
Abstract
An integrated stress response (ISR), identified in several different animal models of inherited retinal degeneration (IRD), is activated following various cellular stresses. The ISR results in the phosphorylation of eIF2α (p-eIF2α) and a consequent halt in protein synthesis. Although generally protective, persistent elevations in p-eIF2α could lead to cell demise. Therefore, we aimed to determine whether ISR activation is associated with diminished translation rates in mice with IRD. Retinal protein extracts from rd16 mice at different time points were analyzed and the retinal levels of protein synthesis were assessed using the SUnSET method. We found that rd16 mice experience persistent ISR activation: p-eIF2α, ATF4, and CHOP were significantly upregulated at P15 and P20. In agreement with ISR activation, we found that rd16 mice experience translational attenuation at P15. Similar to rd16, other IRD models, T17M RHO, and rd10 also demonstrated a decline in protein synthesis, correlating with p-eIF2α elevation. We then assessed the role of PERK and eIF2α in translational attenuation in rd16 using a PERK inhibitor, GSK2606414. We found that while the treatment significantly reduced p-eIF2α, it did not cause a complete recovery in translation. This suggests that eIF2α is not the only or even the primary point of translational control in IRD, and a second node of translational regulation comprising AKT and mTOR should be evaluated. Surprisingly, we found that AKT-mTOR signaling was diminished in rd16 and rd10 retinas, suggesting a potential link between AKT-mTOR and translational inhibition. Therefore, for the first time, this study shows translation attenuation in IRD models, and highlights the potential roles of eIF2α kinases and AKT-mTOR signaling that could grant valuable insight into the potential treatments for IRD.
Similar content being viewed by others


Introduction
Leber congenital amaurosis (LCA) is a class of inherited retinal dystrophies, in which there is currently no cure and generally diagnosed within the first few months of life1,2. In the United States, LCA cases most commonly involve mutations in the gene encoding the centrosomal protein of 290kD (CEP290)2. Various retinal degenerative disorders share cellular signaling pathways including the integrated stress response (ISR)3,4,5. In this study, we analyzed ISR signaling in the retinas of rd16 mice, a model of LCA harboring a 5-exon deletion in Cep290. A variety of stress responses including endoplasmic reticulum (ER) stress, amino acid starvation, and viral infection lead to the activation of one or more kinases that phosphorylate the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) at serine 51. eIF2 is responsible for bringing Met-tRNA to the 43S pre-initiation complex. When eIF2α is phosphorylated, eIF2 can no longer participate in translation, and protein synthesis rates go down. Persistent elevations in p-eIF2α have been shown to lead to cell demise3,6,7,8.
Elevated p-eIF2α levels and translational inhibition are becoming a more common finding in neurodegenerative diseases such as Alzheimer’s disease and tauopathies, but the exact role of the ISR in the pathogenesis of these diseases remains unknown. Recently, restoring translation has shown great promise as a potential therapy in neurodegenerative diseases6,8,9. In a model of Tau neurodegeneration, decreasing the PERK branch of the unfolded protein response (UPR) restored the levels of important synaptic proteins and rescued neurons9. However, the exact role PERK plays in the retina has not been carefully assessed because detecting phosphorylated PERK (p-PERK) in vivo has proven challenging with current methodology. Although p-PERK has not been detected in the retinas of mice with retinal degenration (RD), the activation of the UPR and ISR has been observed3,10,11. Interestingly, a recent study showed a 50% decrease in p-eIF2α in P23H rats following treatment with a PERK inhibitor3, but the correlation between the ISR and translation has not been explored in RD.
In this study, we sought to determine if different retinopathies could experience metabolic imbalance through the inhibition of protein synthesis over the course of RD. We adapted the surface sensing of translation (SUnSET)12 method to measure translation in normal and degenerating retinas. In addition, we treated rd16 mice with a PERK inhibitor to determine if PERK is the major eIF2α kinase in RD, and to understand whether eIF2α is the main point of translational control within the retinas of mice with RD.
Results
The ISR is persistently active in retinas of rd16 mice
Recently, we and other investigators have reported that animal models of varied retinal degenerative disorders share cellular signaling pathways activated over the course of RD11,13. T17M RHO14, P23H RHO15,16, rd1017, and TUPL118 are just a few models of RD with elevated levels of p-eIF2α that are thought to contribute to retinal pathogenesis. Before examining ISR signaling within the retinas of rd16 mice, we first wanted to confirm the rate of photoreceptor (PR) cell loss1,19,20. Rows of nuclei within the outer nuclear layer (ONL), which correspond to the nuclei of PRs, were counted in C57BL/6J (wild type; WT) and rd16 mice on postnatal day (P)15 and P20 (Fig. 1). The number of nuclei did not significantly differ from WT (n = 3) at P15; however, PR nuclei were greatly reduced by P20 (n = 3). To determine if the ISR is active in the retinas of rd16 mice, we assessed the levels of ISR markers at two time points, P15 (n = 3) and P20 (n = 4). Using an antibody specifically against p-eIF2α, we found that it was upregulated at both P15 (Fig. 2a) and P20 (Fig. 2b). We next assessed markers downstream of eIF2α phosphorylation. During the ISR, an alternative translational program is activated and activating transcription factor 4 (ATF4) is selectively translated21. ATF4 regulates the expression of several stress response genes including the genes encoding C/EBP homologous protein (CHOP), growth arrest/DNA damage inducible 34 (GADD34), and Tribbles Homolog 3 (TRB3)21. Both ATF4 and CHOP were elevated at P15 and P20 (Fig. 2), suggesting that the ISR translational program is active in rd16 mice. TRB3 was elevated at P15 while GADD34 was elevated at P20.

Representative H&E-stained eye sections from C57BL/6 and rd16 mice (a), as well as the corresponding chart displaying the loss of photoreceptor nuclei in the ONL in rd16 mice at P20 (b). OS outer segments, ONL outer nuclear layer (PR nuclei), OPL outer plexiform layer, INL inner nuclear layer, IPL inner plexiform layer. Scale bar represents 100 μm. Data are shown as mean ± SEM. n = 3. ****p < 0.0001

Western blots and corresponding graphs of ISR markers in the retinas of rd16 mice at P15 (a) and P20 (b). At P15, p-eIF2α, ATF4, CHOP, and TRB3 are significantly elevated. At P20, levels of p-eIF2α, ATF4, CHOP, and GADD34 are increased. Markers of the UPR are elevated in rd16 retinas. BiP and cleaved (~50 kD) ATF6 are upregulated in the retinas of rd16 mice at P15 (c) and P20 (d). Relative density measurements correspond to the intensities of the immunoblotting bands or lanes normalized to an internal control. Data are shown as mean ± SEM. a.u. arbitrary units. P15 n = 3. P20 n = 4. *p < 0.05, **p < 0.01, ***p < 0.001
We next aimed to determine if ER stress may be contributing to the active ISR in the retinas of rd16 mice. We assessed the levels of two markers of ER stress, binding immunoglobulin protein (BiP) and ATF6. BiP is an ER resident protein chaperone that assists in the folding and clearing of aberrant proteins. Levels of BiP typically go up during ER stress and it is a well-established marker of the UPR. BiP was elevated at both P15 and P20 (Fig. 2c, d) in rd16 retinas. Aside from the beforehand-mentioned PERK pathway, two additional UPR signaling cascades exist. One of these pathways is signaled through ATF6. Following an ER insult, ATF6 travels to the Golgi body where it is cleaved by sites 1 and 2 proteases. The cleaved ATF6 migrates to the nucleus to act on the genes encoding proteins involved in ER-associated degradation21. We assessed levels of cleaved ATF6 (~50 kD) and found that it was elevated at both P15 and P20 (Fig. 2c, d). These results indicate that the UPR may be active in the retinas of rd16 mice.
Protein synthesis is inhibited in retinal degeneration
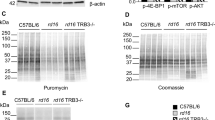
Interestingly, various models of RD experience persistent phosphorylation of eIF2α14,1821,22. For example, T17M RHO mice, which mimic autosomal-dominant retinitis pigmentosa, demonstrate the elevation of p-eIF2α at P15 and P3011,14. rd10 mice, with a point mutation in PDE6β, model autosomal recessive retinitis pigmentosa (ARRP) and have been shown to experience the activation of ISR signaling in their retinas as early as P15, as shown by an increase in p-eIF2α and CHOP levels17. When p-eIF2α is elevated at consequent time points, it may indicate that steady-state translational inhibition is occurring during the course of RD21. Therefore, by adopting the SUnSET method12, we assessed the levels of newly synthesized proteins in rd16 at P15 (Fig. 3). This method is based on immunological detection of the incorporation of puromycin (puro), an aminoacyl tRNA-analog, into newly synthesized polypeptide chains. Mice were injected with puromycin at a dose of 0.04 μmol/g body mass 30 min prior to retina isolation. Puromycinylated proteins were separated by SDS-PAGE, transferred to a membrane, and then detected with an antibody against puromycin. By measuring the relative density of a membrane treated with an anti-puro antibody, one can evaluate the rate of protein synthesis. The puro measurements were then normalized by Coomassie staining (Fig. 3b). Figure 3 demonstrates that rd16 mice experience a ~43% reduction in the rate of protein synthesis at P15. We next wanted to determine if other models of RD could experience translational inhibition. We found that both rd10 and T17M RHO mice have a reduction in translation (Fig S1). Interestingly, rd10 mice, with a slower rate of RD, demonstrated large translational inhibition (Fig S1A) at P20 (n = 4). Meanwhile, T17M RHO (Fig S1B) demonstrated a 40% reduction in protein synthesis at P15, as compared to WT retinas (n = 4).

Measurement of newly synthesized polypeptides using the SUnSET method. Translational attenuation in the retinas of rd16 mice at P15. Anti-puromycin immunoblotting showing diminished translation in rd16 mice (a). Puromycin immunoblotting was normalized to Coomassie-stained proteins (b). Graph comparing levels of translation in rd16 mice at P15 (c). Relative density measurements correspond to intensities of the immunoblotting bands or lanes normalized to an internal control. n = 4. Data are shown as mean ± SEM. a.u. arbitrary units. **p < 0.01
PERK inhibition does not completely restore the translation rates in RD
Four kinases are known to inhibit translation by acting on eIF2α in a variety of conditions23. Determining exactly which kinases are involved can be challenging due to various factors. Studies have shown that multiple kinases can become active in certain conditions23. Therefore, we first aimed to determine if PERK could play a role in translational attenuation in RD, utilizing a cell culture approach. We inhibited PERK in mouse cone-derived 661W cells and assessed its role in the translational inhibition following the induction of ER stress with thapsigargin (Tg), a SERCA channel inhibitor. Cells were pre-treated with a PERK inhibitor (PERK-i), GSK2606414, at a concentration of 3 μM for 30 min before the addition of Tg (3 μM) for 2 h. For the last 30 min of Tg treatment, cells were treated with puromycin at a concentration of 10 μM. We found that following PERK inactivation, p-eIF2α was reduced by ~38%, and protein synthesis was significantly restored (Fig. 4a); however, translation did not return to normal levels, meaning that PERK signaling is not the only factor contributing to diminishing protein synthesis rates, even in a model as specific as ER-stressed cells. We then tested this idea in vivo. To that end, we designed a short experiment to determine how much PERK and eIF2α contribute to the translational attenuation in rd16 mice. Mice were treated with PERK-i (50 mg/kg) by oral gavage twice a day for 2 days. PERK-i has been shown to have excellent central nervous system penetration9. Two hours prior to retina collection, mice were injected intraperitoneally with azidohomoalanine (AHA) at a dose of 1.20 mg/kg. AHA is an amino acid analog modified with an azide moiety. Cells take up AHA and incorporate it into the growing peptide chains. In the presence of copper (I), azides react with the conjugated alkyne resulting in tagged proteins24. Our AHA-tagged proteins were linked to biotin and separated by SDS-PAGE. The membranes were probed with HRP-conjugated streptavidin and normalized to total protein, as detected by Coomassie staining. We found that the retinas of PERK-i-treated rd16 mice had a significant decrease (48%) in p-eIF2α (Fig. 4b), suggesting that PERK plays a role in eIF2α regulation in the retina during RD. This was in agreement with a recent study showing that PERK participates in eIF2α phosphorylation in the retinas of P23H rats, as the treatment with PERK-i caused a ~50% reduction in p-eIF2α levels3. We next aimed to determine what impact the decline in p-eIF2α had on translation rates. We found that the decrease in phosphorylated eIF2α corresponded to a slight (~16%) but significant increase in the translation rates (Fig. 4c). Altogether, this suggests that eIF2α is not the only, or perhaps even the primary, point of translational control contributing to the decline in the protein synthesis rates in rd16, and specific kinase inactivation may not be enough to restore the translation in complex biological systems such as a degenerating retina.

Treatment with GSK2606414 (PERK-i) brings p-eIF2α down to normal levels, but only partially restores the translation in mouse cone-derived 661 W cells subjected to ER stress (a). Cells were treated with 3 μM thapsigargin to induce ER stress (Tg) or DMSO as a control (n = 3 plates of cells). Treatment of rd16 mice (n = 3 for vehicle, n = 4 for PERK-i) with PERK-i (10 mg/kg) for 2 days brings the retinal levels of p-eIF2α down by ~47% (b). Diminished levels of p-eIF2α significantly restores the translation, but only by ~14%. Relative density measurements correspond to intensities of the immunoblotting bands or lanes normalized to an internal control. Data are shown as mean ± SEM. a.u. arbitrary units. *p < 0.05, **p < 0.01, ***p < 0.001, ****P < 0.0001
The AKT-mTOR pathway signals for the decline of translation in mouse models of retinal degeneration
Since the decrease in p-eIF2α levels did not completely restore translation in the retinas of PERK-i-treated rd16 mice, we next explored a second mechanism of translational control. A second mode of translational control occurs when mTOR signaling is altered25. To determine if this second mode of translational inhibition was active in rd16 mice, we assessed specific markers of the mTOR signaling pathway by western blot at P15 (n = 3) (Fig. 5), a time point where all PR nuclei remain. When nutrients are readily available, signaling cascades activate mTOR. However, we found that p-mTOR (Ser2448) was downregulated in the retinas of rd16 mice at P15. The activation of mTOR results in phosphorylation of eIF4E-binding protein 1 (4E-BP1) and therefore, translational stimulation. When hypophosphorylated, 4E-BP1 interacts with eIF4E, preventing its association into the eIF4F 5′-cap binding complex, thus inhibiting translation25. Using an antibody against p-4E-BP1 (Thr37/46), we found that 4E-BP1 phosphorylation was significantly reduced in the retinas of rd16 mice at P15 (Fig. 5), suggesting that 4E-BP1 is able to interact with eIF4E and inhibit translation initiation. Upstream of mTOR are various factors that regulate its activity including AKT. AKT targets TSC2, preventing it from inhibiting mTOR25. We found that p-AKT (Ser473) was downregulated at P15, which is consistent with mTOR inactivation. These data were also in agreement with TRB3 being upregulated at P15 (Fig. 2), a downstream target of ATF4 known to inhibit AKT phosphorylation26. These results indicate that the second mode of translational inhibition is active in the retinas of rd16 mice at P15.

Diminished AKT-mTOR-4E-BP1 signaling in rd16 mice at P15. n = 4. Data are shown as mean ± SEM. a.u. arbitrary units. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
We then assessed whether other models of RD could have both of the translation inhibition elements active. To test the findings in a different mouse model of RD, we utilized western blot analysis to check the levels of p-eIF2α, p-mTOR, and p-AKT in rd10 mice at two time points, P20 and P25 (n = 4). Just as in rd16, p-eIF2α was upregulated in the retinas of rd10 mice at both time points (Fig. S2A and B), which is consistent with the previous reports. In addition, both p-AKT and p-mTOR were downregulated at P20 and P25. Moreover, rd10 mice demonstrate a significant reduction in p-mTOR as early as P15 (Fig. S2C). In addition, in our previous study, we have also demonstrated that a third RD model, T17M RHO, experiences downregulation of p-AKT at P3027, and that this reduction correlates with an increase in p-eIF2α11. Together, these results indicate that similar to rd16, T17M RHO and rd10 undergo translational inhibitory signaling through both ISR and AKT-mTOR signaling.
Discussion
Although a transient elevation in eIF2α phosphorylation is often physiological and overcame by cells, it is well established that a persistent ISR can lead to apoptosis3,828,29. An activated ISR results in the global reduction of protein synthesis23. This study provides a novel mechanism by which retinas expressing mutant or aberrant proteins in PRs could degenerate and highlights potential therapeutic targets that should be validated in future studies. We revealed that degenerating retinas with persistent phosphorylation of eIF2α exhibit translational inhibition. Second, we found that a second mode of translational control associated with AKT-mTOR signaling is weakened in degenerating retinas.
We found that rd16 mice had an elevation of p-eIF2α, CHOP, and ATF4 at two time points, demonstrating that rd16 mice have an activated ISR. Our previous study demonstrated that in degenerating retinas, elevated levels of p-eIF2α are accompanied by the acceleration of RD13. Indeed, eIF2α phosphorylation in rd16 retinas was associated with translational attenuation at P15. The other two mouse models used in this study, T17M RHO and rd10, develop different ocular phenotypes, but also experience an elevation in levels of p-eIF2α. T17M RHO mice demonstrate an increase in eIF2α phosphorylation at P15 and P3011,14. At P15, there is no PR cell loss in T17M RHO retinas;11 however, partial mislocalization of rhodopsin is already observed. rd10 mice also show an elevation in p-eIF2α levels at P20 and P25 (Fig. S2), and another study has shown p-eIF2α elevation as early as P1517. As in rd16 retinas, we found that protein synthesis was attenuated in the retinas of T17M RHO and rd10 mice, suggesting that translational inhibition is a common theme in RD. However, whether inhibition of translation is a defense mechanism for degenerating PRs or whether a decline in protein synthesis could cause PR cell death needs to be addressed in future studies.
Since there are four kinases that can phosphorylate eIF2α in a variety of conditions, pinpointing the causative factor can be challenging. In fact, ER stress-inducing compounds such as Tg have been shown to activate eIF2α kinases, other than PERK23. Our results from in vitro experiments using mouse cone-derived 661W cells and an in vivo study using rd16 mice demonstrated only partial restorations in translation rates following treatment with PERK-i. This was in agreement with a recent study that investigated the role of PERK in P23H rats3. When the rats were treated with PERK-i, eIF2a phosphorylation was reduced by ~50%. This reduction in eIF2α corresponded to a slight increase in cell death, suggesting that eIF2α phosphorylation could be protective in RD. However, the translation rates were not assessed in these rats, and PERK activity have never been directly shown in the retinas of P23H rats. It would have been interesting to observe the translation rates in these rats, especially since we are now showing that PERK inhibition does not completely restore translation in rd16 mice. Since PERK ablation is toxic to the pancreas4, the use of PERK inhibitors may not be the best approach moving forward. Perhaps a study utilizing retinal specific mutations in PERK and eIF2α would better address these questions. However, we showed that eIF2α may not be the primary point of translational regulation in RD.
We used the SUnSET method to estimate the levels of translation in the retinas of mice with RD. The ability of puromycin to incorporate into nascent peptide chains provides investigators with a powerful tool; however, the SUnSET method is not without its limitations. Detecting in vivo translation rates in the retina means that our dosage must meet some basic requirements. One of these requirements is to use the time point when the studied models retain nearly all of their PRs. While rd16 outer segments do not develop properly20, and this could potentially pose a problem when comparing the groups with similar translational rates, large differences (35–47%; Fig. 3 and Fig. S1) are beyond this limitation.
We also found that AKT-mTOR signaling is weakened in mice with RD. The exact contribution of AKT-mTOR signaling on the translation rates in the retinas of mice with RD, and whether there is cross-talk between the ISR and AKT-mTOR signaling in the retina, are topics that need to be evaluated in future studies. It is entirely possible that AKT-mTOR inactivation could occur independently of the ISR, but several links between the two cascades have been outlined in the literature25. For example, TRB3 has been shown to inhibit AKT activation26, which could potentially explain how rd16 mice have reduced p-AKT levels accompanied by an elevation in TRB3 levels at P15. GADD34 is another potential culprit, as it has been shown to result in cytoplasmic retention of AKT, markedly reducing AKT phosphorylation30. In addition, ATF4 regulates the transcription of 4EBP131. Elevated levels of 4E-BP1 would result in more inactivated eIF4E, but our total 4E-BP1 levels did not rise in the retinas of rd16 mice (Fig. 5). Future studies that look at AKT-mTOR signaling in the absence of TRB3 and GADD34 could help determine if one of these proteins is involved in the potential cross-talk between the ISR and AKT-mTOR signaling. Although we found that translational arrest is associated with RD, there is still a question as to whether inhibition of protein synthesis can cause PR cell death. Interestingly, a recent study identified hypomorphic mutations in the gene encoding tRNA nucleotidyl transferase (TRNT1) that causes retinitis pigmentosa32. TRNT1 is required for protein synthesis, and it is likely that mutations in the gene encoding this enzyme lead to RP due to sustained translation attenuation.
T17M RHO mice express a protein known to misfold; however, whether rd16 mice express misfolded CEP290 is not known, which leads to an uncertainty as to why the rd16 mouse model has an activated ISR. Our observation agrees with a previous study that highlighted active PERK signaling in a BBS12 model33. That study utilized an shRNA in ex vivo retinal culture to knockdown BBS12, a ciliary transition zone protein, and the resulting phenotype had an activated UPR. Similar to the retinas of rd16, the BBS12 model did not have a gene known to yield a product that aggregates in the ER lumen, but instead displayed abberant protein trafficking to the outer segment, suggesting an issue with intraflagellar transport (IFT). How an abnormal IFT could promote ISR activation remains to be investigated. For example, a stress sensor within the connecting cilium or inner segment could signal for a halt in translation. Additional studies aiming to elucidate the mechanisms responsible for the reduction in translation rates are needed to gain a better understanding of the stresses involved in the progression of RD.
Therefore, for the first time, chronic inhibition of translation has been observed in various models of RD, highlighting translational attenuation as a common theme and potential therapeutic target in RD. We found that retinas with elevated levels of p-eIF2α have reduced translation rates. In addition, we discovered that eIF2α is likely not the primary target of translational regulation in mice with RD, as PERK inhibition brought p-eIF2α down to normal levels but only partially restored translation. Finally, we found that a decline in AKT-mTOR signaling may signal for translational attenuation in RD. Future studies highlighting roles of eIF2α kinases, AKT-mTOR signaling and other means of translational attenuation in the progression of RD could grant valuable insight into potential treatments for RD.
Materials and methods
Animals
All animal experiments followed a protocol (IACUC#131109793) approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC), and conformed to guidelines set by the Association of Research in Vision Science and Ophthalmology. Rd16, rd10 and C57BL/6J (Wild type; WT) mice were obtained from Jackson Laboratory (Bar Harbor, ME). T17M RHO+/- mice were generated, as previously described14. Mice were housed in a facility with a 12 h light/dark cycle. Mice had free access to a standard diet and water. Sample sizes ranged from 3 to 5. At the time points specified in the following sections and text body, mice were euthanized by CO2 asphyxiation followed by cervical dislocation.
Histology
Eyes were enucleated at P15 or P20 and fixed in 4% paraformaldehyde overnight at 4 °C. The fixed eyes were washed with PBS and immersed in 30% sucrose for at least an hour. The eyes were embedded in tissue-tek O.C.T. compound (VWR: 25608-930) and kept at −80 °C for 30 min. Eyes were then cut to 12 μM sections using a cryostat tissue sectioning system (Leica CM 1510S; Leica, Buffalo Grove, IL, USA). The sections were stained with hematoxylin and eosin. The nuclei in the outer nuclear layer were then counted by a masked investigator. The groups were compared by a two-sample t-test using GraphPad Prism 7 software.
Western blotting
For western blot analyses, mouse retinas were dissected and homogenized in lysis buffer (150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris pH 8.0). Protein concentrations were estimated by the Bio-Rad protein assay (5000001, Hercules, CA, USA). The protein lysate (40–60 μg) was separated by SDS-PAGE and transferred to a PVDF membrane for immunoblotting. Primary antibodies specific to phospho-eIF2α (p-S51, 3398, rabbit, lot: 6), eIF2α (9722, rabbit, lot: 15), p-AKT (p-S473, 4060, rabbit, lot: 14), ATF4 (11815, rabbit, lot: 4), β-actin (4970, rabbit, lot: 4), p-mTOR (p-S2448, 2971, rabbit, lot: 21), AKT (4691, rabbit, lot: 20), mTOR (2983, rabbit, lot: 14), p-4E-BP1 (p-Thr37/46, 2855P, rabbit, lot: 20), and 4E-BP1 (9644, rabbit, lot: 10) were purchased from Cell Signaling Technologies (Danvers, MA, USA). Antibodies against TRB3 (sc-34211, goat, lot: A0615), CHOP (sc-7351, mouse, lot: I2614), GADD34 (sc-8327, rabbit, lot: I0810), and GRP78 (sc-1050, rabbit, lot:J3015) were purchased from Santa Cruz Biotechnologies (Dallas, TX, USA). ATF6 (NBP1-40256, mouse) and Novus Biologicals (Littleton, CO, USA). Secondary antibodies (WesternSure HRP goat anti-mouse IgG: 926-80010, WesternSure HRP goat anti-rabbit IgG: 926-80011, IRDye 800CW Donkey anti-goat: 926-32214 and IRDye 800CW Goat anti-mouse: 925-32210) were purchased from LI-COR (Lincoln, NE, USA). The probed membranes were imaged on a LICOR Fc imaging system. Relative band densities were measured using ImageJ software. Westerns were normalized to β-actin. Phosphorylated proteins were normalized to their non-phosphorylated counterparts. The groups were compared by a two-sample t-test using GraphPad Prism 7 software.
in vivo SUnSET
For in vivo analysis of nascent peptide synthesis as detected by western blot – T17M RHO P15, rd16 P15, rd10 P20, and age matched wild type (C57BL/6) mice – were intraperitoneally injected with puromycin (puromycin dihydrochloride; Santa Cruz Biotechnology, CAS 58-58-2) at a dosage of 0.04 μmol/g body mass. After 30 min of the puromycin injection, the retinas were harvested. Proteins were extracted in RIPA buffer, and the proteins were estimated, as stated above in the western blotting section. Between 60 and 80 μg of protein were separated by SDS-PAGE and transferred to a PVDF membrane. PVDF membranes were probed with an anti-puromycin (MABE343, mouse, lot: 2861354) antibody purchased from EMD Millipore. A secondary antibody specific for IgG2a (goat anti-mouse peroxidase affinipure IgG, Fcγ Subclass 2a Specific: 115-035-206, Jackson Immuno Research Laboratories Inc; West Grove, PA, USA) was used to prevent the detection of the heavy and light chains of the endogenous immunoglobulin. As a loading control, the membranes were stained with coomassie blue R-250 (Bio-Rad: #1610436). After anti-puromycin immunoblotting, the membranes were washed with distilled water and then incubated in coomassie staining solution for 60 s. Membranes were then de-stained (ethanol/acetic/water; 5:1:4 proportion) for 10–15 min and air dried before scanning on a Kyocera Taskalfa copier. The relative densities of entire lanes were measured using ImageJ software. Puromycin densities were normalized to their corresponding coomassie densities. Groups were compared by a two-sample t-test using GraphPad Prism 7 software. For the PERK-i experiment, rd16 mice were fed GSK2606414 (Millipore, 516535; Burlington, MA, USA) by oral gavage (in vehicle: 0.5% HPMC, 0.1% Tween-80 in H2O at pH 4.0) at a dose of 50 mg/kg twice a day for 2 days. The treatment started when the mice were P14. Two hours prior to retinal collection, the mice were intraperitoneally injected with Click-it Azidohomoalanine (ThermoFisher, C10102; Waltham, MA, USA) at a dosage of 1.20 mg/kg. The retinas were collected and 100 µg of protein were subjected to a click-it reaction, following the protocol outlined by the manufacturer (ThermoFisher, C10276). The reaction involved chemically linking AHA to an alkyne (Biotin Alkyne, B10185). After the tagged proteins were precipitated to remove the reaction components, they were re-solubilized in 1× SDS-loading buffer (2% SDS, 10% glycerol, 0.005% bromophenol blue and 5% 2-mercaptoethanol) and heated at 95 °C for 10 min before loading on a gel and separating by SDS-PAGE.
Immunocytochemical SUnSET
Mouse cone-derived 661 W cells were plated at a density of 1.78 × 105 cells/cm2 and grown to confluency at 37 °C in Dulbecco’s modified eagle medium (DMEM; GE healthcare, SH30022; Chicago, IL, USA), supplemented with 10% fetal bovine serum (FBS) as well as penicillin and streptomycin. Cells were then pre-treated with DMEM + 10% FBS containing either 3 μM PERK inhibitor (GSK2606414) or solvent (DMSO) for 30 min. Thapsigargin (at a final concentration of 3 μM), or DMSO was then added to the media for 2 h. For the last 30 min of treatment, puromycin was added at a final concentration of 10 μM. Proteins were extracted using 1× SDS-loading buffer (2% SDS, 10% glycerol, 0.005% bromophenol blue and 5% 2-mercaptoethanol) and heated at 95 °C for 10 min before loading on a gel and separating by SDS-PAGE. The relative densities of entire lanes were measured using ImageJ software. Puromycin densities were normalized to their corresponding coomassie densities. Groups were analyzed by one-way ANOVA using GraphPad Prism 7 software.
References
Subramanian, B., Anand, M., Khan, N. W. & Khanna, H. Loss of Raf-1 kinase inhibitory protein delays early-onset severe retinal ciliopathy in Cep290rd16 mouse. Invest. Ophthalmol. Vis. Sci. 55, 5788–5794 (2014).
Collin, R. W. & Garanto, A. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr. Opin. Ophthalmol. 28, 260–266 (2017).
Athanasiou, D. et al. The role of the ER stress response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 26, 4896–4905 (2017).
Gao, Y. et al. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol. Cell. Biol. 32, 5129–5139 (2012).
Rana, T. et al. An activated unfolded protein response promotes retinal degeneration and triggers an inflammatory response in the mouse retina. Cell Death Dis. 5, e1578 (2014).
Yang, W. et al. Repression of the eIF2alpha kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 41, 19–24 (2016).
Sadleir, K. R., Eimer, W. A., Kaufman, R. J., Osten, P. & Vassar, R. Genetic inhibition of phosphorylation of the translation initiation factor eIF2alpha does not block Abeta-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of Alzheimer’s disease. PLoS ONE 9, e101643 (2014).
Moreno, J. A. et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485, 507–511 (2012).
Radford, H., Moreno, J. A., Verity, N., Halliday, M. & Mallucci, G. R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642 (2015).
Bhootada, Y., Choudhury, S., Gully, C. & Gorbatyuk, M. Targeting caspase-12 to preserve vision in mice with inherited retinal degeneration. Invest. Ophthalmol. Vis. Sci. 56, 4725–4733 (2015).
Bhootada, Y. et al. Limited ATF4 expression in degenerating retinas with ongoing ER stress promotes photoreceptor survival in a mouse model of autosomal dominant retinitis pigmentosa. PLoS ONE 11, e0154779 (2016).
Goodman, C. A. & Hornberger, T. A. Measuring protein synthesis with SUnSET: a valid alternative to traditional techniques? Exerc. Sport. Sci. Rev. 41, 107–115 (2013).
Nashine, S., Bhootada, Y., Lewin, A. S. & Gorbatyuk, M. Ablation of C/EBP homologous protein does not protect T17M RHO mice from retinal degeneration. PLoS ONE 8, e63205 (2013).
Kunte, M. M. et al. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 53, 3792–3800 (2012).
Sizova, O. S., Shinde, V. M., Lenox, A. R. & Gorbatyuk, M. S. Modulation of cellular signaling pathways in P23H rhodopsin photoreceptors. Cell Signal. 26, 665–672 (2014).
Athanasiou, D. et al. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Mol. Genet. 26, 305–319 (2017).
Rodriguez-Muela, N. et al. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 22, 476–487 (2015).
Lobo, G. P., Au, A., Kiser, P. D. & Hagstrom, S. A. Involvement of endoplasmic reticulum stress in TULP1 induced retinal degeneration. PLoS ONE 11, e0151806 (2016).
Murga-Zamalloa, C. A. et al. Accumulation of the Raf-1 kinase inhibitory protein (Rkip) is associated with Cep290-mediated photoreceptor degeneration in ciliopathies. J. Biol. Chem. 286, 28276–28286 (2011).
Chang, B. et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 15, 1847–1857 (2006).
Gorbatyuk, M. & Gorbatyuk, O. Review: retinal degeneration: focus on the unfolded protein response. Mol. Vis. 19, 1985–1998 (2013).
Gorbatyuk, M. S. et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl Acad. Sci. USA 107, 5961–5966 (2010).
Taniuchi, S., Miyake, M., Tsugawa, K., Oyadomari, M. & Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2alpha kinases. Sci. Rep. 6, 32886 (2016).
Wang, J. et al. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat. Protoc. 12, 279–288 (2017).
Preston, A. M. & Hendershot, L. M. Examination of a second node of translational control in the unfolded protein response. J. Cell Sci. 126, 4253–4261 (2013).
Du, K., Herzig, S., Kulkarni, R. N. & Montminy, M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300, 1574–1577 (2003).
Choudhury, S., Bhootada, Y., Gorbatyuk, O. & Gorbatyuk, M. Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis. 4, e528 (2013).
Rajesh, K. et al. Phosphorylation of the translation initiation factor eIF2α at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 6, e1591 (2015).
Raven, J. F. et al. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J. Biol. Chem. 283, 3097–3108 (2008).
Farook, J. M. et al. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis. 4, e754 (2013).
Yamaguchi, S. et al. ATF4-mediated induction of 4E-BP1 contributes to pancreatic beta cell survival under endoplasmic reticulum stress. Cell Metab. 7, 269–276 (2008).
DeLuca, A. P. et al. Hypomorphic mutations in TRNT1 cause retinitis pigmentosa with erythrocytic microcytosis. Hum. Mol. Genet. 25, 44–56 (2016).
Mockel, A. et al. Pharmacological modulation of the retinal unfolded protein response in Bardet-Biedl syndrome reduces apoptosis and preserves light detection ability. J. Biol. Chem. 287, 37483–37494 (2012).
Acknowledgements
The study was supported by NIH Grant R01EY027763 and P30 EY003039.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Starr, C.R., Pitale, P.M. & Gorbatyuk, M. Translational attenuation and retinal degeneration in mice with an active integrated stress response. Cell Death Dis 9, 484 (2018). https://doi.org/10.1038/s41419-018-0513-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0513-1
This article is cited by
-
Tribbles homolog 3-mediated targeting the AKT/mTOR axis in mice with retinal degeneration
Cell Death & Disease (2021)
-
Delineating the role of eIF2α in retinal degeneration
Cell Death & Disease (2019)
