
Abstract
The mitogen-activated protein kinase (MAPK) pathway has been shown to be involved in both neurodevelopment and neurodegeneration. c-Jun N-terminal kinase (JNK), a MAPK important in retinal development and after optic nerve crush injury, is regulated by two upstream kinases: MKK4 and MKK7. The specific requirements of MKK4 and MKK7 in retinal development and retinal ganglion cell (RGC) death after axonal injury, however, are currently undefined. Optic nerve injury is an important insult in many neurologic conditions including traumatic, ischemic, inflammatory, and glaucomatous optic neuropathies. Mice deficient in Mkk4, Mkk7, and both Mkk4 and Mkk7 were generated. Immunohistochemistry was used to study the distribution and structure of retinal cell types and to assess RGC survival after optic nerve injury (mechanical controlled optic nerve crush (CONC)). Adult Mkk4- and Mkk7-deficient retinas had all retinal cell types, and with the exception of small areas of disrupted photoreceptor lamination in Mkk4-deficient mice, the retinas of both mutants were grossly normal. Deficiency of Mkk4 or Mkk7 reduced JNK signaling in RGCs after axonal injury and resulted in a significantly greater percentage of surviving RGCs 35 days after CONC as compared to wild-type controls (Mkk4: 51.5%, Mkk7: 29.1%, WT: 15.2%; p < 0.001). Combined deficiency of Mkk4 and Mkk7 caused failure of optic nerve formation, irregular retinal axonal trajectories, disruption of retinal lamination, clumping of RGC bodies, and dendritic fasciculation of dopaminergic amacrine cells. These results suggest that MKK4 and MKK7 may serve redundant and unique roles in molecular signaling important for retinal development and injury response following axonal insult.
Similar content being viewed by others

Introduction
The mitogen-activated protein kinase (MAPK) pathway is involved in development, neurodegeneration, and the immune response1,2,3,4,5. In the retina, MAPK signaling plays a role in retinal formation and axonal injury-induced retinal ganglion cell (RGC) death6,7,8,9,10,11,12. The MAPK, c-Jun N-terminal kinase (JNK), is regulated by two upstream MAP2Ks: MKK4 and MKK75,13,14. The specific requirements of MKK4 and MKK7 in retinal development and neurodegeneration, however, are currently undefined.
MKK4 and MKK7 are required for normal development15. In the central nervous system, MKK4 and MKK7 and their downstream effector molecules, the JNKs (JNK1–3), play important roles in both development and maintenance of neural structures. MKK4, MKK7, and the JNKs contribute to the regulation of cellular organization and axonal migration through both overlapping and non-redundant mechanisms16,17,18,19,20. JNK signaling has also been shown to contribute to multiple aspects of retinogenesis such as progenitor cell proliferation14,21. The exact contributions of MKK4 and MKK7 to retinal development, however, remain largely unexplored. In the adult, multiple MAPK members have been shown to be key mediators of the apoptotic injury response and RGC death after axonal injury. Specifically, JNKs and their canonical downstream effector molecule, the transcription factor JUN, are important for RGC death after mechanical- and ocular hypertension-induced axonal injury7,8,21,22,23,24. Despite this known involvement, the critical molecular events leading from axonal injury to RGC death are not fully defined. Determining the molecular mechanisms of RGC pro-death signaling after axonal injury is necessary for understanding the molecular underpinnings of diseases such as glaucoma and traumatic optic neuropathies which result in RGC loss.
The importance of JNK signaling for both RGC development and response to axonal injury is well established, but little is known regarding the role of the MAP2Ks upstream of JNK in these processes. Selectively targeting these upstream MAPKs may allow us to define the specific pathological signaling pathway that activates pro-death JNK activation in RGC axons after an insult. Furthermore, understanding the contribution of MKK4 and MKK7 to the injury response and to JUN activation in RGCs will likely have implications for other diseases or traumas involving axonal injury. Here, using conditional null alleles of Mkk4 and Mkk7, we demonstrate their redundant and unique roles in retinal development and injury signaling.
Materials and methods
Mice
All experiments were conducted in adherence with the Association for Research in Vision and Ophthalmology’s statement on the use of animals in ophthalmic and vision research and were approved by the University of Rochester’s University Committee on Animal Resources. Animals were housed on a 12-h light and dark cycle and received chow and water ad libitum. Floxed alleles of Mkk4 and Mkk725,26 were maintained on the C57BL/6J background. To generate animals deficient in Mkk4 or Mkk7, mice carrying the respective floxed allele were bred to animals carrying Six3cre which is first expressed in the optic cup between E9.0 and 9.527. Animals carrying the Cre recombinase and one Mkk4 or Mkk7 floxed allele were intercrossed to generate animals: (1) carrying Cre recombinase and two copies of either Mkk4 or Mkk7 floxed alleles, referred to as Mkk4 deficient (Mkk4−/− or Six3cre+; Mkk4f/f) and Mkk7 deficient (Mkk7−/− or Six3cre+; Mkk7f/f) respectively and (2) animals without floxed alleles and/or without Cre recombinase, referred to as wild-type controls (Six3cre+; Mkk4+/+, Six3cre−; Mkk4+/+, Six3cre−; Mkk4f/+, Six3cre−; Mkk4f/f, Six3cre+; Mkk7+/+, Six3cre−; Mkk7+/+, Six3cre−; Mkk7f/+, or Six3cre−; Mkk7f/f). Animals deficient in both Mkk4 and Mkk7 were generated by breeding animals carrying the floxed alleles and Six3cre together to generate Six3cre+; Mkk4f/f; Mkk7f/f animals (Mkk4/Mkk7-deficient animals).
Retinal histology, immunohistochemistry, and cell counts
Eyes used to evaluate retinal morphology were processed in 2.5% glutaraldehyde and 2% paraformaldehyde (PFA) for 24 h at 4 °C. Eyes were then dehydrated with a series of ethanol washes, embedded in Technovit (Electron Microscopy Services), sectioned at 2.5 μm, and stained with Multiple Stain Solution (Polysciences, Inc). Eyes to be processed for immunohistochemistry and retinal flat mounts were harvested and processed in 4% PFA for 2 h at room temperature prior to storage in 1 M phosphate-buffered saline (PBS). The anterior segment of the eye was dissected away and the posterior segment of the eye was processed for cyrosectioning (14 μm sections) or whole retina flat mounts as previously described7. For optic nerve morphological analysis, 8 additional animals per genotype were perfused with 4% PFA. Brains were then dissected free and the optic nerves were photographed using a stereomicroscope.
For immunohistochemical staining on cryosections, sections were blocked in 10% horse serum with 0.1% Triton X in 1× PBS for 3 h at room temperature and then incubated in primary antibody overnight at 4 °C. Primary antibodies included: goat anti-SOX2 (1:250, Santa Cruz), goat anti-CHaT (1:200, Millipore), rabbit anti-calretinin (1:1000, Millipore), mouse anti-calbindin-D-28K (1:1000, Sigma), rabbit anti-PKCα (1:2000, Sigma), and rabbit anti-pJNK (1:250, Cell Signaling). Cryosections were washed in PBS and incubated in Alexafluor-conjugated secondary antibodies (Invitrogen) for 2 h at room temperature. Cyrosections were then washed, counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and mounted in Fluorogel in TRIS buffer (Electron Microscopy Sciences). A complete list of antibodies used is provided in Table 1.
For immunohistochemistry on flat mounts, floating retinas were blocked in 10% horse serum with 0.3% Triton X in 1× PBS overnight on a shaker at 4 °C and then incubated in primary antibody for 3 days at 4 °C as previously described28. Primary antibodies included: mouse anti-TUJ-1 (1:1000, BioLegend), goat anti-BRN3b (1:250, Santa Cruz), rabbit anti-TH (1:1000 Millipore), rabbit anti-pJUN (1:250, Cell Signaling), rabbit anti-cCASP3 (1:1000, Millipore), and rabbit anti-Neurofilament light chain (1:100; Cell Signaling). Whole retinas were then washed in 1× PBS and incubated in Alexafluor-conjugated secondary antibodies for 2 days at room temperature. Whole retinas were then mounted RGC side up in Fluorogel in TRIS buffer. RGCs were quantified in eight equally spaced 40× fields taken approximately 220 μm from the peripheral edge of the retina as previously described29. Quantification was completed using the cell counter tool in ImageJ. The area of fasciculations in Mkk7-deficient mice was determined by tracing the outside edge of each fasciculation using the outline tool in Axiovision software (Carl Zeiss).
Protein extraction and western blotting
Western blots for pJUN in WT and Mkk4- and Mkk7-deficient mice were completed as previously described7. Nanodrop was used to determine protein concentration from retinal lysates and 30 μg of protein was loaded into each well. Blots containing rabbit anti-pJUN (1:500, Cell Signaling) and rabbit anti-GAPDH (1:2000, Cell Signaling) were incubated overnight at 4 °C. A chemiluminescent kit (Supersignal West Dura Extended Substrate, Pierce 34075, Bio-Rad 170–5070) was used to detect immunoreactive bands followed by densitometric analysis using Quantity One Software (Bio-Rad). Western blots for MKK4 in Mkk4-deficient mice, MKK7 in Mkk7-deficient mice, and pJNK in Mkk4/Mkk7-deficient animals were performed as previously described8. Briefly, retinas were placed in 100 μl ice-cold lysis buffer after dissection (1× RIPA buffer (Santa Cruz, 24948) and protease/phosphatase inhibitor cocktail (Cell Signaling, 5872S)). Tissue was sonicated (Bransa Digital Sonifier) and spun down in a microcentrifuge. Then, 10 μl of supernatant was boiled for 10 min with 10 μl of 2× Laemmli loading buffer (Bio-Rad) and then run on a 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel. Membranes were treated with the Qentix Western Blot Signal Enhancer kit (Thermo Scientific, 21050) after they were transferred to a polyvinylidene difluoride (PVDF) membrane and then blocked and probed overnight at 4 °C with primary antibodies: rabbit anti-pJNK (1:500, Cell Signaling), rabbit anti-MKK4 (1:500, Cell Signaling), rabbit anti-MKK7 (1:500, Cell Signaling), or rabbit anti-GAPDH (1:2000, Cell Signaling). Membranes were then washed and treated with secondary antibody: horseradish peroxidase-conjugated anti-rabbit IgG (1:5000, Bio-Rad). A chemiluminescence kit (Immun-star, Bio-Rad 170–5070) was used to detect immunoreactive bands prior to exposure using either film or digital detection equipment (Azure Biosystems c500). Occasionally, membranes were stripped (buffer: 0.1 M Tris-Cl pH 6.8, 2% SDS, 0.7% β-mercaptoethanol) after development and treated with another primary antibody. ImageJ was used to perform densitometric analyses to determine the relative abundance of protein expressed relative to those of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) loading controls.
Mechanical optic nerve injury
Controlled optic nerve crush (CONC) was performed as previously described28,30. Briefly, animals were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine, the optic nerve was surgically exposed, and then crushed for 5 s with a pair of self-closing forceps immediately behind the globe. A cohort of eyes underwent sham surgery, in which the optic nerve was exposed but not crushed. These eyes along with eyes that had not been manipulated served as experimental controls. Animals were harvested after 2 h, 1 day, 5 days, or 35 days.
Statistical analysis
At least three retinas of each genotype were analyzed for all experimental conditions. Experimenters were masked to genotype and experimental condition for all quantification of RGC counts. Unpaired Student’s t-tests were used to compare differences across two groups. A one-way analysis of variance followed by the Bonferroni post hoc test for group comparisons was used to compare differences across more than two groups at a single time point. A P value < 0.05 was considered statistically significant. Means ± SEM are displayed in graphs.
Results
Deficiency of Mkk4 or Mkk7 leads to mild alterations in retinal structure
To generate retinas deficient in Mkk4 or Mkk7, Six3cre was used to recombine floxed alleles of either Mkk4 or Mkk7 with efficient deletion of both Mkk4 and Mkk7 (>95% and >85% protein reduction, respectively, Fig. S1)27. To determine if deletion of Mkk4 or Mkk7 is necessary for retinal development, sections of adult WT, Mkk4-deficient, and Mkk7-deficient retinas were evaluated (Fig. 1a). The Mkk4-deficient retina and optic nerve head had grossly normal morphology, apart from occasional photoreceptor cell bodies that were misplaced among photoreceptor inner/outer segments and more cells in the inner plexiform layer. The Mkk7-deficient retinas had increased cellularity in the surface nerve fiber layer and prelaminar region of the optic nerve head. Retinal lamination in Mkk7-deficient animals appeared normal, although there was slight disruption in the contour of the inner limiting membrane and ganglion cell layer.

a Representative 3 μm plastic sections stained with H&E demonstrate areas of photoreceptor cell body attachment to retinal pigmented epithelium in Mkk4-deficient retinas (arrowheads). Misplaced cells are also seen in the inner plexiform layer in Mkk4-deficient retinas. Retinal lamination in Mkk7-deficient retinas appeared grossly normal, though regions of the optic nerve head displayed slightly increased cellularity in the prelaminar region. Antibodies against ChAT and calretinin revealed normal development of cholinergic and AII amacrine cells, respectively, in both Mkk4- and Mkk7-deficient retinas (b). Calbindin-D-28K-labeled horizontal cells (c), PKCα-labeled bipolar cells (d), and SOX2-labeled Müller glia (e) in Mkk4 and Mkk7 mutants were furthermore indistinguishable from controls. Merged images with DAPI (blue) are shown below. N ≥ 4 retinas for each genotype. Scale bars: 100 μm
To further examine if Mkk4 and Mkk7 were necessary for retinal development, immunohistochemistry was used to study specific retinal cell types. Antibodies against choline acetyltransferase (ChAT) and calretinin were used to label amacrine cell bodies in the inner nuclear layer and synaptic strata in the inner plexiform layer32. Amacrine cells and inner plexiform lamination in both Mkk4- and Mkk7-deficient retinas appeared to develop normally (Fig. 1b). Horizontal cells and bipolar cells, stained with calbindin-D-28K and protein kinase Cα (PKCα), respectively, also appeared normal (Fig. 1c, d). Finally, there were no observable differences between WT and Mkk4- or Mkk7-deficient retinas in SOX2 staining (Müller glia and a subset of amacrine cells, Fig. 1e). Together, these data suggest that aside from sporadic photoreceptor nuclei directly abutting the retinal pigment epithelium in the outer nuclear layer, all major cell types within the inner nuclear layer appeared to differentiate in Mkk4- and Mkk7-deficient retinas.
Adult Mkk4- and Mkk7-deficient animals have fewer RGCs
The cellularity of the ganglion cell layer appeared less in the Mkk4- and Mkk7-deficient retinas than in controls (Fig. 1a). There also appeared to be some disorganization of the nerve fiber layer or inner limiting membrane in the Mkk7-deficient retina. Therefore, the number of TUJ-1+ RGCs was counted in retinal flat mounts (Fig. 2a). Retinas contained 15% and 25% fewer RGCs in Mkk4- and Mkk7-deficient animals, respectively, as compared to controls (P < 0.05). Furthermore, while no gross alteration was observed in Mkk4-deficient RGCs, Mkk7 deficiency resulted in sporadic clumping and axonal fasciculation (discussed below; experimenters performing cell counts avoided these small areas). To determine whether the decreased RGC density in adult Mkk4- and Mkk7-deficient retinas was due to an early developmental defect, flat mounts were examined at P0, a time point subsequent to RGC birth and determination33. Retinas were stained for BRN3B (POU4F2), another marker for RCGs34,35. RGC cell counts in both mutant mice were normal at this age, suggesting the correct amount of RGCs were born but died at a later stage (Fig. 2b).

a The number of TUJ-1-positive RGCs is significantly reduced in adult Mkk4- and Mkk7-deficient retinal flat mounts. Representative flat mounts are displayed to the right of the graph. (*P ≤ 0.05). N ≥ 10 retinas per genotype. b P0 flat mounts stained with BRN3B reveal normal RGC densities in both Mkk4- and Mkk7-deficient retinas, implying RGC dropout occurs postnatally. Representative flat mounts shown to the right of the graph. N ≥ 4 retinas per genotype. All examined Mkk7-deficient adult retinas displayed areas of abnormal fasciculation in both TUJ-1-positive RGCs (c; N ≥ 12) and tyrosine hydroxylase (TH)-positive amacrine cells (d; N ≥ 4). Scale bars: (a–c) 50 μm; (d) 100 μm. Error bars represent SEM
Deficiency of Mkk7 causes axonal fasciculation in RGCs and dopaminergic amacrine cells
RGC cell body clumping and axonal fasciculation was observed in 100% of Mkk7-deficient retinas examined and never observed in Mkk4-deficient retinas (Fig. 2c). Areas of affected retina in Mkk7-deficient animals comprised only a small portion of the ganglion cell layer (0.91% ± 0.32% of the retina, 0.142 ± 0.05 mm2, n = 8; Fig. S4A). Aside from the few areas of clumping and fasciculation, which appeared to be randomly distributed throughout the retina, RGC structure in Mkk7-deficient retinas appeared similar to WT controls (range: 1–4 areas of fasiculation per retina, mean: 2.5 areas of fasciculation, n = 8).
JNK1 is required for Netrin-1-induced axon outgrowth in the spinal cord36. Down syndrome cell adhesion molecule (DSCAM) has also been implicated in JNK1–Netrin-1-mediated neurite growth, and its loss of function leads to clustering of both RGC axons and cholinergic amacrine cell dendrites36,37. To examine whether the same amacrine cell phenotype resulted from Mkk4 or Mkk7 deficiency, additional retinal flat mounts were stained for tyrosine hydroxylase (TH), which labels a subset of dopaminergic amacrine cells. Similar to Dscam mutant mice, deficiency of Mkk7 resulted in fasciculation of dopaminergic amacrine cell dendrites (Fig. 2d) which was far less pronounced within Mkk4-deficient retinas (data not shown).
Deficiency of Mkk4 or Mkk7 does not prevent JNK–JUN signaling after axonal injury in RGCs
MKK4 and MKK7 are the only two MAP2Ks that activate JNK, which in turn activates its conical target, JUN. JNK–JUN signaling is important for pro-apoptotic signaling after axonal injury6,7,38,39,40,41. MKK4 and MKK7 have been previously reported to serve non-redundant functions in vivo18,26,42,43,44. For example, activation of MKK4 but not MKK7 triggers neuronal death following oxidative stress45 while MKK7 is required for JNK activation caused by pro-inflammatory cytokines (such as tumor necrosis factor-α and interleukin-1)15. Together, these results suggest activation of JNK after axonal injury may be preferentially controlled by a single MAP2K. To test this possibility, JNK–JUN signaling was evaluated in the Mkk4- and Mkk7-deficient animals. Both JNK and JUN are activated following optic nerve injury7,10,46,47. Expression of pJNK was observed in RGC axons entering the optic nerve head and pJUN-positive RGCs were present in WT and Mkk4- and Mkk7-deficient mice (Fig. 3a, b). Levels of pJUN were significantly reduced in both Mkk4- and Mkk7 (P < 0.001, N ≥ 3 for each cohort)-deficient retinas after CONC; however, detectable levels of the protein were found in both cohorts (Fig. 3c, d). No pJUN was detected in the sham condition in any animal cohort (data not shown). Thus, neither Mkk4 nor Mkk7 are independently required for activation of JNK or JUN after axonal injury.

a Representative sections containing the optic nerve head demonstrate robust expression of pJNK 2 h after CONC in WT controls and in Mkk4 and Mkk7 mutants (N ≥ 3 per genotype). b Representative retinal flat mounts from each group reveal robust levels of pJUN in TUJ-1-positive RGCs 1 day after CONC (N ≥ 3 per genotype). Scale bars: 50 μm. c Representative western blots showing levels of pJUN protein in WT and Six3cre driven conditional knockouts after CONC. d pJUN protein levels were significantly reduced by 61.8% in Mkk4- and by 83.1% in Mkk7-deficient mutants compared to littermate WT controls (N ≥ 3 per genotype, *P < 0.001)
Deficiency of Mkk4 or Mkk7 protects RGCs after acute mechanical axonal injury
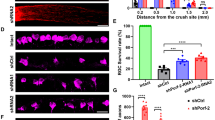
The MAPK signaling family has been previously shown to be an important pro-apoptotic signaling pathway after axon injury 6,7,38,39,40,41. To determine if Mkk4 and/or Mkk7 is critical for axonal injury-mediated RGC death, RGC survival was analyzed in mice deficient in Mkk4 and Mkk7 after CONC. Mkk4- and Mkk7-deficient retinas had significantly fewer dying RGCs (cleaved caspase-3-positive cells) at 5 days after CONC as compared to controls even after correcting for the decreased number of RGCs in Mkk4- and Mkk7-deficient retinas (Fig. 4a, b). In addition, at 35 days after CONC, Mkk4- and Mkk7-deficient retinas had a small but significantly greater percentage of surviving RGCs as compared to controls (Fig. 4c, d; % survival ± SEM: WT, 16.0% ± 1%; Mkk4, 51.5% ± 6.4%; Mkk7, 30.4% ± 1.4%). This protection was not as robust as that previously observed in Jun-deficient mice (~75%) at the same time point, suggesting both Mkk4 and Mkk7 can activate JNK–JUN-dependent RGC death after axonal injury.

a Representative retinal flat mounts stained with cleaved caspase-3 (CASP3) in eyes 5 days after CONC or sham surgery. b Significantly fewer apoptotic cells are present in Mkk4- and Mkk7-deficient animals as compared to WT controls following CONC. N ≥ 4 per genotype per condition; *P ≤ 0.05. c Representative retinal flat mounts stained with TUJ-1 following CONC or sham surgery. d Significantly more cells remain 35 days after CONC in Mkk4- and Mkk7-deficient retinas as compared to WT controls. N ≥ 8 per genotype per genotype and condition. Scale bars: 50 μm. Error bars represent SEM
Combined deficiency of Mkk4 and Mkk7 leads to severe alterations in retinal structure
Deficiency of Mkk4 or Mkk7 alone provided significant protection to RGCs after mechanical axon injury; however, neither deficiency by itself afforded complete protection to RGCs. As MKK4 and MKK7 are the only known molecules acting directly upstream of JNK, MKK4 and MKK7 appear to have redundant roles in RGCs after CONC18,43. Moreover, as either deficiency alone did not cause severe retinal dysgenesis during development, removing both genes, and therefore any possible genetic redundancy, could allow for the role of MAP2Ks in axonal injury-induced RGC death to be tested. To examine these possibilities, retinas deficient in both Mkk4 and Mkk7 were generated using Six3cre. JNK signaling, as measured by pJNK protein levels, was reduced by 86.7% in dual Mkk4/Mkk7-deficient mice (Fig. S3). The remaining pJNK in the Mkk4/7 dual-deficient animals is likely the result of incomplete recombination by Six3cre (known to occur in approximately 20% of RGCs)8. Mkk4/Mkk7-deficient mice had highly atrophic optic nerves (Fig. 5a), disrupted optic nerve head morphology (Fig. 5b), and disorganized ganglion cell layer pathology (Fig. 5c). The lack of an optic nerve prevented analysis of axonal injury-induced death in Mkk4/Mkk7-deficient mice. Surprisingly, despite the lack of an optic nerve, Mkk4/Mkk7-deficient retinas still had TUJ-1+ RGCs. However, there was clear evidence of abnormalities in RGC organization. Some areas of Mkk4/Mkk7-deficient retinas displayed abnormal RGC axonal crossing, while other areas appeared relatively normal (Fig. 6a). The Mkk4/7-deficient animals also displayed an RGC clumping phenotype (both in the periphery and near the optic nerve head) which was more prevalent and severe than the phenotype observed in Mkk7-deficient retinas (Fig. 6b and Fig. S4). Due to the widespread abnormalities in the Mkk4/Mkk7-deficient retinas, direct quantification of total RGC counts or the specific area of RGC fasciculation was not feasible.

a Brains were removed from adult WT and Mkk4/Mkk7 double-deficient mice with optic nerves intact (arrowheads). Near complete loss of both optic nerves was evident in all Mkk4/Mkk7 mutant brains examined (N = 8 per genotype). b Representative 3 μm plastic sections stained with H&E further demonstrate severe optic nerve head abnormalities (asterisk) in Mkk4/Mkk7 double-deficient mice eyes (N = 4 per genotype). c Mkk4/Mkk7 double mutants display extensive disruption of all retinal layers (N = 4 per genotype). Scale bars: 100 μm

Retinal flat mounts were stained with neurofilament (NF; a) and TUJ-1 (b) to assess RGC organization. Mkk4/Mkk7 double-deficient mutants displayed phenotypic variability in RGC structure, in which some areas exhibited axonal overlap (arrowheads in a’) and irregular axonal trajectories (b’), other areas appeared relatively normal (a”), and several areas consisted of RGC clumps (a”’, b”’). N ≥ 4 per genotype. Scale bars: 100 μm
Retinal sections also showed severe abnormalities in retinal lamination (Fig. 5c). These abnormalities included areas of retinal photoreceptor thinning and areas of hyper- and hypoplasia in the inner nuclear layer. Overall, each layer of the retina appeared thinner compared to WT controls, and multiple cell types were misplaced. Interestingly, histological retinal phenotypes did not appear to worsen with age, as 9-month-old Mkk4/Mkk7-deficient mice had similar histological irregularities compared to the younger mice analyzed (Fig. S2). To further investigate the nature of the histological abnormalities, immunohistochemistry was used to identify retinal cell types and overall retinal organization. There were clear abnormalities in CHAT+ and calretinin+ amacrine cells. Particularly, these markers showed that the inner plexiform layer had areas of synaptic disruption (Fig. 7a). Horizontal cells labeled with calbindin-D-28K had a mild disruption of somal and dendritic organization (Fig. 7b). PKCα+ bipolar cells had abnormal somal organization in the inner nuclear layer, occasional bipolar cell nuclei present in the photoreceptor layer, and abnormal bipolar cell termination in the inner plexiform layer (Fig. 7c). SOX2+ Müller glia and amacrine cells were highly irregular in their somal distribution (Fig. 7d). Dopaminergic amacrine cell clumping and dendrite fasciculation was more severe in Mkk4/Mkk7-deficient retinas than in retinas deficient in only Mkk4 or Mkk7 alone as larger areas of clumping occupied all sectors of the retina in the dual Mkk4/7-deficient animals (Fig. 7e and Fig. S4D). These data suggest the involvement of compensatory mechanisms by which Mkk4 and Mkk7 might serve redundant roles in retinal neurodevelopment. Overall, there was clear disruption of inner nuclear layer cell somal and synaptic organization.

Representative sections of WT and Mkk4/Mkk7-deficient retinas stained with specific cell markers show clear disorganization of amacrine cells (a), slight defects in horizontal cells (b), and abnormal distribution and morphology of bipolar cells (c) and Müller glia (d) in retinas of Mkk4/Mkk7 double-deficient mutants. Merged images with DAPI (blue) are shown below. N ≥ 4 per genotype. e Mkk4/Mkk7 double-deficient flat mounts further display clumping and dendritic fasciculation of dopaminergic amacrine cells. N ≥ 4 per genotype. Scale bars: 100 μm
Discussion
MAPK signaling is important for both retinal development and RGC apoptotic signaling after axonal injury. Herein, we demonstrate MKK4 and MKK7 provide both overlapping and divergent roles in retinal development and injury signaling. Single deficiency of Mkk4 or Mkk7 resulted in minor alterations of retinal lamination. RGC counts were significantly lower in adult Mkk4- and Mkk7-deficient retinas as compared to controls. After axonal injury, JNK signaling persisted in Mkk4- and Mkk7-deficient retinas; however, pJUN levels were reduced and RGCs were mildly protected from apoptotic cell death in both mutants. To assess redundancy of MKK4 and MKK7 in development and after injury, animals deficient in both Mkk4 and Mkk7 were developed. Together, MKK4/MKK7 were found to play critical roles in the development and organization of all major retinal cell types. The severity of optic nerve dysgenesis combined with significant alterations in retinal organization in the Mkk4/Mkk7-deficient retinas precluded examination of the contribution of both MKK4/MKK7 to pro-death signaling after axonal injury. Together, these data suggest novel roles for both MKK4 and MKK7 in retinal development and in axonal injury signaling.
Single Mkk4 or Mkk7 deficiency leads to multiple developmental abnormalities in the retina
Deficiency of either Mkk4 or Mkk7 led to significantly decreased RGC number in adult retinas, which was not observed at P0. Throughout retinal development, an excessive number of cells are born that must undergo apoptotic pruning. Developmental RGC pruning occurs from P0 to P7, with a second, smaller wave of cell death occurring around P1548,49,50. Given the postnatal dropout of RGCs in Mkk4- and Mkk7-deficient animals, MKK4 and MKK7 likely contribute to RGC survival during the normal window of programmed RGC death. Previously, the downstream effector molecules of MKK4 and MKK7, the JNKs, have been shown to regulate photoreceptor apoptosis during the final wave of cell death14. Thus, it is possible that MKK4 and MKK7 might play a similar role facilitating pruning of RGCs. Deficiencies in axonal transport or insufficient trophic support within the RGCs themselves may result in excessive loss of RGCs in the mature retina of Mkk4- and Mkk7-deficient mice48,51. Mild disruption of cell adhesion molecules may lead to the axonal fasciculations observed in the Mkk7-deficient retinas. As axonal fasciculations were not observed in the Mkk4-deficient retinas and Mkk7-deficient retinas had significantly fewer RGCs, it is plausible that MKK7 plays a more significant role in axonal guidance and retinal pruning. Determining the overlapping and unique downstream effectors of MKK4 and MKK7 that contribute to retinal development will help define the required molecular cues for proper retinal lamination.
Deficiency in both Mkk4 and Mkk7 causes severe retinal dysgenesis
In contrast to the Mkk4 and Mkk7 single-deficient retinas, the Mkk4/Mkk7 dual-deficient retinas had severe alterations in retinal lamination and axonal projections, as well as optic nerve hypoplasia. These data suggest MKK4 and MKK7 have redundant roles in retinal development and that together these molecules play critical roles in retinogenesis. Previously, JNK signaling has been shown to initiate bone morphogenetic protein-4 (BMP4) and sonic hedgehog (SHH)-mediated control of paired-like homeobox transcription factor (PAX2), which contributes to closure of the optic fissure12,52. Netrin-1 also contributes to optic nerve head formation as deficiency of Netrin-1 leads to failure of RGC axons to exit the eye leading to optic nerve hypoplasia53. Netrins serve as important axonal guidance cues in the nervous system and contribute to proper axon outgrowth and pathfinding54. JNK1 has been shown to be required for netrin-1 signaling and inhibition of JNK1 reduces netrin-1-dependent axonal projections and pathfinding in other neural systems36. DSCAM is a netrin-1 receptor necessary for neurite arborization and prevention of abnormal neural fasciculations55,56. Axonal fasciculations and ectopic photoreceptor phenotypes similar to those in Mkk4/Mkk7-deficient retinas were observed in Dscam-deficient retinas57. Proper fasciculation is necessary for RGC axon pathfinding; however, as axons approach deeper targets in the brain, they must disassociate from their neighbors58. It is possible in Mkk4/Mkk7 mutants, the extrinsic cues directing axon divergence are disrupted, resulting in abnormal fasciculation in the retina. Jnk2/3-deficient mice and Jun-deficient mice do not have aberrant lamination or optic nerve head dysgenesis, which further supports the idea that morphologic differences observed in the Mkk4/Mkk7-deficient mice are likely due to JNK1 or JUN-independent signaling mechanisms7,59. Future study to evaluate the downstream signaling mechanisms of MKK4 and MKK7 will likely reveal key contributors to retinal and optic nerve development. Despite the lack of optic nerve formation in the Mkk4/Mkk7-deficient animals, RGCs survive within the retina. Normal pathfinding and neurotropic support is essential for RGC survival, and thus RGC survival in the absence of optic nerve formation may be due to decreased pro-apoptotic signaling as JNK–JUN signaling contributes to both pro-survival and pro-apoptotic signaling14,21,60,61,62,63,64,65.
MKK4 and MKK7 are both involved in axonal injury-induced RGC death
Pro-apoptotic MAPK signaling is also an important component of molecular signaling after axonal injury in RGCs6,7,8,21,22,23,24,39,40,41. Similar to other MAPK family members, single deficiency of Mkk4 or Mkk7 provides significant protection to RGCs after axonal injury. The level of protection observed, however, does not phenocopy deficiency of other MAPK signaling molecules, supported by the significant, but not complete, reduction of pJUN. Deficiency of the upstream MAP3K, Dlk, and downstream targets, Jnk2/3 and Jun, provided greater protection to RGCs after CONC than that observed in this study, suggesting MKK4 and MKK7 likely have redundant roles in pro-apoptotic signaling after axonal injury6,7,39,40,59,66.
While both MKK4 and MKK7 are known to activate the JNKs, MKK4 additionally regulates p384,67. Activation of p38 occurs after axonal injury in both neurons and glia11,47,68,69,70. Inhibition of p38 signaling has been shown to provide mild protection to RGC somas and reduce axonal transport deficits following axonal injury11,68,69. Activation of the p38 arm of MAPK signaling can also directly activate Ddit3 (DNA damage inducible transcript 3) which encodes the protein CCAAT/enhancer binding homologous protein (CHOP)/GADD153, a key mediator of endoplasmic reticulum (ER) stress71,72. CHOP has been independently shown to be important for pro-apoptotic signaling after axonal injury59,73,74. In order to understand the molecular signaling cascade contributing to RGC death after axonal injury, it will be necessary to parse apart the downstream signaling contributions of MKK4 and MKK7.
Future studies should also evaluate combined Mkk4/Mkk7 deficiency in a temporally controlled conditional knockout animal (allowing for normal retinal and optic nerve development) to determine if deficiency of these molecules together might provide greater protection to RGCs after injury than either alone. It is tempting to consider that dual deficiency of Mkk4/Mkk7 may be more protective of RGCs than deficiency of other downstream targets alone, as combined deficiency of Mkk4/Mkk7 may alter multiple molecular signaling pathways that have been previously shown to be important for RGC death including JNK, p38, and ER stress signaling7,11,59,75.
Future investigation of the unique and overlapping roles of MKK4 and MKK7 must consider the downstream targets of their activation. MKK4 and MKK7 are known to preferentially target different amino acid residues on the JNKs. MKK4 targets the tyrosine residue while MKK7 preferentially phosphorylates the threonine residue76,77. Analysis of the transcriptome of MKK4 and MKK7 is necessary to identify specific downstream targets. Such inquiry would determine if the downstream effectors leading to developmental phenotypes in deficient animals are independent from those downstream effectors mediating pro-apoptotic signaling in RGCs after optic nerve crush.
Conclusion
MKK4 and MKK7 are members of the MAPK pathway important for both retinal development and the injury response following axonal insult. While we have shown MKK4 and MKK7 are required for maintaining RGC survival, future studies should examine the cellular and molecular mechanisms underlying the observed postnatal dropout in single-deficient animals. Single deficiency of Mkk4 and Mkk7 offer mild protection for RGCs against optic nerve injury, though there was likely genetic compensation allowing for pro-apoptotic signaling to proceed. Due to the severity of developmental defects in the double-deficient mice, possible additive protection following mechanical insult was not assessed. Additional experiments employing an inducible double Mkk4/Mkk7 knockout would allow for proper optic nerve formation and subsequent analysis of RGC survival following injury. This would define the aggregate role of these MAP2K molecules and the specificity of the cellular response in a glaucomatous-relevant injury model.
Change history
25 January 2019
There was an error introduced into Figures 4, 5, and 7 during the proofing stage which has since been corrected.
30 January 2019
There was an error introduced into Figures 4, 5, and 7 during the proofing stage. This has been corrected in both the PDF and HTML versions of the Article.
References
Kim, E. K. & Choi, E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 (2010).
Abe, N. & Cavalli, V. Nerve injury signaling. Curr. Opin. Neurobiol. 18, 276–283 (2008).
Chang, L. & Karin, M. Mammalian MAP kinase signalling cascades. Nature 410, 37–40 (2001).
Minden, A. & Karin, M. Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta 1333, F85–F104 (1997).
Jeffrey, K. L., Camps, M., Rommel, C. & Mackay, C. R. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug. Discov. 6, 391–403 (2007).
Fernandes, K. A., Harder, J. M., John, S. W., Shrager, P. & Libby, R. T. DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol. Dis. 69, 108–116 (2014).
Fernandes, K. A. et al. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis. 46, 393–401 (2012).
Syc-Mazurek, S. B., Fernandes, K. A. & Libby, R. T. JUN is important for ocular hypertension-induced retinal ganglion cell degeneration. Cell Death Dis. 8, e2945 (2017).
De Genaro, P., Simon, M. V., Rotstein, N. P. & Politi, L. E. Retinoic acid promotes apoptosis and differentiation in photoreceptors by activating the P38 MAP kinase pathway. Invest. Ophthalmol. Vis. Sci. 54, 3143–3156 (2013).
Isenmann, S. & Bahr, M. Expression of c-Jun protein in degenerating retinal ganglion cells after optic nerve lesion in the rat. Exp. Neurol. 147, 28–36 (1997).
Dapper, J. D., Crish, S. D., Pang, I. H. & Calkins, D. J. Proximal inhibition of p38 MAPK stress signaling prevents distal axonopathy. Neurobiol. Dis. 59, 26–37 (2013).
Weston, C. R. et al. JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev. 17, 1271–1280 (2003).
Davis, R. J. Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000).
Donovan, M., Doonan, F. & Cotter, T. G. Differential roles of ERK1/2 and JNK in retinal development and degeneration. J. Neurochem. 116, 33–42 (2011).
Tournier, C. et al. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 15, 1419–1426 (2001).
Westerlund, N. et al. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat. Neurosci. 14, 305–313 (2011).
Yamasaki, T. et al. Stress-activated protein kinase MKK7 regulates axon elongation in the developing cerebral cortex. J. Neurosci. 31, 16872–16883 (2011).
Wang, X., Destrument, A. & Tournier, C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim. Biophys. Acta 1773, 1349–1357 (2007).
Yamasaki, T., Kawasaki, H. & Nishina, H. Diverse roles of JNK and MKK pathways in the brain. J. Signal Transduct. 2012, 459265 (2012).
Noh, H., Lee, H., Park, E. & Park, S. Proper closure of the optic fissure requires ephrin A5-EphB2-JNK signaling. Development 143, 461–472 (2016).
Ribas, V. T., Goncalves, B. S., Linden, R. & Chiarini, L. B. Activation of c-Jun N-terminal kinase (JNK) during mitosis in retinal progenitor cells. PLoS One 7, e34483 (2012).
Fernandes, K. A., Harder, J. M., Kim, J. & Libby, R. T. JUN regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp. Eye Res. 112, 106–117 (2013).
Kim, B. J. et al. In vitro and in vivo neuroprotective effects of cJun N-terminal kinase inhibitors on retinal ganglion cells. Mol. Neurodegener. 11, 30 (2016).
Lingor, P., Koeberle, P., Kugler, S. & Bahr, M. Down-regulation of apoptosis mediators by RNAi inhibits axotomy-induced retinal ganglion cell death in vivo. Brain 128, 550–558 (2005).
Yoshida, K. et al. Amino-terminal phosphorylation of c-Jun regulates apoptosis in the retinal ganglion cells by optic nerve transection. Invest. Ophthalmol. Vis. Sci. 43, 1631–1635 (2002).
Hubner, A. et al. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc. Natl. Acad. Sci. USA 109, 12046–12051 (2012).
Wang, X. et al. Targeted deletion of the mitogen-activated protein kinase kinase 4 gene in the nervous system causes severe brain developmental defects and premature death. Mol. Cell. Biol. 27, 7935–7946 (2007).
Furuta, Y., Lagutin, O., Hogan, B. L. & Oliver, G. C. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis 26, 130–132 (2000).
Harder, J. M. & Libby, R. T. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol. Neurodegener. 6, 50 (2011).
Harder, J. M., Fernandes, K. A. & Libby, R. T. The Bcl-2 family member BIM has multiple glaucoma-relevant functions in DBA/2J mice. Sci. Rep. 2, 530 (2012).
Libby, R. T. et al. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 1, 17–26 (2005).
Haverkamp, S. & Wassle, H. Immunocytochemical analysis of the mouse retina. J. Comp. Neurol. 424, 1–23 (2000).
Young, R. W. Cell differentiation in the retina of the mouse. Anat. Rec. 212, 199–205 (1985).
Gan, L., Wang, S. W., Huang, Z. & Klein, W. H. POU domain factor Brn-3b is essential for retinal ganglion cell differentiation and survival but not for initial cell fate specification. Dev. Biol. 210, 469–480 (1999). [pii].
Gan, L. et al. POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proc. Natl. Acad. Sci. USA 93, 3920–3925 (1996).
Qu, C. et al. c-Jun N-terminal kinase 1 (JNK1) is required for coordination of netrin signaling in axon guidance. J. Biol. Chem. 288, 1883–1895 (2013).
Li, S. et al. DSCAM promotes refinement in the mouse retina through cell death and restriction of exploring dendrites. J. Neurosci. 35, 5640–5654 (2015).
Coffey, E. T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 15, 285–299 (2014).
Watkins, T. A. et al. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. USA 110, 4039–4044 (2013).
Welsbie, D. S. et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. USA 110, 4045–4050 (2013).
Yang, J. et al. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176 (2015).
Asaoka, Y. & Nishina, H. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J. Biochem. 148, 393–401 (2010).
Haeusgen, W., Herdegen, T. & Waetzig, V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur. J. Cell Biol. 90, 536–544 (2011).
Merritt, S. E. et al. The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J. Biol. Chem. 274, 10195–10202 (1999).
Kim, S. D., Moon, C. K., Eun, S. Y., Ryu, P. D. & Jo, S. A. Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochem. Biophys. Res. Commun. 328, 326–334 (2005).
Levkovitch-Verbin, H. et al. The transcription factor c-jun is activated in retinal ganglion cells in experimental rat glaucoma. Exp. Eye Res. 80, 663–670 (2005).
Tezel, G., Chauhan, B. C., LeBlanc, R. P. & Wax, M. B. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest. Ophthalmol. Vis. Sci. 44, 3025–3033 (2003).
Valenciano, A. I., Boya, P. & de la Rosa, E. J. Early neural cell death: numbers and cues from the developing neuroretina. Int. J. Dev. Biol. 53, 1515–1528 (2009).
Young, R. W. Cell death during differentiation of the retina in the mouse. J. Comp. Neurol. 229, 362–373 (1984).
Linden, R. & Pinto, L. H. Developmental genetics of the retina: evidence that the pearl mutation in the mouse affects the time course of natural cell death in the ganglion cell layer. Exp. Brain Res. 60, 79–86 (1985).
Yang, X. J. Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Semin. Cell. Dev. Biol. 15, 91–103 (2004).
Gregory-Evans, C. Y., Wallace, V. A. & Gregory-Evans, K. Gene networks: dissecting pathways in retinal development and disease. Prog. Retin. Eye. Res. 33, 40–66 (2013).
Deiner, M. S. et al. Netrin-1 and DCC mediate axon guidance locally at the optic disc: loss of function leads to optic nerve hypoplasia. Neuron 19, 575–589 (1997).
Lai Wing Sun, K., Correia, J. P. & Kennedy, T. E. Netrins: versatile extracellular cues with diverse functions. Development 138, 2153–2169 (2011).
Fuerst, P. G., Koizumi, A., Masland, R. H. & Burgess, R. W. Neurite arborization and mosaic spacing in the mouse retina require DSCAM. Nature 451, 470–474 (2008).
Ly, A. et al. DSCAM is a netrin receptor that collaborates with DCC in mediating turning responses to netrin-1. Cell 133, 1241–1254 (2008).
Fernandes, K. A. et al. Novel axon projection after stress and degeneration in the Dscam mutant retina. Mol. Cell. Neurosci. 71, 1–12 (2016).
Marcos, S. et al. Secreted frizzled related proteins modulate pathfinding and fasciculation of mouse retina ganglion cell axons by direct and indirect mechanisms. J. Neurosci. 35, 4729–4740 (2015).
Syc-Mazurek, S. B., Fernandes, K. A., Wilson, M. P., Shrager, P. & Libby, R. T. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol. Neurodegener. 12, 71 (2017).
Svensson, C. et al. Pro-survival effects of JNK and p38 MAPK pathways in LPS-induced activation of BV-2 cells. Biochem. Biophys. Res. Commun. 406, 488–492 (2011).
Yu, C. et al. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol. Cell 13, 329–340 (2004).
Granato, M. et al. JNK and macroautophagy activation by bortezomib has a pro-survival effect in primary effusion lymphoma cells. PLoS One 8, e75965 (2013).
Dhanasekaran, D. N. & Reddy, E. P. JNK signaling in apoptosis. Oncogene 27, 6245–6251 (2008).
Dakubo, G. D. et al. Control of glial precursor cell development in the mouse optic nerve by sonic hedgehog from retinal ganglion cells. Brain Res. 1228, 27–42 (2008).
Reese, B. E. Development of the retina and optic pathway. Vision Res. 51, 613–632 (2011).
Welsbie, D. S. et al. Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron 94, 1142–1154.e6 (2017).
Brancho, D. et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17, 1969–1978 (2003).
Kikuchi, M., Tenneti, L. & Lipton, S. A. Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J. Neurosci. 20, 5037–5044 (2000).
Manabe, S. & Lipton, S. A. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Invest. Ophthalmol. Vis. Sci. 44, 385–392 (2003).
Hashimoto, K. et al. Long-term activation of c-Fos and c-Jun in optic nerve head astrocytes in experimental ocular hypertension in monkeys and after exposure to elevated pressure in vitro. Brain Res. 1054, 103–115 (2005).
Wang, X. Z. & Ron, D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science 272, 1347–1349 (1996).
Ferreiro, I. et al. Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC Genom. 11, 144 (2010).
Hu, Y. et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 73, 445–452 (2012).
Yang, L. et al. Rescue of glaucomatous neurodegeneration by differentially modulating neuronal endoplasmic reticulum stress molecules. J. Neurosci. 36, 5891–5903 (2016).
Hu, Y. Axon injury induced endoplasmic reticulum stress and neurodegeneration. Neural Regen. Res. 11, 1557–1559 (2016).
Lawler, S., Fleming, Y., Goedert, M. & Cohen, P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr. Biol. 8, 1387–1390 (1998).
Kishimoto, H. et al. Different properties of SEK1 and MKK7 in dual phosphorylation of stress-induced activated protein kinase SAPK/JNK in embryonic stem cells. J. Biol. Chem. 278, 16595–16601 (2003).
Acknowledgements
The authors would like to thank Drs. C. Tournier and R.J. Davis for generously providing the Mkk4 and Mkk7 floxed alleles. This work was supported by EY018606 (to R.T.L.), EY026301 (to R.L.R.), EY007125 (to R.L.R.), and Research to Prevent Blindness, an unrestricted grant to the Department of Ophthalmology at the University of Rochester Medical Center. The funding agencies had no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Yaron
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Syc-Mazurek, S.B., Rausch, R.L., Fernandes, K.A. et al. Mkk4 and Mkk7 are important for retinal development and axonal injury-induced retinal ganglion cell death. Cell Death Dis 9, 1095 (2018). https://doi.org/10.1038/s41419-018-1079-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-1079-7
This article is cited by
-
BAX activation in mouse retinal ganglion cells occurs in two temporally and mechanistically distinct steps
Molecular Neurodegeneration (2023)
-
Endothelin 1-induced retinal ganglion cell death is largely mediated by JUN activation
Cell Death & Disease (2020)
-
DDIT3 (CHOP) contributes to retinal ganglion cell somal loss but not axonal degeneration in DBA/2J mice
Cell Death Discovery (2019)
