
Abstract
Glycine N-methyltransferase (GNMT) is the most abundant methyltransferase in the liver and a master regulator of the transmethylation flux. GNMT downregulation leads to loss of liver function progressing to fibrosis, cirrhosis, and hepatocellular carcinoma. Moreover, GNMT deficiency aggravates cholestasis-induced fibrogenesis. To date, little is known about the mechanisms underlying downregulation of GNMT levels in hepatic fibrosis and cirrhosis. On this basis, microRNAs are epigenetic regulatory elements that play important roles in liver pathology. In this work, we aim to study the regulation of GNMT by microRNAs during liver fibrosis and cirrhosis. Luciferase assay on the 3ʹUTR-Gnmt was used to confirm in silico analysis showing that GNMT is potentially targeted by the microRNA miR-873-5p. Correlation between GNMT and miR-873-5p in human cholestasis and cirrhosis together with miR-873-5p inhibition in vivo in different mouse models of liver cholestasis and fibrosis [bile duct ligation and Mdr2 (Abcb4)-/- mouse] were then assessed. The analysis of liver tissue from cirrhotic and cholestatic patients, as well as from the animal models, showed that miR-873-5p inversely correlated with the expression of GNMT. Importantly, high circulating miR-873-5p was also detected in cholestastic and cirrhotic patients. Preclinical studies with anti-miR-873-5p treatment in bile duct ligation and Mdr2-/- mice recovered GNMT levels in association with ameliorated inflammation and fibrosis mainly by counteracting hepatocyte apoptosis and cholangiocyte proliferation. In conclusion, miR-873-5p emerges as a novel marker for liver fibrosis, cholestasis, and cirrhosis and therapeutic approaches based on anti-miR-873-5p may be effective treatments for liver fibrosis and cholestatic liver disease.
Similar content being viewed by others


Introduction
Glycine N-methyltransferase (GNMT) is the most important and abundant S-adenosylmethionine (SAMe)-dependent methyltransferase in the liver. GNMT is predominantly expressed in hepatocytes although it is also found in other cell types such as cholangiocytes, the epithelial cells of the bile duct1,2,3. Lack of GNMT induces undesired methylation reactions leading to proliferative, inflammatory, and profibrogenic responses that culminate in liver disease4,5. GNMT expression is reduced in different liver diseases including liver cirrhosis of diverse etiology1,6, chronic cholestatic liver disease7, hepatocellular carcinoma (HCC)1, and cholangiocarcinoma2. In agreement, Gnmt-deficient (Gnmt-/-) mice, characterized by elevated SAMe levels, develop liver fibrosis spontaneously at the age of 3 months and HCC at 8 months8. Indeed, an aberrant DNA methylation signature has been identified in the Gnmt-/- mouse8, underscoring the importance of this protein as an epigenetic regulator. Although GNMT promoter hypermethylation causes GNMT downregulation in some HCC patients9, other mechanisms are likely to be involved in the regulation of this gene in liver diseases such as fibrosis or cirrhosis.
Hepatic fibrosis is the result of the wound-healing response of the liver to repeated injury that occurs in most types of chronic liver diseases. The injury can be caused by the accumulation of lipids in the liver, as occurs during non-alcoholic fatty liver disease (NAFLD), can be a result of a toxicant or viral insult, such as excessive alcohol consumption or hepatitis, respectively, or the accumulation of bile acids (BA), as in chronic liver cholestasis. The finding of potential molecular and pathway targets for reverting or halting the progression of liver fibrosis or cholestasis to cirrhosis and HCC is an emerging field.
MicroRNAs (miRNAs) are highly conserved, small non-coding RNAs that post-transcriptionally regulate gene expression10 of essential biological processes, as well as cellular responses11,12. In the liver, miRNA signature has been implicated in NAFLD, cirrhosis, and liver cancer13. The biological significance and therapeutic potential of miRNAs in liver disease management is a rapidly growing field.
In this study, we show that liver fibrosis progression is associated with the repression of GNMT, which is targeted by miR-873-5p in two preclinical models of liver fibrosis and cholestasis: the bile duct ligation (BDL) and the Mdr2 (Abcb4)-deficient mice (Mdr2-/-), respectively. Targeting miR-873-5p in these models resulted in a reduction of liver damage affecting mainly hepatocyte apoptosis and cholangiocyte proliferation, through a GNMT-dependent epigenomic mechanism. Of note, an inverse correlation between hepatic GNMT and miR-873-5p was identified in a cohort of cirrhotic patients with diverse etiology. Importantly, increased circulating miR-873-5p was also detected in serum samples from cirrhotic and cholestatic patients. A negative correlation was found between hepatic GNMT expression and serum levels of miR-873-5p in a cohort of cholestatic patients according with the fibrotic stage.
Overall, miR-873-5p emerges as a novel druggable target and a marker for liver fibrosis, cholestasis and cirrhosis.
Results
MiR-873-5p targets Gnmt expression in mouse hepatocytes
Understanding the mechanism underlying GNMT repression is essential for the development of new therapeutic approaches in liver fibrosis and cholestatic diseases. From three independent unbiased approaches employed (www.targetscan.org, www.ebi.ac.uk, and www.microrna.org) only miR-873-5p appears as a common microRNA targeting GNMT (Suppl. Fig. 1a). For functional analysis of miR-873-5p, primary mouse hepatocytes, characterized by high levels of GNMT, were transfected with pmir-GLO and pmir-GLO-Gnmt-3ʹUTR both fused to a luciferase reporter gene. Transfection with a miR-873-5p mimic (mimic-miR-873-5p) results in a 50% reduction of 3ʹUTR Gnmt reporter activity vs. those transfected with miR-Control (Fig. 1a). Moreover, mimic-miR-873-5p efficiently reduced mRNA and protein GNMT levels in cultured hepatocytes (Fig. 1b). Altogether, these data support miR-873-5p as a post-transcriptional repressor of Gnmt.

(a) Luciferase reporter assay of Gnmt 3ʹUTR expression in hepatocytes transfected with mimic-miR-873-5p (N = 8). (b) GNMT and miR-873-5p expression at mRNA and protein levels in primary hepatocyte after mimic-miR-873-5p transfection. Gnmt-miR-873-5p expression and correlation and WB analysis of GNMT at different time points of BDL (N = 3) (c) and in Mdr2WT and Mdr2-/- mice at different ages (3,4,9,12,17 months each column and 3▲, 4▼, 9♦, 12●, and 17■ months in the correlation panel) (N = 3) (d). Densitometry analyses of WB are shown in Supp. Fig. 1B, D, E. Data normalized as fold change vs. control. Error bars represent the means ± SEM. Statistical significance was determined by the Student’s t-test or ANOVA when more than two groups were compared. p < 0.05*; p < 0.01**; p < 0.001***
MiR-873-5p inversely correlates with GNMT expression in liver fibrosis and cholestasis
The inverse association between GNMT and miR-873-5p expression was confirmed in different preclinical models of liver fibrosis. Administration of carbon tetrachloride (CCl4) for 6 weeks induced a reduction in Gnmt expression, associated with the induction of miR-873-5p (Suppl. Fig. 1C). Additionally, in two models of cholestatic liver disease induced by BDL after 3 and 7 days of surgery14 and the Mdr2-/- mouse, a model of inflammation-induced cholestatic liver injury, fibrosis, and cancer15, we found an inverse correlation between GNMT levels and miR-873-5p expression (Fig. 1c, d).
Likewise, an inverse correlation between hepatic GNMT expression and miR-873-5p was identified in a group of 16 cirrhotic patients of diverse etiology (Suppl. Table 1, Fig. 2a). These results prompted us to assay circulating miR-873-5p levels in cirrhotic patients. Blood samples were collected at the screening visit in 35 cirrhotic patients diagnosed with cirrhosis or advanced scaring (F4) with diverse etiology and levels of circulating miR-873-5p were evaluated and compared to nine healthy controls. Higher miR-873-5p levels in serum were observed in cirrhotic patients (Fig. 2b). Subsequently, we analyzed circulating miR-873-5p levels in a cohort of cholestatic patients primary biliary cholangitis (PBC)/ primary sclerosing cholangitis (PSC) (n = 41) and compared to healthy patients with antimitochondrial antibodies (AMA + ) but without abnormal liver test (Suppl.Table II). In these patients, whose circulating miR-873-5p levels are increased relative to healthy controls, a positive correlation was also found with fibrosis grade (Fig. 2c). Furthermore, a reduction of hepatic GNMT expression was associated with the fibrosis grade in a cohort of 64 liver cholestasis patients (Suppl. Table III and Fig. 2d). Finally, a significant correlation between reduced expression of hepatic GNMT and increased levels of miR-873-5p in the serum of a cohort of patients with early and advance cholestatic liver disease was detected in 19 paired samples (Fig. 2e). Fibrotic stage positively correlated with miR-873-5p and negatively correlated with GNMT levels (Fig. 2f). Overall, miR-873-5p expression is augmented and inversely correlated with hepatic GNMT expression in cirrhosis and cholestatic disease.

Hepatic GNMT-miR-873-5p expression and correlation in a cohort of cirrhotic patients (N = 16) (a) and circulating miR-873-5p in cirrhotic patients (N = 35) (b). (c) Circulating miR-873-5p levels in cholestatic (PBC/PSC) patients (41) and its correlation with fibrosis (LSM, kPa). (d) GNMT levels in cholestatic patients (N = 64). (e) Correlation between circulating miR-873-5p and hepatic GNMT in cholestatic patients (N = 19). (f) Correlation between circulating miR-873-5p and hepatic GNMT with fibrotic stage in cholestatic patients, respectively. Data normalized as fold change vs. control. Error bars represent the means ± SEM. Statistical significance was determined by the Student’s t-test or ANOVA when more than two groups were compared. p < 0.05*; p < 0.01**; p < 0.001***
Anti-miR-873-5p attenuates liver injury after bile duct ligation
The potential benefit of targeting miR-873-5p was evaluated in the BDL animal model of cholestasis. Thus, we performed BDL surgery for one week in Gnmt wild-type (WT) mice and injected anti-miR-873-5p or a miR-Ctrl i.v. at day 3 and 5, corresponding to the initial phase of hepatic fibrogenesis14 (Fig. 3a and Suppl Fig. 2A) and sacrificed animals at 7 days after BDL. MiR-873-5p inhibition (Fig. 3b) resulted in GNMT restoration levels (Fig. 3c). No effect on miR-873-5p and Gnmt mRNA levels was detected in other tissues where Gnmt is usually expressed, such as pancreas and kidney, after miR-873-5p inhibition (data not shown). Anti-miR-873-5p treatment was associated with reduced serum transaminases (ALT/AST), caspase 3 activity and PARP cleavage as well as parenchymal disruption as detected by H&E staining, all readouts of cell death (Fig. 3d–f).

(a) Time scheme of the BDL mouse model with anti-miR-873-5p (time points indicated). MiR-873-5p expression (b) and GNMT mRNA and protein expression (c) in the liver of miR-Ctrl-BDL and anti-miR-873-5p-BDL mice. (d) Serum transaminases (ALT and AST) levels in miR-Ctrl and anti-miR-873-5p-treated-mice at 7 days of BDL. (e) Caspase 3 activity and WB analysis of apoptosis mediated by PARP cleavage. (f) H&E, F4/80, Sirius Red, αSMA, and CK19 staining in liver sections from miR-Ctrl and miR-873-5p-inhibited-mice at 7 days of BDL. (g) qPCR analysis of indicated genes in total liver extracts at 7 days of BDL. (h) WB analysis with indicated Ab in total liver of WT, BDL-Ctrl and anti-miR-873-BDL mice at 7 days. Densitometry analyses of WB are shown in Supp. Fig. 2B. Data normalized as fold change vs. control. Error bars represent the means ± SEM (N = 4). Statistical significance was determined by the Student’s t-test or ANOVA when more than two groups were compared. p < 0.05*; p < 0.01**; p < 0.001***
In the BDL mouse model, cholangiocyte proliferation and liver inflammation play essential roles during the progression of fibrosis that is linked to BA-induced hepatocyte cell death. Anti-miR-873-5p treatment in vivo significantly reduced the proliferation of cholangiocytes as evaluated by cytokeratin 19 (CK19) levels (Fig. 3f) and counteracted the expression of markers related with cholangiocyte activation, such as monocyte chemoattractant protein (Mcp-1) and epithelial cell adhesion molecule (Epcam) (Fig. 3g). Moreover, miR-873-5p inhibition minimized inflammatory markers such as F4/80, inducible nitric oxide synthase (iNos), acute phase response gene serum amyloid A1 (Saa1), interleukin-6 (Il-6), and JNK phosphorylation (Fig. 3f–h). Indeed, tumor necrosis factor (Tnf) and Il-6 reduction (Fig. 3g) modulated STAT3 activation after miR-873-5p inhibition (Fig. 3h). Finally, in miR-873-5p-inhibited BDL mice, fibrogenic indicators such as Sirius red, alpha smooth muscle actin (α-SMA), metalloproteinase 9 (Mmp9), transforming growth factor beta (Tgfb), and Smad2/3 phosphorylation were significantly reduced in comparison to those mice treated with miR-Control (Fig. 3f–h).
RNA sequencing analysis further retrieved a completely different gene ontology (GO) profile in the BDL model after miR-873-5p inhibition. During this initial phase of hepatic fibrogenesis, biological processes related to cell proliferation, natural killer cell activation, and immune response processes were downregulated in anti-miR-873-5p vs. miR-Ctrl mice. On the other hand, anti-miR-873-5p-treated-mice showed increased gene expression related to biological processes implicated in steroid and fatty acid metabolism and in the respiratory electron transport chain compared to miR-ctrl mice (Supp. Fig. 3A and Supp. Table IV and V).
Of relevance, the treatment of Gnmt-/- mice after 3 and 5 days of BDL with anti-miR-873-5p failed to counteract both inflammation and fibrosis (Supp Fig. 4a), indicating that Gnmt is one of the principal targets that mediates the anti-miR-873-5p effect in vivo, even though miR-873-5p could affect many different mRNAs besides Gnmt.
Overall, these data show that miR-873-5p inhibition is hepatoprotective and reduces cell death, affects ductular reaction and prevents the generation of liver inflammation and fibrosis after BDL, which is suggested to take place by enhancing GNMT expression.
MiR-873-5p directly regulates hepatocyte apoptosis and cholangiocyte proliferation
In the pathology of liver fibrosis and in cholestatic liver injury different types of hepatic cells including hepatocytes, cholangiocytes, inflammatory macrophages (Kupffer cells)16 and Natural Killer7,17,18,19 cells, and hepatic stellate cells (HSC)20 are known to mediate different effects, regulating the development and the progression of the disease. During initiation of fibrogenesis, hepatocyte apoptosis and cholangiocyte proliferation are the primary events driving disease progression14. Although GNMT in the liver is known to be mainly found in hepatocytes, it has been described to be expressed in other cell types such as cholangiocytes, where its expression is associated with a differentiated and a non-proliferative phenotype of these cells2. Thus, we have analyzed the targeting of Gnmt by miR-873-5p in these cells and its potential contribution to cholestatic liver injury.
Firstly, primary isolated murine hepatocytes were cultured with the toxic bile acid deoxycholic acid (DCA, 100 µM) for 2 h to induce hepatocyte apoptosis, an in vitro cell model that mimics in vivo BDL. Under these circumstances, DCA treatment was associated with reduced Gnmt expression together with the induction of miR-873-5p and hepatocyte apoptosis, suggesting the importance of the regulation of GNMT-miR-873-5p axis in hepatocytes (Fig. 4a). The potential role of miR-873-5p in the induction of BA-induced apoptosis in hepatocytes through the repression of Gnmt was studied by inhibiting miR-873-5p directly in these cells. MiR-873-5p inhibition in hepatocytes induced GNMT levels (Fig. 4b), resulting in attenuation of BA-induced apoptosis measured by Caspase 3 activity, TUNEL assay and JNK phosphorylation (Fig. 4c, d). Moreover, miR-873-5p inhibition increased the expression of different genes related to BA-induced apoptosis [B-Cell CLL/Lymphoma 2 (Bcl2) and the Hepatocyte nuclear factor (Hnf) Hnf1α and Hnf4α] and, more importantly, genes implicated in BA metabolism and regulation, such as the master regulator of BA metabolism Fxr and different BA exporters, such as Bsep, Abcg5 and some Mdr/Mrp (Fig. 4e).

(a) Gnmt and miR-873-5p expression in cultured hepatocytes treated with deoxycholic acid (DCA) for 2 h (N = 3). (b) GNMT expression analysis by qPCR and WB after anti-miR-873-5p and DCA treatment. Cell death was measured by C3 activity (2 h) TUNEL and JNK phosphorylation (15’/30’) at different time points of DCA treatment (c, d). (e) Gene expression analysis by qPCR of indicated genes implicated in BA-induced apoptosis, metabolism and transport after 2 h of DCA and anti-miR.873-5p treatment (N = 3). Densitometry analyses of WB are shown in Supp. Fig. 5A. (f) MiR-873-5p expression in normal human cholangiocytes (NHC) after 48 h of Interleukin-6 (IL-6) treatment (N = 6). Cell proliferation measured by MTT assay and analysis of GNMT and indicated genes measured by qPCR in anti-miR-873-5p cholangiocytes treated with IL-6 (N = 4). Data normalized as fold change vs. control. Error bars represent the means ± SEM. Statistical significance was determined by the Student’s t-test or ANOVA when more than two groups were compared. p < 0.05*; p < 0.01**; p < 0.001***
Then, GNMT targeting by miR-873-5p was evaluated in cholangiocyte cell lines. It has been reported that inflammatory mediators like IL-6 (significantly augmented after BDL and modulated in the presence of anti-miR-873-5p (Fig. 3g)) induces cholangiocyte proliferation. Indeed, the normal human cholangiocyte cell line NHC incubated with IL-6 show increased miR-873-5p expression (Fig. 4f). MiR-873-5p inhibition reduced cholangiocyte proliferation measured by MTT assay and the levels of cyclin D1 (CD1) and proliferating cell nuclear antigen (PCNA) (Fig. 4f). Furthermore, anti-miR-873-5p increased GNMT expression and minimized the levels of growth factors (TGFβ and platelet derived growth factor (PDGF)) implicated in cholangiocyte contribution to BDL-induced fibrosis (Fig. 4f).
Overall, these data suggest that anti-miR-873-5p has broad effects in liver cells affecting fibrosis progression, mainly mediating anti-apoptotic activity in hepatocytes and anti-proliferative effect in cholangiocytes contributing to reduce liver injury and fibrosis progression.
Anti-miR-873-5p prevents liver injury in Mdr2-/- mice
Mice lacking the ATP-binding cassette ABCB4 protein encoded by the Mdr2 gene (Mdr2-/-) provide a model for the study of cholestasis in the context of chronic inflammation as a result of increased BA accumulation15. The increase in miR-873-5p and the low expression levels of GNMT at the different stages of hepatic fibrogenesis in the Mdr2-/- mice (Fig. 1d) prompted us to investigate the impact that anti-miR-873-5p treatment could have in this model. Four-month old Mdr2-/- mice, with advanced fibrosis and biliary proliferation (Suppl Fig. 6a), were treated once a week with anti-miR-873-5p during 4 weeks (Fig. 5a). Anti-miR-873-5p treatment rescued GNMT (Fig. 5b, c) and reduced ALT/AST transaminases, total serum BA levels (Fig. 5d, e), cholangiocytes proliferation by CK19, hepatocyte death assessed by caspase 3 activity and hepatic ammonia content21 (Fig. 5f). Moreover, Mdr2-/- mice treated with anti-miR-873-5p showed lower cell death receptor Dr5 and higher biliary acids transporters of the ABC superfamily Mdr1, Mrp3, and Mrp5 (Suppl Fig. 6B), consistent with the reduction of BA content, BA-induced apoptosis and markers related to cholangiocyte activation (Suppl Fig. 6B). These changes were accompanied by a reduced inflammatory response as determined by F4/80 and Il-6, Ccl1 and iNos expression, as well as less fibrosis evaluated through Sirius red staining and the levels of profibrogenic (Tgfb, Timp1, and Timp2) genes (Fig. 5f and Suppl Fig. 6b). Altogether, these data suggest that anti-miR-873-5p targeting in the Mdr2-/-cholestasis animal model reduces cholangiocyte proliferation, increases the export of bile acids and ameliorates BA-induced hepatocyte cell death and inflammatory response.

(a) Time scheme of the anti-miR-873-5p treatment in Mdr2-/- mice (time points indicated). MiR-873-5p (b) and GNMT expression (c) in the liver of Mdr2-/- and anti-miR-873-Mdr2-/- mice. (d) Serum transaminases levels expressed as fold change from the initial values and (e) bile acid content in Mdr2-/- mice. (f) CK19, Cleaved Caspase 3, ammonia, F4/80, and Sirius red staining in liver sections from miR-Ctrl and miR-873-5p-inhibited Mdr2 -/- mice. Densitometry analyses of WB are shown in Supp. Fig. 6C. Data normalized as fold change vs. control. Error bars represent the means ± SEM (N = 5). Statistical significance was determined by the Student’s t-test or ANOVA when more than two groups were compared p < 0.05*; p < 0.01**; p < 0.001 ***. ----- line in d indicates initial levels of the measurements
MiR-873-5p regulation results in GNMT-dependent epigenomic modulation
DNA (CpG) methylation acts as a blueprint for global alterations in the epigenome that drive liver injury22 among them hepatic fibrogenesis23,24,25,26. BDL and Mdr2-/- mice under anti-miR-873-5p treatment showed improved hepatic SAMe metabolism with a decrease in the ratio SAMe/S-adenosylhomocysteine (SAH) consistent with the re-expression of GNMT (Table 1). This effect in the methylation flux correlated with a reduction in the global DNA methylation content as detected after miR-873-5p inhibition (Fig. 6a). DNA methylation is mediated by two groups of DNA methyltransferases (DNMTs), DNMT1 that is implicated in maintenance of DNA methylation and DNMT3a/3b that are responsible for de novo DNA methylation27. According to the changes in DNA methylation levels in the liver, anti-miR-873-5p treatment reduced Dnmt3a expression in both fibrotic animal models (Fig. 6b). Indeed, the inhibition of miR-873-5p during BDL and in Mdr2-/- mice increased the expression of other genes of the methionine cycle besides Gnmt, such as Methionine adenosyltransferase (Mat1a) and S-adenosylhomocysteine hydrolase (Sahh) (Suppl. Fig. 7A).

(a) DNA CpG (5mC) methylation in Ctrl and BDL/anti-miR-873-BDL livers (N = 4). qPCR and WB analysis of genes implicated in DNA methylation (b). qPCR analysis of indicated genes susceptible of promoter hypermethylation related to cholangiocyte proliferation and bile acid metabolism (c-e) in miR-Ctrl and anti-miR-873-5p BDL/Mdr2-/- mice (N = 4 (BDL) and N = 5 (Mdr2-/-). (f) WB analysis of EZH2 in anti-miR-873-5p-BDL/Mdr2-/- mice (N = 4 (BDL) and N = 5 (Mdr2-/-). Densitometry analyses of WB are shown in Supp. Fig. 7C. Data normalized as fold change vs. control. Error bars represent the means ± SEM. Statistical significance was determined by the Student’s t-test or ANOVA when more than two groups were compared. p < 0.05*; p < 0.01**; p < 0.001***
Different genes could be regulated by methylation of their CpG islands. The activity of STAT3, a key player in liver inflammation, was repressed in BDL under miR-873-5p inhibition (Fig. 3g), correlating with the re-expression of its inhibitor Protein inhibitor of activated STAT (Pias) Pias 1/3 (Fig. 6c), that has been associated to its promoter methylation status28. Similarly, the promoter regulation of RAS-association domain family (RASSF) (inhibitors of RAS signaling), has been reported to be susceptible of hypermethylation in the absence of GNMT8 and in early HCCs28. We have found that the expressions of RASSF genes (Rassf1a/Rassf4a) are upregulated in Mdr2-/- mice after blockade of miR-873-5p (Fig. 6c).
Regulation of cholangiocyte proliferation and BA metabolism have both been described to be under epigenetic control29,30. SRY-Box 17 (SOX17) regulates the phenotype of normal human cholangiocytes acting as a tumor suppressor in cholangiocarcinoma, occurring its downregulation through DNA methylation31. Under anti-miR-873-5p treatment, Sox17 expression was significantly upregulated in BDL and Mdr2-/- mice (Fig. 6d). Finally, inhibiting miR-873-5p also restored the expression of genes methylated under pathological conditions, such as Abcg5 and Bsep, as well as the nuclear receptors Shp and Fxr, implicated in BA transport and metabolism29 (Fig. 6e).
Besides DNA, SAMe can methylate histones at different residues. Histone modification is also involved in establishing patterns of gene repression during liver injury. Methylation of histones and DNA are linked to the activation of HSCs32. Importantly, miR-873-5p inhibition significantly reduced the protein levels of EZH2, a profibrogenic histone lysine methyltransferase, after BDL and in Mdr2-/- mice (Fig. 6f). Also, the mRNA level of the histone methyltransferase Ash1 is reduced after anti-miR-873-5p treatment, correlating with the decrease in the methylation level of its target H3K4, which has been related with TGFβ induction in fibrosis (Suppl. Fig. 6B)33.
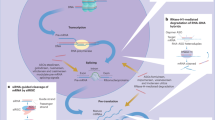
Overall, these data suggest that the prompt recuperation of hepatic GNMT expression through anti-miR-873-5p treatment could lead to the restoration of SAMe metabolism and the epigenetic regulation of specific genes that are implicated directly in BA homeostasis, cholangiocyte proliferation and inflammatory and fibrogenic pathways (Fig. 7).

MiR-873-5p targets GNMT expression in the liver mediating global epigenomic changes and contributing to liver fibrosis progression
Discussion
GNMT is considered a major regulator of the transmethylation flux in the liver controlling liver homeostasis and health4,5. A decrease in GNMT levels occurs in NAFLD, at early stages of fibrosis, cirrhosis, and HCC1,6,7,34,35. Data highlighting that attenuation of GNMT was found at early stages of liver disease, where fibrosis is still not prominent, points to GNMT deficiency as a contributor/mediator of fibrogenesis rather than a consequence. Even though GNMT repression in some primary tumors has been associated with aberrant promoter methylation9, hypermethylation does not seem to be the unique mechanism for GNMT regulation, and it may coexist with other regulatory pathways9.
miRNAs regulate a wide variety of biological functions in the liver and are directly implicated in inflammation, cirrhosis, and malignant transformation13. These small molecules are, therefore, emerging as viable therapeutic tools. Three independent unbiased approaches revealed miR-873-5p as a potential microRNA targeting GNMT. In here, we have shown that miR-873-5p is modulated in several in vitro and in vivo models of liver fibrosis where GNMT levels are modified. Furthermore, in the clinical setting, we have found high levels of circulating miR-873-5p in the serum of cirrhotic patients as well as a negative correlation between hepatic GNMT and miR-873-5p in human cirrhosis and cholestasis. Altogether, this evidence allowed us to identify for the first time GNMT as a direct target of miR-873-5p.
Although liver fibrosis was historically thought to be an irreversible process, the understanding of its molecular pathogenesis has stimulated the development of novel antifibrotic therapies, still in an experimental phase36. Increased miR-873-5p levels have been observed in hepatocytes and in proliferative cholangiocytes under cholestatic/fibrogenic situations. Anti-miR-873-5p treatment in vitro decreased the BA-induced hepatocyte cell death as well as cholangiocyte proliferation. Importantly, anti-miR-873-5p treatment in Gnmt wild-type mouse submitted to BDL surgery blocked the overwhelming inflammatory profile associated to fibrosis. Of relevance, no effects under miR-873-5p inhibition were detected in the Gnmt-/- mice, indicating that GNMT activity is the hub of the regulatory network modulated by miR-873-5p.
In the Mdr2-/- cholestatic mouse model inhibition of miR-873-5p for only 4 weeks starting at the time at which an overt inflammation, cholestasis, and severe fibrosis is observed (4 months old mice), resulted in reduced BA-related cell death and cholangiocyte proliferation, with mild blunted inflammation and decreased fibrosis related markers.
DNA methylation and hydroxymethylation are epigenetic mechanisms that modulate gene expression and play a critical role in the development of liver fibrosis22,24,25,26. We have identified that miR-873-5p changes methyltransferase signaling and DNA methylation state during BDL-mediated hepatic injury and in the chronic model of Mdr2-/- mice. Anti-miR-873-5p therapy restores the transmethylation flux to normal levels in both animal models of liver damage. This miR-873-5p-dependent modulation of the methylome reduces global DNA methylation by decreasing Dnmt3a expression that functions as a de novo methyltransferase24. DNA methylation changes that occur in the livers of mice under anti-miR-873-5p treatment are associated with altered expression of specific genes. Indeed, in BDL mouse, anti-miR-873-5p restores Pias 1,3 levels, genes highly sensible to methylation changes and regulators of STAT3 phosphorylation with the concomitant regression of the inflammatory response. The reduction in STAT3 activation observed in BDL mouse after anti-miR-873-5p treatment, could also have an important effect in the amelioration of fibrosis, due to its direct role in promoting HSC, the major fibrogenic cell type37. In Mdr2-/- mice, the upregulation of the RASSF tumor suppressor, also susceptible to epigenetic modification linked to miR-873-5p treatment, blocks the Ras/Raf/MEK/ERK pathway, related to tumor formation38.
Hepatic fibrosis is also related to methylation changes in BA metabolism and cholangiocyte proliferation29. BAs are highly cytotoxic, their synthesis, transport, and pool size are tightly regulated under physiological conditions. FXR, implicated in every step of BA homeostasis and susceptible to be regulated by methylation29, was upregulated under anti-miR-873-5p treatment. FXR has been shown to induce BSEP and MRP2 in hepatocytes39. Importantly, the inhibition of miR-873-5p counteracts the repression of BSEP and the basolateral drug transporters MRP3 and MRP4 in our cholestatic models. Thus, the stimulation of basolateral BA efflux may be an important protective response against the cholestatic liver injury detected in BDL and Mdr2-/- that could also be regulated by DNA methylation of different genes. Regarding cholangiocyte proliferation, SOX17 promoter was previously found to be hypermethylated and downregulated in cholangiocarcinoma31. In addition, downregulation of SOX17 is implicated in cholangiocyte damage and fibrosis40. MiR-873-5p therapy is able to re-express Sox17 in both cholestatic animal models potentially regulating cholangiocyte proliferation.
Finally, the activation of HSC to myofibroblast is a key process in liver fibrosis mediated by epigenetic mechanism41. We now provide evidence that SAMe flux recovery mediated by anti-miR-873-5p treatment and GNMT rescue reduced EZH2 levels. This histone methyltransferase is one of the major regulators for profibrogenic factors42. Moreover, other signatures of histone methylation are observed in the BDL model, including changes in the histone methyltransferase Ash1, correlating with methylation of the histone H3K4, which has been related with TGFβ induction of fibrosis33.Therefore, it is tempting to hypothesize that changes in global methylation seen after GNMT recovery could exert a negative feedback in the transdifferentiation and activation of HSC.
Our results show that miR-873-5p inhibition directly regulates in vivo BA metabolism, cholangiocyte proliferation and activation and hepatocyte cell death. Modulation of hepatocyte cell death by inhibiting miR-873-5p further reduces inflammatory responses and ameliorates the fibrotic phenotype in diverse liver injury mouse models, possibly associated with epigenetic changes (Fig. 7). Indeed, there is an emerging field in the screening of epi-drugs in the fibrosis disease.
Summing up, the significant inverse correlation between hepatic GNMT and miR-873-5p expression both in cirrhotic and cholestatic patients targeting the miR-873-5p/GNMT axis may provide a novel therapeutic approach to treat liver fibrosis.
Materials and methods
Human studies
All the studies were performed in agreement with the Declaration of Helsinki, and with local and national laws. The Human Ethics Committee of Valdecilla Hospital and of the University of Navarra approved the study procedures.
Cirrhotic patients
Cohort 1
Sixteen liver samples from cirrhotic patients (grade F4) of diverse etiologies were included in this study. The characteristics of these patients are summarized in Suppl. Table I. Healthy liver samples (from individuals with normal or minimal changes in the liver) were collected at surgery of digestive tumors or from percutaneous liver biopsy performed because of mild alterations of liver function. All samples were obtained from the Biobank of the University of Navarra (Pamplona, Spain).
Cohort 2
Thirty-five serum samples from cirrhotic patients (grade F4) (45.7% women and 54.3% men, mean age 63.9 ± 13.4 years) were included in this study from patients recruited ate the Marqués de Valdecilla University Hospital (MVUH, Santander, Spain). Cirrhosis diagnosis was established by clinical or histological data. Etiology was varied: 43% hepatitis C, 31% alcoholic, 17% autoimmune hepatitis, 6% primary biliary cholangitis and 3% hepatitis B. Serum samples (n = 9; five women and four men, mean age 32.4 ± 6.4 years) from healthy volunteer subjects were used as controls and recruited through the Gastroenterology and Hepatology Department of MVUH, Santander, Spain.
Cholestatic patients
Cohort 3
Forty-one serum samples from cholestatic patients (PBC = 36; PSC = 5) with different grade of fibrosis (early = 34; advanced = 7) were included in the study (90% women and 10% men, mean age 59.5 ± 12.2 years). PBC/PSC diagnosis was established in MVUH and based on clinical and biochemical data, immunological markers, imaging, liver histology, and exclusion of other possible causes of liver injury. Healthy human serum samples (n = 13) (92% women and 8% men, mean age 45.5 ± 10.5) from subjects with AMA in serum and without abnormal liver test were included and provided by the Gastroenterology and Hepatology Department MVUH, Santander, Spain. Characteristics of these patients are summarized in Suppl. Table II.
Cohort 4
Sixty-four liver samples from patients with cholestatic liver disease, PBC (n = 60) and PSC (n = 4) were included in this study (90% women and 10% men, mean age 51.64 ± 10.65 years). PBC/PSC diagnosis, established in Marqués de Valdecilla University Hospital (MVUH, Santander, Spain), was based on clinical and biochemical data, immunological markers, imaging, liver histology, and exclusion of other possible causes of liver injury. Patients were classified for early or advance fibrosis based on histological analysis and according to Ludwig classification stage (Grade I–IV). Characteristics of these patients are summarized in Suppl. Table III. Healthy human liver samples (n = 12; 42% women and 58% men, mean age 63.16 ± 13.01 years) from organ-transplant donor were used as controls and recruited through the Gastroenterology and Hepatology Department of MVUH, Santander, Spain.
Animal studies
Three-month-old male (C57BL6), Gnmt wild type (WT), Gnmt-knockout (Gnmt-/-) and Mdr2-/- and Mdr2WT mice were used15,43. Animal procedures were approved by CIC bioGUNE’s Animal Care and Use Committee and the competent authority (Diputación de Bizkaia, Spain).
In vivo miR-873-5p inhibition after BDL
Three-month-old male Gnmt WT and Gnmt-/- mice were injected in the tail vein with miRIDIAN microRNA Hairpin Inhibitor anti-miR-873-5p or miR-Control (60 µg/mouse) (Dharmacon, USA) at 3 days after BDL and repeated on day 5 using Invivofectamine 3.0, following the manufacturer’s instructions (Invitrogen, USA). Mice were sacrificed at day 7, blood withdrawn and livers removed and snap frozen in liquid nitrogen or fixed in formalin for subsequent analysis.
In vivo miR-873-5p inhibition in Mdr2-/- mice
Four-month-old male Mdr2-/- mice were injected in the tail vein with miRIDIAN anti-miR-873-5p or miR-Control (60 µg/mouse) once a week during 4 weeks using Invivofectamine 3.0, following the manufacturer’s instructions (Invitrogen, USA). Animals were then sacrificed, blood withdrawn and livers were removed and snap frozen in liquid nitrogen or fixed in formalin.
In vitro miR-873-5p inhibition
Mouse primary hepatocytes were isolated from male Gnmt WT mice via collagenase perfusion as described44. All adhered cells were maintained in MEM with 10% fetal bovine serum (FBS). NHC cholangiocytes and primary mouse hepatocytes were transfected with anti-miR-873-5p or a miR-Control using DharmaFECT transfection reagent (Dharmacon) following the manufacturer’s procedure.
SAMe and SAH measurement
Hepatic SAMe and SAH were determined by Liquid-chromatography/mass spectrometry (LC/MS) using a Waters ACQUITY-UPLC system coupled to a Waters Micromass LCT Premier Mass Spectrometer equipped with a Lockspray ionization source as described previously45.
Global DNA methylation measurement
Global DNA methylation (5mC) analyses were performed following the method previously described46.
Bile acids (BA) measurement
Total bile acids in serum of Mdr2-/- mice were measured using the Total Bile Acid Assay Kit (Cell Biolabs, Inc, USA) following the manufacturer’s instructions.
Statistical analysis
Data are represented as mean ± SEM. mRNA/RNA data is normalized as fold change vs. control. Statistical significance was determined by the Student’s t-test when two groups were compared and when comparing three groups one-way analysis of variance (ANOVA) was used. A p < 0.05 was considered significant.
References
Avila, M. A. et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J. Hepatol. 33, 907–914 (2000).
Huang, Y. -C. et al. Glycine N-methyltransferase is a favorable prognostic marker for human cholangiocarcinoma. J. Gastroenterol. Hepatol. 23, 1384–1389 (2008).
Chen, C. -Y. et al. Deficiency of glycine N-methyltransferase aggravates atherosclerosis in apolipoprotein E–null mice. Mol. Med. 18, 744–752 (2012).
Heady, J. E. & Kerr, S. J. Purification and characterization of glycine N-methyltransferase. J. Biol. Chem. 248, 69–72 (1973).
Farrar, C. & Clarke, S. Altered levels of S-adenosylmethionine and S-adenosylhomocysteine in the brains ofl-isoaspartyl (d-Aspartyl)O-methyltransferase-deficient mice. J. Biol. Chem. 277, 27856–27863 (2002).
Chen, Y. M. et al. Characterization of glycine-N-methyltransferase-gene expression in human hepatocellular carcinoma. Int. J. Cancer J. Int. Cancer 75, 787–793 (1998).
Fernández-Álvarez, S. et al. TRAIL-producing NK cells contribute to liver injury and related fibrogenesis in the context of GNMT deficiency. Lab. Investig. J. Tech. Methods Pathol. 95, 223–236 (2015).
Martínez-Chantar, M. L. et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatol. Baltim. Md. 47, 1191–1199 (2008).
Huidobro, C. et al. A DNA methylation signature associated with the epigenetic repression of glycine N-methyltransferase in human hepatocellular carcinoma. J. Mol. Med. Berl. Ger. 91, 939–950 (2013).
Filipowicz, W., Jaskiewicz, L., Kolb, F. A. & Pillai, R. S. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr. Opin. Struct. Biol. 15, 331–341 (2005).
Meltzer, P. S. Cancer genomics: small RNAs with big impacts. Nature 435, 745–746 (2005).
Croce, C. M. Oncogenes and cancer. N. Engl. J. Med. 358, 502–511 (2008).
Michelotti, G. A., Machado, M. V. & Diehl, A. M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 10, 656–665 (2013).
Alpini, G. et al. Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation. Am. J. Physiol. 272, G289–G297 (1997).
Popov, Y., Patsenker, E., Fickert, P., Trauner, M. & Schuppan, D. Mdr2 (Abcb4)-/- mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J. Hepatol. 43, 1045–1054 (2005).
Zubiete-Franco, I. et al. Deregulated neddylation in liver fibrosis. Hepatol. Baltim. Md. 65, 694–709 (2017).
Kahraman, A. et al. TRAIL mediates liver injury by the innate immune system in the bile duct–ligated mouse. Hepatology 47, 1317–1330 (2008).
Gao, B., Radaeva, S. & Jeong, W. -I. Activation of natural killer cells inhibits liver fibrosis: a novel strategy to treat liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 1, 173–180 (2007).
Gomez-Santos, L. et al. Inhibition of NK cells protects the liver against acute injury in the absence of GNMT. Hepatol. Baltim. Md. 56, 747–759 (2012).
Mederacke, I. et al. Fate-tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its etiology. Nat. Commun. 4, 2823 (2013).
Gutiérrez-de-Juan, V. et al. A morphological method for ammonia detection in liver. PLoS ONE 12, e0173914 (2017).
Mann, D. A. Epigenetics in liver disease. Hepatol. Baltim. Md. 60, 1418–1425 (2014).
Yang, Y.-L., Wang, F.-S., Li, S.-C., Tiao, M.-M. & Huang, Y.-H. MicroRNA-29a Alleviates Bile Duct Ligation Exacerbation of Hepatic Fibrosis in Mice through Epigenetic Control of Methyltransferases. Int. J. Mol. Sci. 18, (2017).
Page, A. et al. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 64, 661–673 (2016).
Moran-Salvador, E. & Mann, J. Epigenetics and liver fibrosis. Cell. Mol. Gastroenterol. Hepatol. 4, 125–134 (2017).
Hardy, T. & Mann, D. A. Epigenetics in liver disease: from biology to therapeutics. Gut 65, 1895–1905 (2016).
Okano, M., Bell, D. W., Haber, D. A. & Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999).
Calvisi, D. F. et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 117, 2713–2722 (2007).
Schiöth, H. B. et al. A targeted analysis reveals relevant shifts in the methylation and transcription of genes responsible for bile acid homeostasis and drug metabolism in non-alcoholic fatty liver disease. BMC Genom. 17, 462 (2016).
Braconi, C., Huang, N. & Patel, T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 51, 881–890 (2010).
Merino-Azpitarte, M. et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 67, 72–83 (2017).
Bian, E. -B. et al. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol. Appl. Pharmacol. 264, 13–22 (2012).
Perugorria, M. J. et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatol. Baltim. Md. 56, 1129–1139 (2012).
Moylan, C. A. et al. Hepatic gene expression profiles differentiate pre-symptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatol. Baltim. Md. 59, 471–482 (2014).
Murphy, S. K. et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 145, 1076–1087 (2013).
Bataller, R. & Brenner, D. A. Liver fibrosis. J. Clin. Invest. 115, 209–218 (2005).
Jiang, J. X., Mikami, K., Venugopal, S., Li, Y. & Török, N. J. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-κB-dependent pathways. J. Hepatol. 51, 139–148 (2009).
Baghdasaryan, A. et al. Curcumin improves sclerosing cholangitis in Mdr2−/− mice by inhibition of cholangiocyte inflammatory response and portal myofibroblast proliferation. Gut 59, 521–530 (2010).
Ananthanarayanan, M., Li, S., Balasubramaniyan, N., Suchy, F. J. & Walsh, M. J. Ligand-dependent activation of the farnesoid X-receptor directs arginine methylation of histone H3 by CARM1. J. Biol. Chem. 279, 54348–54357 (2004).
Waisbourd-Zinman, O. et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatol. Baltim. Md. 64, 880–893 (2016).
Zeybel, M. et al. A proof-of-concept for epigenetic therapy of tissue fibrosis: inhibition of liver fibrosis progression by 3-deazaneplanocin A. Mol. Ther. J. Am. Soc. Gene Ther. 25, 218–231 (2017).
Mann, J. et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138, 705–714 (2010). 714–4.
Luka, Z., Capdevila, A., Mato, J. M. & Wagner, C. A Glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic. Res. 15, 393–397 (2006).
Embade, N. et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatol. Baltim. Md. 55, 1237–1248 (2012).
Martínez-López, N. et al. Hepatoma cells from mice deficient in glycine N-methyltransferase have increased RAS signaling and activation of liver kinase B1. Gastroenterology 143, 787–798.e13 (2012).
Le, T., Kim, K. -P., Fan, G. & Faull, K. F. A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Anal. Biochem. 412, 203–209 (2011).
Acknowledgements
This work was supported by grants from NIH (US Department of Health and Human services)- R01AT001576 (S.C.L., J.M.M., and M.L.M.-C.), Ministerio de Economía, Industria y Competitividad: SAF2017-87301-R (M.L.M.-C.), Gobierno Vasco-Departamento de Salud 2013111114 (to M.L.M.-C.), BIOEF (Basque Foundation for Innovation and Health Research: EiTB Maratoia BIO15/CA/016/BD (M.L.M.-C.), ELKARTEK 2016, Departamento de Industria del Gobierno Vasco (to M.L.M.-C.), Asociación Española contra el Cáncer (to T.C.D, P.F.-T. and M.L.M.-C.), Mitotherapeutix (to M.L.M.-C.), Daniel Alagille award from EASL (to T.C.D), Fundación Científica de la Asociación Española Contra el Cancer (AECC Scientific Foundation) Rare Tumor Calls 2017 (to M.L.M, J.M.B., and M.A), La Caixa Foundation Program (to M.L.M, J.M.B.), FIS: PI15/00892 (to M.F.F), FIS: PI15/01132 and FIS PI18/01075 (to J.M.B.), (to J.M.B.), FIS: PI14/00399 and PI17/00022 (to M.J.P.), Miguel Servet Program (CON14/00129 to J.M.B.), Ramon y Cajal Program (RYC-2015-17755 to M.J.P.), GRUPIN14-052 (to M.F.), PI14/00962 Instituto Carlos III (to J.B.), AECC Scientific Foundation (to J.B.), World Wide Cancer Research (to J.B.), Programma di Ricerca Regione-Università 2007-2009 and 2011-2012, Regione Emilia-Romagna (to E.V.), “Diputación Foral Gipuzkoa” (DFG15/010, DFG16/004 to J.M.B.), BIOEF (Basque Foundation for Innovation and Health Research: EiTB Maratoia BIO15/CA/016/BD to J.M.B.), Department of Health of the Basque Country (2013111173 to J.M.B.; 2015111100 to M.J.P.). Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III. “Hepacare Project” from Fundación La Caixa, Spain (to M.A.A and C.B.), BiO-Eusko Fundazioa project BIO15/CA/011 (to M.A.A and C.B.). We thank this work produced with the support of a 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation (to M.V.R.). We thank MINECO for the Severo Ochoa Excellence Accreditation (SEV-2016-0644).
Author information
Authors and Affiliations
Contributions
D.F.-R.: Acquisition of data; analysis, and interpretation of data, critical revision of the manuscript. P.F.-T.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript. F.L.-O.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript. V.G.d.J.: Acquisition of data, analysis and interpretation of data, statistical analysis. N.N.: Acquisition of data, analysis and interpretation of data, statistical analysis. L.B.-T.: Analysis and interpretation of data, critical revision of the manuscript. I.Z.-F.: Analysis and interpretation of data, critical revision of the manuscript. J.S.: Analysis and interpretation of data, critical revision of the manuscript. A.F.F.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript. A.A.: Material support, critical revision of the manuscript. A.M.A.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript. J.L.L.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript. N.B.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript. M.J.P.: Material support, critical revision of the manuscript, obtained funding. J.M.B.: Material support, critical revision of the manuscript, obtained funding. E.V.: Critical revision of the manuscript, obtained funding. M.F.F.: Acquisition of data, analysis and interpretation of data, critical revision of the manuscript, obtained funding. J.A.: Critical revision of the manuscript, obtained funding. M.A.Á.: Material and animal support, critical revision of the manuscript, obtained funding. C.B.: Material support, critical revision of the manuscript. P.I.: Material support, critical revision of the manuscript. J.C.: Material support, critical revision of the manuscript. S.C.L.: Critical revision of the manuscript, obtained funding. M.V.-R.: Analysis and interpretation of data, critical revision of the manuscript, obtained funding. J.M.M.: Critical revision of the manuscript, obtained funding. T.C.D.: Acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript, obtained funding. M.L.M.-C.: Study concept and design, analysis and interpretation of data, study supervision, drafting of the manuscript, obtained funding.
Corresponding authors
Ethics declarations
Conflict of interest
M.L. Martínez-Chantar: Consultant/Advisory Board for Mitotherapeutix LLC. J. Mato: Consultant/Advisory Board for Abbott and Galmed, owns stocks for OWL. The other authors declare that they have no conflict of interest.
Additional information
Transcript profiling: ACCESSION NUMBERS: the NCBI Gene Expression Omnibus accession number for the RNA-seq data reported in this paper is GSE77840.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Giannelli
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernández-Ramos, D., Fernández-Tussy, P., Lopitz-Otsoa, F. et al. MiR-873-5p acts as an epigenetic regulator in early stages of liver fibrosis and cirrhosis. Cell Death Dis 9, 958 (2018). https://doi.org/10.1038/s41419-018-1014-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-1014-y
This article is cited by
-
The novel miR-873-5p-YWHAE-PI3K/AKT axis is involved in non-small cell lung cancer progression and chemoresistance by mediating autophagy
Functional & Integrative Genomics (2024)
-
Anti-miR-518d-5p overcomes liver tumor cell death resistance through mitochondrial activity
Cell Death & Disease (2021)
-
MiR-873, as a suppressor in cervical cancer, inhibits cells proliferation, invasion and migration via negatively regulating ULBP2
Genes & Genomics (2020)
