
Abstract
The autoimmune-mediated beta-cell death in type 1 diabetes (T1DM) is associated with local inflammation (insulitis). We examined the role of MCPIP1 (monocyte chemotactic protein–induced protein 1), a novel cytokine-induced antiinflammatory protein, in this process. Basal MCPIP1 expression was lower in rat vs. human islets and beta-cells. Proinflammatory cytokines stimulated MCPIP1 expression in rat and human islets and in insulin-secreting cells. Moderate overexpression of MCPIP1 protected insulin-secreting INS1E cells against cytokine toxicity by a mechanism dependent on the presence of the PIN/DUB domain in MCPIP1. It also reduced cytokine-induced Chop and C/ebpβ expression and maintained MCL-1 expression. The shRNA-mediated suppression of MCPIP1 led to the potentiation of cytokine-mediated NFκB activation and cytokine toxicity in human EndoC-βH1 beta-cells. MCPIP1 expression was very high in infiltrated beta-cells before and after diabetes manifestation in the LEW.1AR1-iddm rat model of human T1DM. The extremely high expression of MCPIP1 in clonal beta-cells was associated with a failure of the regulatory feedback-loop mechanism, ER stress induction and high cytokine toxicity. In conclusion, our data indicate that the expression level of MCPIP1 affects the susceptibility of insulin-secreting cells to cytokines and regulates the mechanism of beta-cell death in T1DM.
Similar content being viewed by others


Introduction
Type 1 diabetes (T1DM) is an autoimmune disease characterized by a selective death of pancreatic beta-cells, mediated by an inflammatory process in the pancreatic islets (insulitis)1,2,3,4. Beta-cell destruction is mediated by CD8+ T cell killing5 and by the action of proinflammatory cytokines1,2,6,7. Proinflammatory cytokines released by activated immune cells infiltrating the islets activate various signaling pathways in beta-cells1,2,6,7 and can lead to an increase in MHC class I on the surface of beta-cells8. The typically secreted cytokines IL-1β, TNFα and IFNγ influence transcription, translation and cause posttranscriptional and posttranslational modifications. These changes eventually lead to nitrooxidative stress and generation of proinflammatory mediators, causing mitochondrial and ER stress responses that result in beta-cell dysfunction and damage9,10,11,12,13,14,15.
MCPIP1 (monocyte chemotactic protein–induced protein 1) is a novel antiinflammatory protein, discovered in human blood monocytes stimulated with MCP-116 and in human monocyte-derived macrophages stimulated in vivo with IL-1β17. MCPIP1-knockdown mice suffer from severe inflammation18. MCPIP1 possesses a PIN-like domain with RNase and deubiquitinase properties (PIN/DUB) and is able to affect mRNA decay of several targets, including transcripts for proinflammatory cytokines (IL-1β, IL-6, IL-8) and proapoptotic proteins19,20,21,22,23,24. Recent studies have suggested that MCPIP1 can regulate mRNA degradation by an ARE-independent manner by binding to the stem-loop structure formed in the 3’UTR region of the targeted mRNAs21. MCPIP1 negatively regulates cellular inflammatory responses not only through its RNAse function, but also by deubiquitination of TRAF proteins (TRAF2, TRAF3, TRAF6) and interfering with the NFκB signaling25,26. MCPIP1 and NFκB regulate each other’s activity via a tight regulatory feedback-loop mechanism24. Targeted myocardial MCPIP1 overexpression resulted in inhibition of NFκB activity and a decrease of LPS-induced proinflammatory cytokine production, iNOS expression and caspase-3 activation27. Thus MCPIP1 seems to be a powerful negative regulator of inflammation.
The role of MCPIP1 in cytokine-mediated toxicity to pancreatic beta-cells in T1DM is unknown. Taking into account the important role of this protein in inflammatory processes, we decided to characterize its function in cytokine-mediated beta-cell death.
Materials and methods
Chemicals
Biotherm Taq polymerase was from GeneCraft (Münster, Germany) and Phusion High-Fidelity DNA polymerase from Thermo Fisher Scientific (Braunschweig, Germany). Cytokines were obtained from PromoCell (Heidelberg, Germany). Membranes and the ECL detection system were from Amersham Biosciences (Freiburg, Germany) and Milipore (Bedford, MA, USA). Other reagents were from Sigma Chemicals (München, Germany).
Animal and tissues
Pancreatic islets and other tissues were obtained from 250–300 g adult male Lewis rats bred in the Central Animal Facility of Hannover Medical School according to the principles of laboratory care approved by the Local Institutional Animal Care and Research Advisory Committee of Hannover Medical School and the Lower Saxony State Office (AZ: 2014/56). Islets were isolated by collagenase digestion, separated by Ficoll gradient, and hand-picked under a stereomicroscope. Pancreatic sections were obtained from healthy and diabetic LEW.1AR1-iddm rats28.
Cell culture, cytokine incubation, qRT-PCR, and RNA sequencing
INS1E cells (a kind gift of Prof.C.Wollheim, Geneva) and human EndoC-βH1 beta-cells (ENDOCELLS SARL, Paris, France;29) were cultured in a humidified atmosphere at 37 °C and 5% CO211,13,30. IL-1β was used at 600 U/ml. The cytokine mixture comprised IL-1β (60 U/ml), TNFα (185 U/ml), and IFNγ (14 U/ml). Double concentrations were used with human EndoC-βH1 beta-cells, as these cells are less sensitive to cytokine-mediated toxicity13. The incubation time for cytokine toxicity analysis for rat INS1E cells was 24, 48 or 72-h and for human EndoC-βH1 beta-cells 7 days, based on our earlier experience4,10,11,13 and time-dependency experiments (Fig. S1A and Fig. 7). To analyze an impact of cytokines on gene expression cells were incubated for 6, 12 or 24-h. In each series of experiments control cells and cells with modified expression of MCPIP1 were incubated with cytokines from various batches, which was responsible for various cytokine activities between different experiments.
Total RNA was isolated using peqGOLD Total RNA columns (Peqlab, Erlangen, Germany). After quality control, 1 µg of RNA was reverse transcribed using random hexamer primers. The QuantiTect SYBR GreenTM technology (Qiagen) was employed13. For absolute quantification of MCPIP1 gene expression an external standard curve was prepared for each target sequence (MCPIP1, β-actin, GAPDH) by cloning the specific cDNA into the pCR 4-TOPO vector (Thermo Fisher Scientific, Darmstadt, Germany). The gene expression of Bip, C/ebpβ, Chop, iNOS, Mcl-1 and MnSOD was normalized to housekeeping genes as indicated in the figure legends. Gene expression was measured by qBasePlus system (Biogazelle, Zwijnaarde, Belgium). Primer sequences are given in supplementary Table S2.
Five human islet preparations (detailed information is provided in31) were exposed to IL-1β (50 U/ml) + IFNγ (1000 U/ml) for 48-hrs and RNA was sequenced as described31. Raw data were deposited at the Gene Expression Omnibus, submission number GSE35296.
Genetic manipulation of MCPIP1 expression
INS1E cells were stably transfected with pcDNA3 vectors containing human wild-type MCPIP1, mutant ΔPIN/DUB MCPIP1 (a kind gift from Prof. J. Jura, Krakow, Poland23) or with splice variants 4 and 5 of MCPIP1 (synthesized by GeneCust, Luxembourg), or empty vector (mock) using the lipofectamine transfection method. MCPIP1 is a conserved protein in its function over various species32. Overexpression of human MCPIP1 in rat cells results in the generation of a fully functional MCPIP1 protein, and enables an easy tracking of the overexpression efficiency vs. induction of endogenous rat MCPIP1. This is an important control tool which allowed to distinguish the efficiency of overexpression in healthy cells from the infection/stress-induced endogenous MCPIP1 expression. Positive clones were selected against G418. MCPIP1 expression was determined by qRT-PCR and Western blotting. For inducible expression of MCPIP1 in human EndoC-βH1 beta-cells, a doxycycline-dependent overexpression Tet-ON system was used at a concentration of 25 ng/ml. Lentiviral particles for the pLVX-Tet3G vector (containing the transactivator protein) and pLVX-TRE3G-huMCPIP1 vector (containing the MCPIP1 gene under the TRE3G promoter) were generated using the Lenti-XTM Tet-On® 3G system (Clontech, Saint-Germain-en-Laye, France), according to the manufacturer’s manual. For MCPIP1 suppression cells were infected with lentiviral particles containing rat or human MCPIP1 short hairpin RNA (sc-156178-SH, sc-78944-SH, Santa Cruz Biotechnology, Heidelberg, Germany). shRNA-lentivirus particles encoding a scrambled shRNA sequence were used as a negative control (shQ). Positive clones were selected using puromycin (5 µg/ml; InvivoGen, Toulouse, France) and verified by qRT-PCR and Western blotting.
Cytokine toxicity
Cell viability was determined after a 24-h, 48-h or 72-h incubation of INS1E cells with cytokines using a microplate-based 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at 562/650 nm33. Caspase-3 or −12 activation was quantified using a green caspase-3 or red caspase-12 staining kit (PromoCell, Heidelberg, Germany) according to the instruction manual, followed by data analysis by FlowJo software (Tree Star, Ashland, OR)3,34. In the case of human EndoC-βH1 beta-cells the MTT assay does not provide reproducible results and therefore the PI method was used as in the case of other studies using this human beta-cell line35,36,37. The percentage of dead human EndoC-βH1 beta-cells (at least 500 cells per each condition) was determined after a 15-min incubation with the DNA-binding dye propidium iodide (PI) (50 µg/ml). The proliferation rate was quantified by using the Cell Proliferation BrdU–ELISA (Roche, Mannheim, Germany)3.
Western blot analyses
Cells were homogenized in ice-cold PBS containing protease inhibitor (Roche, Mannheim, Germany) using short bursts (Braun-Sonic 125 Homogenizer, Quigley-Rochester, Rochester, NY, USA). Protein content was determined by the BCA assay (Pierce). 40 μg of total protein was resolved by SDS polyacrylamide gel electrophoresis and then electroblotted onto nitrocellulose (iNOS) or PVDF membranes (MCPIP1, P-IKKα/β and MCL-1). Immunodetection was performed using specific primary antibodies followed by secondary antibody incubation (supplementary Table S3). The hybrids were visualized using the enhanced chemiluminescence detection kit and captured by the INTAS chemiluminescence detection system (Intas Science Imaging Instruments, Göttingen, Germany).
Nitrooxidative stress, NFκB activity, and ATP measurements
Nitrooxidative stress was detected after preincubation with 10 μM dichlorodihydrofluorescein diacetate DCFDA-H2 (30 min, 37 °C) followed by incubations with the test compounds and fluorescence measurements10. Nitrite accumulation was determined spectrophotometrically at 562 nm by the Griess reaction38. The pSEAP-NFκB construct and the Phospha-LightTM System kit (Thermo Fisher Scientific) were used to estimate NFκB activation39. ATP content was determined using the ATPlite Detection Assay System (PerkinElmer Life Sciences, Zaventem, Belgium) and normalized to protein content40.
Immunostaining
Immunofluorescence staining in INS1E cells was performed onto collagen-coated glass slides after an overnight fixation with 4% paraformaldehyde in PBS13,34. Images were captured and analyzed using a CellR/Olympus IX81 inverted microscope system (Olympus, Hamburg, Germany). Pancreatic sections from LEW.1 AR1-iddm rats were stained with the double immunofluorescence technique41. The beta-cells (stained in green by Cy2) were quantified in 10 islets per animal and subsequently analyzed for a possible co-localization of MCPIP1 (stained in red by Cy3) using an Olympus microscope BX61. The antibodies used are listed in the Table S3.
Data analysis
All data are expressed as means ± SEM. Statistical analyses were performed using the Prism analysis program as indicated in the figure legends (Graphpad, San Diego, CA).
Results
Expression of MCPIP1 in rat and human insulin-secreting cells
Gene expression of Mcpip1 was analyzed in several rat tissues (supplementary Table S4). Mcpip1 was strongly expressed in intestine, spleen, kidney, lung and liver, in contrast to skeletal muscle where its expression was very low (~3% of expression in liver) (supplementary Table S4). The Mcpip1 expression in rat insulin-secreting INS1E cells and in rat islets, as well as in brain was in the range of one third to half of that found in liver, respectively (supplementary Table S4).
The Mcpip1 gene expression was strongly induced by a mixture of proinflammatory cytokines (60 U/ml IL-1β, 185 U/ml TNFα and 14 U/ml IFNγ) in rat INS1E cells and rat islets (Fig. 1a). The induction of MCPIP1 was observed already after a 6-h incubation with cytokines and was continuously present after longer incubation times (12 and 24-h) (Fig. S1B). In the human EndoC-βH1 beta-cells the basal expression was around 2.3-times higher than in rat insulin-secreting INS1E cells as revealed by comparison of the number of copies of MCPIP1 normalized to the number of copies of the house-keeping gene β-actin (INS1E cells: 0.0005 vs. EndoC-βH1 beta-cells: 0.0013, n = 4). Furthermore, comparison of RNA sequencing data from human islets31 with data from primary rat beta-cells42 indicates a much higher expression of MCPIP1 in human islets (20.2 ± 3.6; data shown as RPKM; n = 5) than in rat beta cells (0.9 ± 0.1; data shown as RPKM; n = 3); The cytokine-mediated increase of MCPIP1 expression was smaller in human beta-cells than in rat beta-cells (Fig. 1a), like it was in the case of human vs. rat islets. Thus, though the basal expression of MCPIP1 was higher in human beta-cells than in rat beta-cells, a significant increase in MCPIP1 expression was observed in all analyzed beta-cell samples exposed to proinflammatory cytokines.

Real-time qRT-PCR analysis of MCPIP1 expression after 24-h incubation with a cytokine mixture (60 U/ml IL-1β, 185 U/ml TNFα, 14 U/ml IFNγ) in a Rat INS1E cells (relative gene expression of untreated cells: 0.023 ± 0.004, n = 5), rat pancreatic islets (relative gene expression of untreated cells: 0.018 ± 0.005, n = 4); human EndoC-βH1 beta-cells (relative gene expression of untreated cells: 2.175 ± 0.88, n = 5), data are means*1000 ± SEM, and human pancreatic islets (relative gene expression of untreated cells: 12.65 ± 3.89, n = 5, data from RNAseq31). b Immunostaining of MCPIP1 expression in untreated INS1E cells and after 24-h incubation with a cytokine mixture; green: MCPIP1, blue (DAPI): nuclei. Images were captured and analyzed using an Olympus fluorescence microscope with a 60 × oil objective. Overexpression of MCPIP1 in INS1E cells was established by stable transfection with pcDNA3-MCPIP1 vector and expression of MCPIP1 was measured by c qRT-PCR and d. Western blotting. Insulin-secreting control and MCPIP1-overexpressing INS1E cells were incubated for 24-h with IL-1β (600 U/ml) or a cytokine mixture (60 U/ml IL-1β, 185 U/ml TNFα, 14 U/ml IFNγ). e Cell viability was estimated by the MTT assay (n = 12–19); f Caspase-3 activation was estimated by the green caspase-3 staining kit and analyzed by FACS (n = 8–14); g Proliferation rate was analyzed by BrdU ELISA (n = 6–8); h ATP content was analyzed by ATPlite chemiluminescence assay (n = 5–6). Data are expressed as a percentage of the values in untreated cells. Data are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 vs. untreated, ##p < 0.01; ###p < 0.001 vs. control cells treated the same way, ANOVA followed by Bonferroni
Overexpression of MCPIP1 in INS1E cells
To analyze the role of MCPIP1 in insulin-secreting cells we generated a number of cell clones either overexpressing or suppressing MCPIP1 in rat INS1E cells (Fig. 2a and S2). The expression level of overexpressed human MCPIP1 varied significantly in the cell clones and had a differential influence on the susceptibility of the cells to cytokines as measured by MTT assay. The clones with a moderate MCPIP1 overexpression were protected against cytokine toxicity, while the clones with the highest overexpression were more sensitive to cytokines (Fig. 2a). The EC50 value for MCPIP1 expression after which a toxic, undesirable effect was observed was 0.35 ± 0.02 (n = 6) for IL-1β and 0.37 ± 0.03 (n = 6) for the cytokine mixture. The detailed analysis of MCPIP1 protein expression in the generated clones revealed that MCPIP1 protein bands of unexpected size appeared in the clones with the highest expression level (Fig. 2b), strongly suggesting the presence of inactive misfolded protein. To check this possibility we measured cytokine-mediated NFκB activation, since NFκB is a main target of MCPIP1 activity24. The results revealed that cytokine-induced NFκB activation was not downregulated in the clones with the highest expression of MCPIP1 (Fig. 2c). Moreover, the feedback-loop mechanism by which MCPIP1 regulates its own transcript24 was also dysregulated in the clones with the highest expression of MCPIP1 (Fig. 2d). The clones with the highest expression of MCPIP1 showed clear signs of an ER overload with misfolded protein as indicated by induction of the ER stress marker CHOP expression and caspase-12 activation (Fig. 2e, f). Therefore only the clones with the overexpression below the EC50 value were chosen for further analysis (Fig. 1c, d). Suppression of MCPIP1 did not significantly influence the cytokine-mediated toxicity in INS1E cells (Fig. S2).

Overexpression of MCPIP1 in INS1E cells was achieved by stable transfection with the pcDNA3-MCPIP1 vector. Cells were incubated for 24-h with IL-1β (600 U/ml) or a cytokine mixture (60 U/ml IL-1β, 185 U/ml TNFα, 14 U/ml IFNγ). a Cell viability was estimated by MTT assay; b Human MCPIP1 protein expression was analyzed by Western blotting; c NFκB activity was measured by a SEAP-reporter gene assay; d Rat Mcpip1 gene expression was analyzed by qRT-PCR and e Caspase-12 activation was estimated by the red caspase-12 staining kit and analyzed by FACS; f Representative immunostaining, green: MCPIP1, red: CHOP, blue (DAPI): nuclei. Images were captured and analyzed using an Olympus fluorescence microscope, using a 60 × oil objective. Open bars, untreated; grey bars, IL-1β; black bars, cytokine mixture. Data are means ± SEM of 6–8 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 vs. untreated, #p < 0.05; ###p < 0.001 vs. control cells treated in the same way; ANOVA followed by Bonferroni
MCPIP1 prevented the cytokine-mediated toxicity in INS1E cells
In control insulin-secreting INS1E cells (transfected with an empty vector) IL-1β and the cytokine mixture (IL-1β, TNFα and IFNγ) led to 50% loss of cell viability and strong caspase-3 activation after 24-h incubation (Fig. 1e, f). Overexpression of MCPIP1 prevented cytokine-mediated loss of cell viability and caspase-3 activation (Fig. 1e, f). The protective effect of MCPIP1 was present even in the case of a prolonged exposure to proinflammatory cytokines, however to a lesser extent as in the case of the 24-h incubation (Fig. S1A). MCPIP1 overexpression also resulted in a mild counteraction of the cytokine-induced proliferation rate decrease (Fig. 1g). The protective effect of MCPIP1 overexpression was not related to the ATP content (Fig. 1h), it seemed however to correlate with a decreased expression of MHC class I on the cell surface (Fig. S3).
MCPIP1 inhibited the NFκB-iNOS-NO signaling pathway in INS1E cells
Proinflammatory cytokines stimulated the transcription factor NFκB activation in control INS1E cells (Fig. 3a). Overexpression of MCPIP1 significantly reduced cytokine-mediated NFκB activation, which correlated with decreased phosphorylation of IKKα/β (Fig. 3a, c). Consequently, the cytokine-induced iNOS gene expression was inhibited by MCPIP1 overexpression as compared to control INS1E cells (Fig. 3b). This went along with diminished iNOS protein expression and reduced accumulation of nitrite in cytokine-treated MCPIP1-overexpressing INS1E cells as compared to control cells (Fig. 3c, d). In contrast, the expression of MnSOD was not significantly downregulated by MCPIP1 overexpression (Fig. 3e). The measurement of overall nitrooxidative stress revealed a weaker increase of DCF fluorescence in the MCPIP1-overexpressing cells than in control INS1E cells (Fig. 3f).

Insulin-secreting control and MCPIP1-overexpressing cells were incubated for 24-h with IL-1β (600 U/ml) or a cytokine mixture (60 U/ml IL-1β, 185 U/ml TNFα, 14 U/ml IFNγ). a NFκB activity was measured by a SEAP-reporter gene assay (n = 6–10); b iNOS gene expression was analyzed by qRT-PCR (relative gene expression of untreated mock cells: 0.061 ± 0.039, mean*1000 ± SEM, n = 6) (n = 6–7); c Representative blots of iNOS and p-IKKα/β protein expression were obtained by Western blotting after a 24-h or 30 min incubation, respectively (n = 4–6); d Nitrite accumulation was measured by Griess assay (n = 8–15); e MnSOD gene expression was analyzed by qRT-PCR (relative gene expression of untreated mock cells: 18 ± 2, mean*1000 ± SEM, n = 8) (n = 6–9); f Overall nitrooxidative stress was analyzed by DCF fluorescence (n = 8–12). Open bars, untreated; grey bars, IL-1β; black bars, cytokine mixture. Data are means ± SEM. ***p < 0.001 vs. untreated, #p < 0.05; ##p < 0.01; ###p < 0.001 vs. control cells treated the same way, ANOVA followed by Bonferroni
MCPIP1 downregulated cytokine-induced CHOP and C/EBPβ expression in INS1E cells
A short time (6-h) exposure to proinflammatory cytokines did not influence the expression of C/ebpβ, Chop and Bip (Fig. 4a). A longer incubation (12 and 24-h) with IL-1β or a cytokine mixture led to a strong increase of C/ebpβ expression in control INS1E cells (Fig. 4a). In contrast, MCPIP1 overexpressing INS1E cells showed a significantly lower expression level of C/ebpβ, which was only mildly affected by proinflammatory cytokines (Fig. 4a). Proinflammatory cytokines induced the expression of Chop and slightly decreased the expression of Bip in insulin-secreting INS1E cells after a 12-h incubation, with a more pronounced effect observed after a 24-h incubation (Fig. 4a). Overexpression of MCPIP1 reduced cytokine-mediated CHOP induction in INS1E cells (Fig. 4a, b), but did not influence the cytokine-mediated Bip decrease (Fig. 4a).

Insulin-secreting control and MCPIP1-overerexpressing cells were incubated for 6, 12 and 24-h with IL-1β (600 U/ml) or a cytokine mixture (60 U/ml IL-1β, 185 U/ml TNFα, 14 U/ml IFNγ). a C/ebpβ gene expression (relative gene expression of untreated mock cells at 24-h: 0.005 ± 0.001, mean*1000 ± SEM, n = 6) (n = 6–7); Chop gene expression (relative gene expression of untreated mock cells at 24-h: 6.052 ± 0.333, mean*1000 ± SEM, n = 8) (n = 6–10); Bip gene expression (relative gene expression of untreated mock cells at 24-h: 409 ± 44, mean*1000 ± SEM, n = 8) (n = 6–10) were measured by qRT-PCR. b Representative immunostaining, green: MCPIP1, red: CHOP, blue (DAPI): nuclei. Images were captured and analyzed using an Olympus fluorescence microscope, using a 60xoil objective. Open bars, untreated; grey bars, IL-1β; black bars, cytokine mixture. Data are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 vs. untreated, #p < 0.05; ##p < 0.01; ###p < 0.001 vs. control cells treated in the same way; ANOVA followed by Bonferroni
MCPIP1-mediated protection against cytokine-induced toxicity depended on the presence of the PIN/DUB domain
In order to confirm the importance of the NFκB-iNOS-NO signaling pathway inhibition for the protection against cytokine toxicity in MCPIP1-overexpressing INS1E cells, we generated a construct with a deletion of the PIN/DUB domain and overexpressed it in INS1E cells (Fig. 5a). The successful deletion of the PIN/DUB domain was counterchecked by qRT-PCR measurements using specific primers, which enabled a distinction between native and mutant MCPIP1 forms (Fig. 5a).

a Schematic presentation of the MCPIP1 domains in the wild type and mutant ΔPIN/DUB MCPIP1 protein. DNA gel electrophoresis of qRT-PCR end-products for two MCPIP1 specific pairs of primers at two different binding sites on the MCPIP1 sequence. b Cell viability was measured by MTT assay (n = 6–8); c Caspase-3 activation was estimated by the green caspase-3 staining kit and analyzed by FACS (n = 6–9); d Overall nitrooxidative stress was measured by DCF fluorescence (n = 4–6); e Nitrite accumulation was analyzed by Griess assay (n = 4–7); f Chop gene expression was analyzed by qRT-PCR (relative gene expression of untreated mock cells: 6.052 ± 0.333, mean*1000 ± SEM, n = 8) (n = 5–8); g Bip gene expression was analyzed by qRT-PCR (relative gene expression of untreated mock cells: 409 ± 44, mean*1000 ± SEM, n = 8) (n = 5–8). INS1E insulin-secreting cells were incubated for 24-h with IL-1β (600 U/ml) or a cytokine mixture (60 U/ml IL-1β, 185 U/ml TNFα, 14 U/ml IFNγ). Open bars, untreated; grey bars, IL-1β; black bars, cytokine mixture Data are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 vs. untreated, #p < 0.05; ##p < 0.01; ###p < 0.001 vs. control cells treated the same way; ANOVA followed by Bonferroni
The experiments showed that the PIN/DUB domain is crucial for the protective effect provided by MCPIP1 overexpression (Fig. 5). Overexpression of the mutant MCPIP1 did not counteract cytokine-mediated cell viability loss, caspase-3 activation and nitrooxidative stress (Fig. 5b–e). Also, the induction of Chop gene expression by cytokines was not affected by mutant MCPIP1 overexpression as compared to control cells (Fig. 5f). Overexpression of the mutant MCPIP1 decreased basal and cytokine-mediated Bip expression in INS1E cells (Fig. 5g).
Overexpression of various splice variants of MCPIP1 in INS1E cells
RNA-seq analysis of human pancreatic islets exposed to the proinflammatory cytokines IL-1β + IFN-γ for 48 h, as described in31, revealed the presence of alternatively spliced forms of MCPIP1 (Fig. 6a). The splice variant 4 lacks the sequence coding for the PIN/DUB domain while the splice variant 5 partially lacks the PIN/DUB and ZF domains (Fig. 6b). In order to examine the function of splice variants 4 and 5, constructs containing the spliced forms were transfected into INS1E cells in which the endogenous MCPIP1 expression was suppressed. The cells were than exposed to cytokines and their viability and overall nitrooxidative stress were analyzed (Fig. 6c, d). The cytokine sensitivity of cells expressing various splice variants was done in comparison with control cells transfected with an empty vector used for splice variant expression together with silencing of the wild type of MCPIP1 (Fig. 6d). These control cells differed in their viability from the control cells transfected only with an empty vector (Fig. 1e and Fig. 6c, d). Overexpression of the splice variant 4 enhanced cytokine-mediated loss of cell viability and aggravated nitrooxidative stress (Fig. 6c). The overexpression of variant 5 led only to minor changes in response to cytokines (Fig. 6d).

a Expression of the MCPIP1 variants 4 and 5 in human islets after exposure to a mixture of proinflammatory cytokines (IL-1β 50 U/ml and IFNγ 1000 U/ml) using RNAseq (29); b Scheme of the MCPIP1 splice variants 4 and 5 in comparison to the full length MCPIP1; c Overexpression of the MCPIP1 variant 4 in insulin-secreting INS1E cells and its effects on cytokine-mediated cell viability and induction of overall nitrooxidative stress; d Overexpression of the MCPIP1 variant 5 in insulin-secreting INS1E cells and its effects on cytokine-mediated cell viability and induction of overall nitrooxidative stress. Cell viability was measured by MTT assay. Nitrooxidative stress was estimated by DCF fluorescence. Open bars, untreated; grey bars, IL-1β; black bars, cytokine mixture. Data are means ± SEM of 4–6 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 vs. untreated, #p < 0.05; ##p < 0.01; ###p < 0.001 vs. control cells treated in the same way; ANOVA followed by Bonferroni
High basal expression of MCPIP1 made the human EndoC-βH1 beta-cells less susceptible to cytokine toxicity
The expression of MCPIP1 in untreated human EndoC-βH1 beta-cells was significantly higher than that measured in rat insulin-secreting INS1E cells (approximately 2.3-fold higher in EndoC-βH1 vs. INS1E cells). However, it was lower than in INS1E cell clones with the highest MCPIP1 expression, in which the deleterious effects of MCPIP1 were observed as described above. It is known that human beta-cells are less sensitive to proinflammatory cytokines than rat beta-cells and the cytokine toxicity develops over a longer time as compared to rat beta-cells13. To investigate the role of MCPIP1 in the differential susceptibility of beta-cells to cytokine toxicity we generated human EndoC-βH1 beta-cell clones overexpressing or suppressing MCPIP1 (Fig. 7a, b). We chose the PI staining for the analysis of cytokine-mediated toxicity in this cell line, since this method provides the most reproducible results and is typically used by other groups working with these cells35,36,37. The analysis of cell death of EndoC-βH1 beta-cells by PI staining revealed that shRNA-mediated suppression of MCPIP1 led to a significant 2-fold increase in cell death already in untreated cells (Fig. 7a). The cell death rate after incubation with cytokines was significantly higher after MCPIP1 suppression (Fig. 7a). Interestingly, and in contrast to control EndoC-βH1 beta-cells, there was a significant activation of NFκB after cytokine exposure in human beta-cells with a MCPIP1 knock-down (Fig. 7a). Importantly, a very mild overexpression of MCPIP1 (doxycycline doses up to 15 ng/ml) was not protective to human EndoC-βH1 cells (data not shown). Induction of MCPIP1 by a slightly higher dose of 25 ng/ml of doxycycline led to increased cell death (Fig. 7b). Thus the basal expression of MCPIP1 in untreated human EndoC-βH1 beta-cells was around twice as high as it was in untreated rat INS1E cells and this correlated with reduced NFκB activation and lower susceptibility towards proinflammatory cytokines.

a Effects of MCPIP1 suppression (by means of shRNA transfection) on cytokine-mediated cell death and NFκB activation. b Effects of MCPIP1 overexpression (by stable transfection with the pLVX-EF1a-Tet3-ZsGreen-MCPIP1 vector, in the presence of 25 ng/ml doxycycline). c Gene and protein expression of MCL-1 in rat INS1E and human EndoC-βH1 beta-cells after exposure to cytokines (24-h). Gene expression was analyzed by qRT-PCT. Protein expression was estimated by Western blotting (n = 4). Cytokine-mediated cell death was analyzed by PI staining, a well-established method of human beta-cell death measurement35,36. NFκB activation was measured by a SEAP-reporter gene assay. Human EndoC-βH1 beta-cells were incubated for 7 days with IL-1β (1200 U/ml) or a cytokine mixture (120 U/ml IL-1β, 370 U/ml TNFα, 280 U/ml IFNγ). This longer exposure time and higher concentrations of cytokines were used due to the weaker sensitivity of this cell line to cytokines13. Open bars, untreated; grey bars, IL-1β; black bars, cytokine mixture. Data are means ± SEM of 6–8 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 vs. untreated, #p < 0.05; ##p < 0.001; ###p < 0.001 vs. control cells treated the same way; ANOVA followed by Dunnett
MCPIP1 maintained a high expression of the antiapoptotic MCL-1 protein
MCL-1 is one of the most important antiapoptotic proteins in beta-cells36. The gene expression of Mcl1 was increased by IL-1β alone and the cytokine mixture in insulin-secreting INS1E cells (Fig. 7c), thus confirming earlier observations43. MCPIP1 overexpressing INS1E cells showed a slightly lower endogenous gene expression of Mcl1 as compared to control cells (Fig. 7c). On the protein level the expression of MCL-1 was not significantly influenced in INS1E cells by 24-h incubation with 600 U/ml IL-1β or a cytokine mixture (Fig. 7c). Interestingly, the protein expression of MCL-1 was significantly higher in INS1E cells overexpressing MCPIP1 than in control INS1E cells (Fig. 7c).
Moreover, human EndoC-βH1 beta-cells were characterized by a strong and significantly higher basal expression of MCL-1 as compared to rat INS1E cells (Fig. 7c). Incubation with IL-1β alone or a mixture of proinflammatory cytokines slightly decreased the MCL-1 protein expression in EndoC-βH1 beta-cells (Fig. 7c). Control experiments with the same cytokine concentrations and combinations as by36 reduced significantly MCL-1 protein expression (data not shown), thus confirming earlier observations. Thus, a moderate expression of MCPIP1 correlated with a strong expression of the antiapoptotic protein MCL-1.
MCPIP1 expression fluctuated during diabetes development in the LEW.1AR1-iddm rat, a model of human T1DM
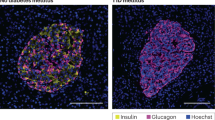
The Mcpip1 expression was analyzed also in islets from the IDDM rat (LEW.1AR1-iddm rat), a model of human T1DM (supplementary Table S1 and Fig. 8). In each islet around 50 beta-cells were analyzed, with exception of the rats with overt diabetes, in which a concomitant loss of insulin-positive beta-cells was observed (Table S1, Fig. 8). The number of MCPIP1-positive beta-cells differed significantly in healthy vs. prediabetic IDDM rats with normoglycaemia and signs of islet infiltration as well as in diabetic IDDM rats with hyperglycaemia and severe islet infiltration (Table S1, Fig. 8). In normoglycaemic rats without any signs of islet infiltration only 12% of beta-cells were MCPIP1-positive with a relatively faint expression level (Table S1, Fig. 8). In the prediabetic phase with islet infiltration, the number of double positive cells increased significantly to 35% (Table S1, Fig. 8). In hyperglycaemic rats with severe signs of islet infiltration and beta-cell loss the number of MCPIP1-positive beta-cells was around 39%, also significantly higher than in normoglycaemic rats (Table S1; Fig. 8).

Pancreatic islets from the IDDM rat were analyzed for MCPIP1 expression under various metabolic conditions (detailed information in Table S1). a Representative pictures of immunostaining, green: insulin, red: MCPIP1, blue (DAPI): nuclei. Images were captured and analyzed using an Olympus fluorescence microscope, using a 60x oil objective. White asterix: erythrocyte, white arrow: MCPIP1-positive beta-cell, white V: MCPIP1-postive non-beta-cell. b Quantification of the absolute number of MCPIP1-positive beta-cells in pancreatic islets of IDDM rats. Four animals and ten islets per animal in each group were analyzed. Data are presented as means±SEM. *p < 0.05 vs. MCPIP1-positive beta-cells in normoglycaemic without islet infiltration
Discussion
The novel cytokine-induced protein MCPIP1 is an interesting candidate for future antiinflammatory therapies in the context of several inflammatory disorders19,32,44,45,46. To date there has been no information about the role of MCPIP1 in pancreatic beta-cells and its significance for T1DM.
To uncover the role of MCPIP1 in beta-cells we employed genetically modified insulin-secreting cell lines of rat and human origin. Interestingly, we observed a higher basal expression of MCPIP1 in human than in rat insulin-secreting cells. This correlated with the lower susceptibility to proinflammatory cytokines and reduced cytokine-mediated MCPIP1 induction in human vs. rat beta-cells. The transcription factor NFκB plays a crucial role in cytokine-induced cell death of rat beta-cells6,47 by a mechanism strongly dependent on the induction of the iNOS-NO pathway and nitrooxidative stress-mediated disturbances of mitochondria and ER3,9,10,11,12,13,14,15. In contrast, proinflammatory cytokines fail to induce the iNOS pathway in human beta-cells, an effect accounted for the weaker activation of the NFκB in human beta-cells10,13,35. Our data indicate that this difference between human and rat beta-cells could be explained at least partially by the higher basal MCPIP1 expression in human beta-cells, reducing the activation of NFκB by a regulatory feedback-loop as described in hepatoma cells24.
Proinflammatory cytokines induced a rapid and strong MCPIP1 expression in insulin-secreting cells of various origins as well as in primary rat and human islets. Importantly, we confirmed these in vitro observations in a rat model of human T1DM, the IDDM rat (LEW.1AR1-iddm rat)28. Upon infiltration of islets, but before hyperglycaemia, the number of MCPIP1-positive beta-cells as well as the magnitude of MCPIP1 expression significantly increased and were maintained at a high level after diabetes manifestation. These data suggest that MCPIP1 might be involved in the regulation of the pancreatic beta-cell fate during T1DM development. Further analyses in pancreatic sections of T1DM patients at various onsets of the disease are however needed to confirm a role of MCPIP1 in human diabetes.
Interestingly, in rat INS1E cells the cytokine-mediated cell viability loss and caspase-3 activation were prevented by moderate overexpression of MCPIP1, but potentiated by its very strong overexpression. Suppression of MCPIP1 in INS1E cells did not affect cytokine toxicity since the basal MCPIP1 expression in rat insulin-secreting cells was already very low. The protective effect of moderate MCPIP1 overexpression in INS1E cells was strongly dependent on the inhibition of the cytokine-mediated phosphorylation of IKKα/β, reduction of NFκB activation and decrease of iNOS gene and protein expression, resulting in a reduced nitrite accumulation. In human beta-cells NO generation is not induced by cytokines13,35, and this correlates with a slower development of cell death. Indeed suppression of MCPIP1 resulted in a potentiation of cytokine toxicity in human EndoC-βH1 beta-cells together with a parallel increase of NFκB activation after cytokine exposure. Interestingly, a weak overexpression of MCPIP1 in human EndoC-βH1 beta-cells did not influence cytokine toxicity, while a stronger overexpression had similar effects to the high, deleterious overexpression in rat INS1E cells. This observation suggests that the basal expression of MCPIP1 in human EndoC-βH1 beta-cells, which are characterized by a weaker sensitivity to cytokine-mediated toxicity than rat INS1E cells, resembles the protective moderate overexpression of MCPIP1 in the rat beta-cell line. The toxic effects of a high overexpression of MCPIP1 in rat INS1E cells were attributed to the accumulation of misfolded MCPIP1 protein and disturbances in the ER. In contrast, moderate MCPIP1 overexpression resulted in a reduced cytokine-mediated Chop expression in INS1E cells, indicative of ER stress inhibition since CHOP induction is a central event in cytokine-induced ER stress48.
The protein C/EBPβ has been shown to be involved in ER stress activation via binding to ATF4 and CHOP49. Here we showed that C/ebpβ expression was significantly induced by proinflammatory cytokines in control-INS1E cells, while its basal expression was significantly lower and not affected by cytokines after moderate MCPIP1 overexpression. This is in line with previous findings showing a cleavage of C/EBPβ by MCPIP122.
The protective effect of moderate MCPIP1 overexpression in rat INS1E cells as well as the lower susceptibility of human EndoC-βH1 beta-cells towards proinflammatory cytokines strongly correlated with the MCL-1 expression level. MCL-1 has been recently shown to be a crucial antiapoptotic protein in rat and human beta-cells and is markedly downregulated in human islets of T1DM patients36,43. MCL-1 turnover depends on the ubiquitination status36. MCPIP1 contains a PIN/DUB domain which has RNAse and deubiquitynation properties as well an UBA domain which participates in the ubiquitination regulation32. Transfections of mutant or spliced forms of MCPIP1 revealed that the protective effects of MCPIP1 can be attributed to the function of the PIN/DUB domain. The PIN/DUB domain was responsible for the reduction of NFκB-iNOS pathway induction as well as the prevention of ubiquitination and degradation of the key antiapoptotic protein MCL-1. We observed also a reduced expression of the ER chaperone Bip after overexpression of the mutant MCPIP1 lacking the PIN/DUB domain activity, as it was shown in a recent study in renal carcinoma cells50. Our data suggest that splice variants of MCPIP1 could be involved in the fine tuning of the MCPIP1 action in beta-cells, a process likely to be dysregulated under unresolved inflammation during T1DM development, which may lead to uncontrolled expression of MCPIP1 and increased cytokine toxicity. These results strengthen the recently expressed view that modified alternative splicing is an important mechanism by which inflammation contributes to beta-cell dysfunction and death51.
Our in vitro data show an antiapoptotic, antiinflammatory potential of MCPIP1 in rat beta-cells upon a moderate overexpression and a toxic effect in the case of a very strong overexpression, a situation which might occur in the case of severely infiltrated islets in the IDDM rat. Noteworthy, in hyperglycaemic IDDM rats beta-cells show a higher expression of caspase-3 and perforin in severely infiltrated islets52. The main difference between faintly and severely infiltrated islets, besides the number and localization of the immune cells, was the dissociated islet architecture that can lead to new interactions between beta-cells and other endocrine as well as stromal cells41. Changes in MCPIP1 expression in islets upon infiltration could also influence the T-cell receptor expression on the activated T-cells within the islet infiltrate, thereby changing the entire milieu as recently shown for psoriasis44. Fluctuations of MCPIP1 expression could also affect the macrophage polarization at the site of insulitis, as shown in other inflammatory disorders53 or MHC class I expression on beta-cell surface as shown in our study.
In conclusion, MCPIP1 is a novel, powerful cytokine-induced protein in beta-cells that regulates beta-cell cytokine susceptibility by affecting a number of cytokine-sensitive pathways. Since MCPIP1 expression undergoes fluctuations in islets during T1DM development in the IDDM rat and regulates the beta-cell fate upon exposure to proinflammatory cytokines, manipulations of its expression and activity might open new perspectives for T1DM treatment.
References
Santin, I. & Eizirik, D. L. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and beta-cell apoptosis. Diabetes Obes. Metab. 15, 71–81 (2013).
Eizirik, D. L., Colli, M. L. & Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 5, 219–226 (2009).
Gurgul-Convey, E. & Lenzen, S. Protection against cytokine toxicity through endoplasmic reticulum and mitochondrial stress prevention by prostacyclin synthase overexpression in insulin-producing cells. J. Biol. Chem. 285, 11121–11128 (2010).
Hahn, C., Tyka, K., Saba, J. D., Lenzen, S. & Gurgul-Convey, E. Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 292, 20292–20304 (2017).
Morgan, N. G., Leete, P., Foulis, A. K. & Richardson, S. J. Islet inflammation in human type 1 diabetes mellitus. IUBMB Life 66, 723–734 (2014).
Eizirik, D. L. & Grieco, F. A. On the immense variety and complexity of circumstances conditioning pancreatic beta-cell apoptosis in type 1 diabetes. Diabetes 61, 1661–1663 (2012).
Eizirik, D. L. & Mandrup-Poulsen, T. A choice of death-the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44, 2115–2133 (2001).
Richardson, S. J. et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 59, 2448–2458 (2016).
Lenzen, S. Oxidative stress: the vulnerable beta-cell. Biochem. Soc. Trans. 36, 343–347 (2008).
Gurgul-Convey, E., Mehmeti, I., Lortz, S. & Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 89, 785–798 (2011).
Kacheva, S., Lenzen, S. & Gurgul-Convey, E. Differential effects of proinflammatory cytokines on cell death and ER stress in insulin-secreting INS1E cells and the involvement of nitric oxide. Cytokine 55, 195–201 (2011).
Jörns, A. et al. Islet infiltration, cytokine expression and beta cell death in the NOD mouse, BB rat, Komeda rat, LEW.1AR1-iddm rat and humans with type 1 diabetes. Diabetologia 57, 512–521 (2014).
Gurgul-Convey, E., Mehmeti, I., Plotz, T., Jörns, A. & Lenzen, S. Sensitivity profile of the human EndoC-betaH1 beta cell line to proinflammatory cytokines. Diabetologia 59, 2125–2133 (2016).
Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic beta-cells. Biochim. Biophys. Acta 1861, 1929–1942 (2017).
Eizirik, D. L., Miani, M. & Cardozo, A. K. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56, 234–241 (2013).
Zhou, L. et al. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 98, 1177–1185 (2006).
Jura, J. et al. Identification of interleukin-1 and interleukin-6-responsive genes in human monocyte-derived macrophages using microarrays. Biochim. Biophys. Acta 1779, 383–389 (2008).
Matsushita, K. et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458, 1185–1190 (2009).
Bugara, B. et al. MCPIP1 contributes to the inflammatory response of UVB-treated keratinocytes. J. Dermatol. Sci. 87, 10–18 (2017).
Dobosz, E. et al. MCPIP-1, Alias Regnase-1, Controls Epithelial Inflammation by Posttranscriptional Regulation of IL-8 Production. J. Innate Immun. 8, 564–578 (2016).
Li, M. et al. MCPIP1 down-regulates IL-2 expression through an ARE-independent pathway. PLoS ONE 7, e49841 (2012).
Lipert, B., Wilamowski, M. & Gorecki, A. Jura J. MCPIP1, alias Regnase-1 binds and cleaves mRNA of C/EBPbeta. PLoS ONE 12, e0174381 (2017).
Mizgalska, D. et al. Interleukin-1-inducible MCPIP protein has structural and functional properties of RNase and participates in degradation of IL-1beta mRNA. Febs. J. 276, 7386–7399 (2009).
Skalniak, L. et al. Regulatory feedback loop between NF-kappaB and MCP-1-induced protein 1 RNase. Febs. J. 276, 5892–58905 (2009).
Liang, J. et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J. Exp. Med. 207, 2959–2973 (2010).
Niu, J. et al. USP10 inhibits genotoxic NF-kappaB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 32, 3206–3219 (2013).
Niu, J., Wang, K., Graham, S., Azfer, A. & Kolattukudy, P. E. MCP-1-induced protein attenuates endotoxin-induced myocardial dysfunction by suppressing cardiac NF-small ka, CyrillicB activation via inhibition of Ismall ka, CyrillicB kinase activation. J. Mol. Cell Cardiol. 51, 177–186 (2011).
Lenzen, S. et al. The LEW.1AR1/Ztm-iddm rat: a new model of spontaneous insulin-dependent diabetes mellitus. Diabetologia 44, 1189–1196 (2001).
Ravassard, P. et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Invest. 121, 3589–3597 (2011).
Gurgul-Convey, E., Kaminski, M. T. & Lenzen, S. Physiological characterization of the human EndoC-betaH1 beta-cell line. Biochem. Biophys. Res. Commun. 464, 13–19 (2015).
Eizirik, D. L. et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552 (2012).
Jura, J., Skalniak, L. & Koj, A. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim. Biophys. Acta 1823, 1905–1913 (2012).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983).
Hanzelka, K., Skalniak, L., Jura, J., Lenzen, S. & Gurgul-Convey, E. Effects of the novel mitochondrial protein mimitin in insulin-secreting cells. Biochem. J. 445, 349–359 (2012).
Brozzi, F. et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 58, 2307–2316 (2015).
Meyerovich, K. et al. MCL-1 Is a key antiapoptotic protein in human and rodent pancreatic beta-cells. Diabetes 66, 2446–2458 (2017).
Plötz, T. et al. The monosaturated fatty acid oleate is the major physiological toxic free fatty acid for human beta cells. Nutr. Diabetes 7, 305 (2017).
Tiedge, M., Lortz, S., Munday, R. & Lenzen, S. Protection against the co-operative toxicity of nitric oxide and oxygen free radicals by overexpression of antioxidant enzymes in bioengineered insulin-producing RINm5F cells. Diabetologia 42, 849–855 (1999).
Azevedo-Martins, A. K. et al. Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-kappaB activation in insulin-producing cells. Diabetes 52, 93–101 (2003).
Gurgul-Convey, E., Hanzelka, K. & Lenzen, S. Mechanism of prostacyclin-induced potentiation of glucose-induced insulin secretion. Endocrinology 153, 2612–2622 (2012).
Jörns, A. et al. Immune cell infiltration, cytokine expression, and beta-cell apoptosis during the development of type 1 diabetes in the spontaneously diabetic LEW.1AR1/Ztm-iddm rat. Diabetes 54, 2041–2052 (2005).
Villate, O. et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 42, 11818–11830 (2014).
Allagnat, F. et al. Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event contributing to beta-cell apoptosis. Cell Death Differ. 18, 328–337 (2011).
Monin, L. et al. MCPIP1/Regnase-1 restricts IL-17A and IL-17C-dependent skin inflammation. J. Immunol. 198, 767–775 (2017).
Cheng, Y. et al. Mmu-miR-27a-5p-dependent upregulation of MCPIP1 inhibits the inflammatory response in LPS-induced RAW264.7 macrophage cells. Biomed. Res. Int. 2015, 607692 (2015).
Miao, R. et al. Targeted disruption of MCPIP1/Zc3h12a results in fatal inflammatory disease. Immunol. Cell Biol. 91, 368–376 (2013).
Ortis, F. et al. Induction of nuclear factor-kappaB and its downstream genes by TNF-alpha and IL-1beta has a pro-apoptotic role in pancreatic beta cells. Diabetologia 51, 1213–1225 (2008).
Allagnat, F. et al. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells. Cell Death Differ. 19, 836–846 (2012).
van der Krieken, S. E., Popeijus, H. E., Mensink, R. P. & Plat, J. CCAAT/enhancer binding protein beta in relation to ER stress, inflammation, and metabolic disturbances. Biomed. Res. Int. 2015, 324815 (2015).
Lichawska-Cieslar, A. et al. RNA sequencing reveals widespread transcriptome changes in a renal carcinoma cell line. Oncotarget 9, 8597–8613 (2018).
Juan-Mateu, J., Villate, O. & Eizirik, D. L. Mechanisms in endocrinology: alternative splicing: the new frontier in diabetes research. Eur. J. Endocrinol. 174, R225–R238 (2016).
Jörns, A. et al. TNF-alpha antibody therapy in combination with the T-cell-specific antibody anti-TCR reverses the diabetic metabolic state in the LEW.1AR1-iddm rat. Diabetes 64, 2880–2891 (2015).
Kolattukudy, P. MCPIP: a key player in macrophage polarization. Oncotarget 6, 28531–28532 (2015).
Acknowledgements
The work was supported by a grant from DAAD (German Academic Exchange Service) to K.T. and the EFSD Albert Renold Fellowship to E.G.C. The excellent technical assistance of M. Funck, M. Wirth and Ch. Schwab is acknowledged. We thank very much Prof. J. Jura (Department of General Biochemistry, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Krakow, Poland) for pcDNA3-MCPIP1 and MCPIP1ΔPIN vectors and MCPIP1 polyclonal antibodies.
Author information
Authors and Affiliations
Contributions
K.T. performed experiments, analyzed the data, prepared figures and tables, participated in writing the manuscript; A.J. performed experiments in the IDDM rat, analyzed these data and made a table, discussed data and reviewed the manuscript; J.V.T. and D.L.E. performed and analyzed RNA sequencing of human islets, discussed the data and reviewed the manuscript, S.L. analyzed the data and reviewed the manuscript; E.G.C. designed the study, performed experiments, analyzed the data, wrote the manuscript, coordinated the project and is the guarantor of this work, as such, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by K. Schroder
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tyka, K., Jörns, A., Turatsinze, JV. et al. MCPIP1 regulates the sensitivity of pancreatic beta-cells to cytokine toxicity. Cell Death Dis 10, 29 (2019). https://doi.org/10.1038/s41419-018-1268-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-1268-4
This article is cited by
-
Mitofusin-2 regulates inflammation-mediated mouse neuroblastoma N2a cells dysfunction and endoplasmic reticulum stress via the Yap-Hippo pathway
The Journal of Physiological Sciences (2019)
