. Proteomics 11, 1510–1522 (2012).
Lundby, A. et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419–431 (2012).
Wang, Q. et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 (2010).
Zhao, S. et al. Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 (2010).
Aka, J. A., Kim, G. W. & Yang, X. J. K-acetylation and its enzymes: overview and new developments. Handb. Exp. Pharmacol. 206, 1–12 (2011).
Newsholme, P., Gaudel, C. & McClenaghan, N. H. Nutrient regulation of insulin secretion and beta-cell functional integrity. Adv. Exp. Med. Biol. 654, 91–114 (2010).
Nolan, C. J., Madiraju, M. S., Delghingaro-Augusto, V., Peyot, M. L. & Prentki, M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 55(Suppl 2), S16–S23 (2006).
Larsen, L. et al. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia 50, 779–789 (2007).
Lundh, M. et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 55, 2421–2431 (2012).
Moynihan, K. A. et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell. Metab. 2, 105–117 (2005).
Haigis, M. C. et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954 (2006).
Caton, P. W. et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 56, 1068–1077 (2013).
Xiong, X. et al. Sirtuin 6 regulates glucose-stimulated insulin secretion in mouse pancreatic beta cells. Diabetologia 59, 151–160 (2016).
Zhou, X., Qian, G., Yi, X., Li, X. & Liu, W. Systematic analysis of the lysine acetylome in Candida albicans. J. Proteome Res. 15, 2525–2536 (2016).
Liu, Z. et al. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 42, D531–D536 (2014).
Schuit, F. et al. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J. Biol. Chem. 272, 18572–18579 (1997).
Henquin, J. C. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52, 739–751 (2009).
Guay, C., Madiraju, S. R., Aumais, A., Joly, E. & Prentki, M. A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J. Biol. Chem. 282, 35657–35665 (2007).
Xu, J., Han, J., Long, Y. S., Epstein, P. N. & Liu, Y. Q. The role of pyruvate carboxylase in insulin secretion and proliferation in rat pancreatic beta cells. Diabetologia 51, 2022–2030 (2008).
MacDonald, M. J. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J. Biol. Chem. 270, 20051–20058 (1995).
Ronnebaum, S. M. et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J. Biol. Chem. 281, 30593–30602 (2006).
Otter, S. & Lammert, E. Exciting times for pancreatic islets: glutamate signaling in endocrine cells. Trends Endocrinol. Metab. 27, 177–188 (2016).
Prentki, M. & Madiraju, S. R. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol. Cell. Endocrinol. 353, 88–100 (2012).
Chen, S. et al. More direct evidence for a malonyl-CoA-carnitine palmitoyltransferase I interaction as a key event in pancreatic beta-cell signaling. Diabetes 43, 878–883 (1994).
Roduit, R. et al. A role for the malonyl-CoA/long-chain acyl-CoA pathway of lipid signaling in the regulation of insulin secretion in response to both fuel and nonfuel stimuli. Diabetes 53, 1007–1019 (2004).
Uchida, Y., Izai, K., Orii, T. & Hashimoto, T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J. Biol. Chem. 267, 1034–1041 (1992).
Kim-Muller, J. Y. et al. FoxO1 deacetylation decreases fatty acid oxidation in beta-cells and sustains insulin secretion in diabetes. J. Biol. Chem. 291, 10162–10172 (2016).
Feldman, J. L., Baeza, J. & Denu, J. M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356 (2013).
Rardin, M. J. et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl Acad. Sci. USA 110, 6601–6606 (2013).
He, W., Newman, J. C., Wang, M. Z., Ho, L. & Verdin, E. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 23, 467–476 (2012).
Hirschey, M. D. et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 (2010).
Gremlich, S. et al. Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology 146, 375–382 (2005).
Zhao, S. et al. alpha/beta-hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell. Metab. 19, 993–1007 (2014).
Zhou, Y. et al. Sirt3 deficiency increased the vulnerability of pancreatic beta cells to oxidative stress-induced dysfunction. Antioxid. Redox Signal. 27, 962–976 (2017).
Kim, M. et al. SIRT3 overexpression attenuates palmitate-induced pancreatic beta-cell dysfunction. PLoS. One. 10, e0124744 (2015).
Peterson, B. S. et al. Remodeling of the acetylproteome by SIRT3 manipulation fails to affect insulin secretion or beta cell metabolism in the absence of overnutrition. Cell Rep. 24, 209–223 e206 (2018).
Hirschey, M. D. et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190 (2011).
Boden, G., Chen, X. & Iqbal, N. Acute lowering of plasma fatty acids lowers basal insulin secretion in diabetic and nondiabetic subjects. Diabetes 47, 1609–1612 (1998).
Shanik, M. H. et al. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 31(Suppl 2), S262–S268 (2008).
Qiu, X., Brown, K., Hirschey, M. D., Verdin, E. & Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell. Metab. 12, 662–667 (2010).
Fukushima, A. & Lopaschuk, G. D. Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta 1862, 2211–2220 (2016).
Molven, A. et al. The hypoglycemic phenotype is islet cell-autonomous in short-chain hydroxyacyl-CoA dehydrogenase-deficient mice. Diabetes 65, 1672–1678 (2016).
Tyni, T. et al. Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency with the G1528C mutation: clinical presentation of thirteen patients. J. Pediatr. 130, 67–76 (1997).
Acknowledgements
We thank Dr. Weili Shen for generously providing SIRT3 knockout mice. This work was funded by grants from the National Natural Science Foundation of China (81270910, 81370876, 81471030, 81570693, and 81770767).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Zhou, F., Bai, M. et al. The pivotal role of protein acetylation in linking glucose and fatty acid metabolism to β-cell function. Cell Death Dis 10, 66 (2019). https://doi.org/10.1038/s41419-019-1349-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-019-1349-z
This article is cited by
-
Hepatic steatosis is associated with dysregulated cholesterol metabolism and altered protein acetylation dynamics in chickens
Journal of Animal Science and Biotechnology (2023)
-
Regulation of insulin secretion in mouse islets: metabolic amplification by alpha-ketoisocaproate coincides with rapid and sustained increase in acetyl-CoA content
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Chemical Modification of the Amino Groups of Human Insulin: Investigating Structural Properties and Amorphous Aggregation of Acetylated Species
The Protein Journal (2023)
-
Protein acylation: mechanisms, biological functions and therapeutic targets
Signal Transduction and Targeted Therapy (2022)
-
Proteome analysis identified proteins associated with mitochondrial function and inflammation activation crucially regulating the pathogenesis of fatty liver disease
BMC Genomics (2021)
