
Abstract
Inter-organellar communication often takes the form of Ca2+ signals. These Ca2+ signals originate from the endoplasmic reticulum (ER) and regulate different cellular processes like metabolism, fertilization, migration, and cell fate. A prime target for Ca2+ signals are the mitochondria. ER–mitochondrial Ca2+ transfer is possible through the existence of mitochondria-associated ER membranes (MAMs), ER structures that are in the proximity of the mitochondria. This creates a micro-domain in which the Ca2+ concentrations are manifold higher than in the cytosol, allowing for rapid mitochondrial Ca2+ uptake. In the mitochondria, the Ca2+ signal is decoded differentially depending on its spatiotemporal characteristics. While Ca2+ oscillations stimulate metabolism and constitute pro-survival signaling, mitochondrial Ca2+ overload results in apoptosis. Many chemotherapeutics depend on efficient ER–mitochondrial Ca2+ signaling to exert their function. However, several oncogenes and tumor suppressors present in the MAMs can alter Ca2+ signaling in cancer cells, rendering chemotherapeutics ineffective. In this review, we will discuss recent studies that connect ER–mitochondrial Ca2+ transfer, tumor suppressors and oncogenes at the MAMs, and chemotherapy.
Similar content being viewed by others

Facts
-
Ca2+ fluxes between ER and mitochondria affect several cancer hallmarks, including apoptosis resistance, migration, and invasion.
-
Oncogenes and tumor suppressors residing at the MAMs execute part of their cellular function by altering ER–mitochondrial Ca2+ transfer, thereby promoting or preventing cancer cell survival.
-
Dependent on the cancer type and cancer stage, ER–mitochondrial Ca2+ transfer can either exert anti-tumorigenic effects like restoring apoptosis sensitivity or exert pro-tumorigenic effects like promoting metastatic behavior.
-
Different chemotherapeutics rely on a Ca2+-signaling component to induce cancer cell death.
-
Ca2+ signaling modulation can (re)sensitize or increase the responsiveness of cancer cells towards chemotherapeutics.
Open questions
-
How can Ca2+ signaling at ER–mitochondrial contact sites be modulated in a cancer-specific manner to fight cancer cell survival?
-
Can ER–mitochondrial Ca2+ signaling events overcome dysregulated cell survival/apoptosis sensitivity in cells with altered oncogene and/or tumor suppressor function?
-
What processes/regulation pathways underlie or control differences between ER–mitochondrial Ca2+ transfer in cancer cells vs. normal cells?
-
How can Ca2+-signaling modulation be applied to increase responsiveness and sensitivity to existing therapies and to induce cancer cell-specific cell death while sparing normal cells?
-
Can Ca2+ signaling be applied in a cancer stage-specific manner, thereby promoting cell death and avoiding metastasis?
-
What other molecular mechanisms, like the generation of ROS, exchange of lipids or alterations in protein composition, or ER–mitochondrial tethering at the MAMs impact or cooperate with Ca2+ signaling in anti-cancer chemotherapeutic actions?
Introduction: ER–mitochondrial Ca2+ signaling in cell death and survival
Mitochondria do not only fulfill the function of powerhouse of the cell but their function also encompasses more than merely providing the cell with ATP1,2. Currently, mitochondrial function has been implicated in apoptosis, autophagy, cell proliferation, cellular senescence, and migration3,4,5,6. Furthermore, mitochondrial function is impacted by the “state” of the mitochondrial network, which can range from highly connected to fragmented7. Nevertheless, mitochondria do not act as sole orchestrators of cellular processes. In fact, the mitochondrial network rather functions as a highly versatile signaling platform, closely connected to other cell organelles, like the endoplasmic reticulum (ER)8 and peroxisomes9. To allow for inter-organellar cross-talk, the different organelles are often located in close proximity to each other10,11, like the ER and the mitochondria, which are connected through mitochondria-associated ER membranes (MAMs). These MAMs are defined as ER membranes that are in close apposition (10–50 nm) to the mitochondria and were first isolated as a distinct entity in the early 1990s11,12,13. In recent years, MAMs were shown to contribute to various cellular functions like metabolism, autophagy, lipid synthesis but also cell survival and cell death8,14,15,16,17,18. In this sense, the MAMs, like mitochondria, are highly dynamic signaling hubs where signals from different cellular pathways converge and are integrated15,19,20,21.
One of the signals transferred between ER and mitochondria at the MAMs is the ubiquitous second messenger Ca2+22,23. While [Ca2+] in the cytosol is maintained at low levels under resting conditions, the bulk of intracellular Ca2+ is confined in the ER22. Ca2+ is predominantly released from the ER via the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R), which is gated by IP324, or the ryanodine receptor (RyR)25. However, Ca2+ accumulation into the mitochondrial matrix requires Ca2+ transport across the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM). At the OMM, Ca2+ transport is mediated via the high-conductance voltage-dependent anion channel 1 (VDAC1), while at the IMM, Ca2+ transport is mediated via the mitochondrial Ca2+ uniporter (MCU), the pore-forming unit in the MCU complex, consisting of MCU itself and its regulators26,27,28. For a detailed description of MCU regulation we would like to refer to refs. 29,30. The MAMs play an important role in mitochondrial Ca2+ uptake, since they provide a Ca2+ micro-domain, where Ca2+ levels are higher than in the bulk cytosol1,15. This is necessary to sustain ER–mitochondrial Ca2+ signaling since the MCU has a low affinity for Ca2+. Thus, the MAMs allow for efficient, ‘quasi-synaptic’ mitochondrial Ca2+ uptake upon ER Ca2+ release through the formation of a micro-domain1,15,31. This emphasizes the importance of the MAMs as a signaling hotspot.
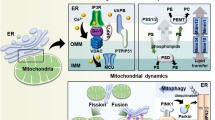
Mitochondrial Ca2+ signals are decoded differentially depending on their spatiotemporal characteristics (see Fig. 1). For example, cytosolic Ca2+ oscillations, efficiently transferred to the mitochondria through these contacts sites, drive mitochondrial metabolism. Moreover, several mechanisms account for the dynamic interplay between Ca2+ signals and mitochondrial metabolism. Ca2+ increases the activity of several rate-limiting enzymes of the tricarboxylic acid (TCA) cycle, including pyruvate, isocitrate, and α-ketoglutarate dehydrogenases32, while MCU transcription is controlled by the cAMP-responsive element binding protein, a Ca2+-dependent transcription factor33. Cells can also fine tune the level of Ca2+ oscillations that drive mitochondrial bioenergetics via a redox-nano-domain34. Mitochondrial Ca2+ uptake triggers K+- and H2O-influx into the mitochondrial matrix, leading to cristae compression and H2O2 release at the MAMs. This provides positive feedback on IP3Rs, enhancing their ability to sustain Ca2+ oscillations34. Other contributions of Ca2+ to cell metabolism are the stimulation of complex III of the electron transport chain, as well as stimulation of the ATP synthase and the adenine nucleotide translocase35.

Arrow-headed lines indicate a stimulatory or consequential effect. The ER is the main intracellular Ca2+ storage organelle. The release of Ca2+ from this organelle is mediated by the IP3R, gated by the intracellular messenger IP3. Ca2+ then travels via VDAC1, which is physically coupled to the IP3R through GRP75, and MCU to the mitochondrial matrix. Ca2+ oscillations targeted to the mitochondria are able to stimulate mitochondrial metabolism in several ways. Firstly, the TCA cycle has three rate-limiting enzymes that are regulated by Ca2+: pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase. Furthermore, both the ATP synthase and complex III of the electron transport chain (ETC) are stimulated by Ca2+. In addition, the adenine nucleotide translocase (ANT) is activated as well. Interestingly, positive feedback mechanisms exist to ensure Ca2+ feeding into the mitochondria. One of these mechanisms is dependent on a redox-nano-domain at the MAMs: Ca2+ influx into the mitochondrial matrix activates Ca2+-activated K+ channels and parallel H2O uptake in the mitochondria. This results in cristae compression (indicated by the red arrows) and H2O2 extrusion, which stimulates IP3R activity. In short, by stimulating cellular metabolism, Ca2+ oscillations contribute to cell survival. However, excessive Ca2+ uptake in the mitochondria causes mitochondrial Ca2+ overload. This results in opening of the mPTP, either by a direct action of Ca2+ on the mPTP or by Ca2+ binding to cardiolipin, thereby disrupting complex II of the ETC and subsequent ROS production. mPTP opening leads to mitochondrial swelling, rupture of the OMM, and release of pro-apoptotic factors like cytochrome c and ultimately apoptosis
Ca2+ oscillations in the cytosol can also modify mitochondrial metabolism indirectly by activating ARALAR, a mitochondrial glutamate/aspartate transporter playing a central role in the malate/aspartate shuttle, which is strongly dependent on cytosolic Ca2+ signaling36. Ca2+ binds to ARALAR and activates ARALAR-mediated glutamate and NAD(P)H transport into the mitochondria36,37,38. Also, pyruvate production is Ca2+ dependent and is linked to the malate/aspartate shuttle, providing pyruvate to the mitochondria39,40. In addition, there is tissue-specific regulation, which determines the threshold for mitochondrial Ca2+ influx and its effects41. Liver cells and cardiomyocytes contain a high and a low ratio of mitochondrial Ca2+ uptake 1 (MICU1)/MCU, respectively, with MICU1 (together with MICU2) being an MCU regulator which affects the cooperativity of mitochondrial Ca2+ uptake42,43. Therefore liver cells display a high cooperativity of mitochondrial Ca2+ uptake, while cardiac cells display a low cooperativity. This prevents the occurrence of mitochondrial Ca2+ signals in response to short-lasting Ca2+ transients and thus avoids mitochondrial Ca2+ overload even when the heart beats at high frequency41. Recently, it was shown that apart from affecting the cooperativity of mitochondrial Ca2+ uptake, MICU1, together with its paralog MICU2, inhibits the MCU at cytosolic Ca2+ concentrations lower than ~600 nm, thereby determining the relatively high threshold for MCU-mediated Ca2+ uptake44. Compared to wild-type cells, loss of MICU1, which also results in loss of MICU2, lowers the threshold for MCU-mediated mitochondrial Ca2+ uptake to about 200 nM Ca2+. Compared to mitochondria lacking both MICU1 and MICU2, mitochondria containing MICU1 but not MICU2 display a higher threshold for mitochondrial Ca2+ uptake (~350 nM Ca2+), indicating that MICU1 by itself can inhibit MCU. These observations indicate that MICU1 does not only control MCU cooperativity43, but can also function as a gatekeeper of MCU44. Moreover, MICU2 requires MICU1 to regulate MCU. MICU1’s function as a gatekeeper is also important in vivo to prevent mitochondrial Ca2+ overload, as evidenced in MICU1-knockout animals, developing ataxia and muscle fatigue associated with elevated mitochondrial Ca2+ levels and reduced ATP levels45. Finally, MICU1 and MICU2 were shown to bind the mitochondrial lipid cardiolipin, facilitating membrane anchoring of the complex and the fine-tuned Ca2+-dependent regulation of the MCU by MICU1 and associated factors, like EMRE44.
On the other hand, massive Ca2+ release from the ER causes mitochondrial Ca2+ overload, resulting in opening of the mitochondrial permeability transition pore (mPTP)46,47, mitochondrial swelling, and subsequent cell death (Fig. 1)48,49,50,51. Several mechanisms may account for this. For instance, mitochondrial Ca2+ influx results in Ca2+ binding to cardiolipin, causing the disintegration of complex II and the release of the functionally active catalytic subunits in the mitochondrial matrix, providing a source of reactive oxygen species (ROS) that triggers the opening of the mPTP52. In addition to this, recent evidence emerged that mitochondrial Ca2+ could directly target the mPTP, resulting in a conformational change leading to its opening and to subsequent mitochondrial swelling53.
Regulation of ER–mitochondrial Ca2+ signaling at the MAMs
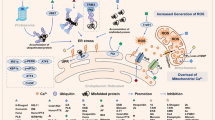
Given its critical role in cell fate and survival, the composition and functional properties of the MAMs have to be carefully regulated and controlled. This occurs through a distinct set of proteins with a variety of cell biological properties and functions. Some of these proteins are directly involved in Ca2+ signaling at the MAMs, e.g. the IP3R and VDACs49,54,55, whereas others alter ER–mitochondrial Ca2+ signaling by acting through modulation of these Ca2+-transport systems, e.g. promyelocytic leukemia protein (PML)56 or phosphatase and tensin homolog (PTEN)57. Other proteins modify the characteristics of the MAMs (e.g. their distance to the mitochondria), like the ER–mitochondria tethers mitofusin-2 (Mfn-2)58,59, the protein kinase RNA-like endoplasmic reticulum kinase (PERK)60, and the spacer protein fetal and adult testis expressed 1 (FATE1)61. We will focus on the proteins involved in ER–mitochondrial Ca2+ transfer at the MAMs, yet for a more extensive list of proteins present at the MAMs, we refer to ref. 13. For further insights on the role of Ca2+-transport systems in cell death and survival, we refer to ref. 62. All the MAM components discussed in this section are schematically depicted in Fig. 2.

Arrow-headed full lines indicate a stimulatory effect, while bar-headed full lines indicate an inhibitory effect. Dot-headed lines indicate a tethering or a spacing effect, depending on the inward- or outward-facing dots, respectively. ER–mitochondrial Ca2+ transfer at the MAMs can be altered by different fine-tuning mechanisms. These mechanisms often involve oncogenes and tumor suppressors. An important tool to regulate the activity of the Ca2+-signaling proteins at the MAMs is phosphorylation. Akt/PKB-mediated phosphorylation is known to suppress Ca2+ release via the IP3R. However, this phosphorylation is counteracted by both PTEN, in a direct way, and PP2A, in an indirect way. This indirect mechanism consists of recruitment of PP2A to the IP3R via PML. In turn, PP2A deactivates Akt/PKB by dephosphorylation and this relieves IP3R inhibition. In addition, Ca2+-signaling proteins like IP3R, VDAC1, and SERCA interact with other proteins, which change their Ca2+-signaling properties. Bcl-2-protein family members are among these proteins altering Ca2+ signals at the MAMs. Bcl-2 binds to the IP3R, inhibiting pro-apoptotic Ca2+ signaling, while Bcl-Xl interacts with VDAC1, inhibiting Ca2+ uptake through the OMM. p53, a master regulator of cell fate, on the other hand, interacts with SERCA, changing its oxidation state, thereby enhancing reuptake of Ca2+ in the ER. A third category of proteins that modifies Ca2+ signaling at the MAMs are those proteins that change the properties of the MAMs, e.g. ER-to-mitochondria distance at the MAMs. Mfn-2 in the ER is able to interact with Mfn-1 or Mfn-2 at the OMM, thereby tethering both organelles. On the other hand, there is FATE1 which has the opposite effect, namely spacing the mitochondria and the ER. The distance between ER and mitochondria at the MAMs is determined by these tethers and spacers and this, in turn, regulates the efficiency of ER–mitochondrial Ca2+ transfer
Since ER–mitochondrial Ca2+ transfer occurs at the MAMs, it is not surprising that several intracellular Ca2+-transport systems reside at the MAMs. Both the IP3R and VDAC1 can be found at the MAMs, where they are connected to each other via glucose-related protein of 75 kDa (GRP75)55. Interestingly, isoform-specific functions for these channels at the MAMs have been discovered. Overexpression of VDAC1, VDAC2, as well as VDAC3 increased mitochondrial Ca2+ uptake, yet pro-apoptotic Ca2+ signals were only enhanced by VDAC1 overexpression63. Moreover, VDAC1 is the only VDAC isoform that co-immunoprecipitated with the IP3R63. Similarly, IP3R3 seems to be the isoform that preferentially transmits pro-apoptotic Ca2+ signals to the mitochondria via the MAMs64,65. Not only Ca2+-release channels are located at the MAMs, the sarco-/endoplasmic reticulum Ca2+ ATPase (SERCA) pump localizes to the MAMs as well66,67,68. SERCA pumps can influence the properties of the MAMs since their activity determines how fast Ca2+ is cleared from the micro-domain69. Moreover, SERCA pumps critically affect apoptotic sensitivity by controlling the steady-state filling level of the ER Ca2+ stores, which is determined by the balance between ER Ca2+ leak and ER Ca2+ uptake66,69,70,71,72. Also for SERCA, isoform-specific functions exist: ER stress causes induction of SERCA1T, a truncated splice-variant of SERCA1. SERCA1T is a determinant of ER leakiness, thereby promoting Ca2+ transfer to the mitochondria and thus supporting pro-apoptotic Ca2+ signaling73,74.
As will be discussed further on, expression levels of the Ca2+-signaling proteins critically determine the cell’s Ca2+-signaling properties. Furthermore, ER–mitochondrial Ca2+ signaling is fine-tuned by various oncogenes and tumor suppressors75,76. These proteins may induce post-translational modifications that alter the Ca2+-signaling properties of proteins at the MAMs77. Particularly, phosphorylation of the IP3R is a critical factor: Akt/protein kinase B (PKB) suppresses IP3R-mediated Ca2+ release through phosphorylation78, while tumor suppressors PTEN (direct dephosphorylation of IP3R)57 and PML (indirect dephosphorylation via sequestration of protein phosphatase 2A (PP2A) and subsequent Akt/PKB inhibition)56 counteract this. Also, SERCA is a target for post-translational modification: p53 changes SERCA’s oxidative state, promoting its ER Ca2+-uptake activity and thus altering the net flux of Ca2+ released from the ER66,79,80.
In addition, complex formation between Ca2+-transport proteins like IP3Rs and VDAC1 at the MAMs and tumor suppressors or oncogenes influences ER–mitochondrial Ca2+ transfer. Notably, several Bcl-2-protein family members, critical regulators of apoptosis, were shown to be present at the MAMs81,82 and can modify IP3R-mediated Ca2+-release77,83,84. Bcl-2, an anti-apoptotic protein of the Bcl-2-protein family, inhibits the IP3R directly by binding with its Bcl-2 homology (BH) 4 domain to the central, modulatory domain of the IP3R85,86,87. The BH4 domain also participates in overall stability of the Bcl-2 protein, affecting its IP3R-inhibitory properties88. Bcl-2’s transmembrane domain is necessary for efficient in cellulo IP3R inhibition89. Indirectly, Bcl-2 changes IP3R activity by providing a docking place at the IP3R for protein phosphatase 1 (PP1), which inhibits IP3R function by dephosphorylating the receptor90. Bcl-2 also regulates IP3R function by docking dopamine- and cAMP-regulated phosphoprotein of 32 kDa, a PP1 inhibitor, and the protein phosphatase calcineurin in a complex on the IP3R91. Thus, IP3R activity is regulated by a negative feedback mechanism that prevents excessive, pro-apoptotic Ca2+ release from the ER. Besides Bcl-2, also Bcl-Xl and Mcl-1 can modulate IP3R activity92,93,94. An extended discussion on the modulation of Ca2+ signaling by Bcl-2-protein family members can be found elsewhere82,95.
Besides the IP3Rs, Bcl-2-family members can also target mitochondrial Ca2+-transport systems, including VDAC1. VDAC1/Bcl-2 complex formation inhibits VDAC1’s function in mitochondrial Ca2+ transport96,97. The BH4 domain of Bcl-2 appeared to play a critical role in VDAC1 regulation98. Follow-up work revealed that the BH4 domain of Bcl-Xl is more effective in targeting and modulating VDAC1 than the BH4 domain of Bcl-281. Consequently, while both the BH4 domains of Bcl-2 and Bcl-Xl could prevent mitochondrial Ca2+ uptake, BH4-Bcl-2 acted at the level of IP3Rs, while BH4-Bcl-Xl acted at the level of VDAC181. To summarize, ER–mitochondrial Ca2+ transfer at the MAMs can be influenced through IP3R-mediated Ca2+ release, VDAC1-mediated mitochondrial Ca2+ uptake, or modulation of the SERCA activity.
In addition to this, ER–mitochondrial Ca2+ transfer is altered by the number of MAMs and the distance between the ER and the OMM at the MAMs99,100. Proteins that hold both membranes together are typically called tethers. An example of a tether at the MAMs is the GTPase Mfn-2 involved in mitochondrial fusion. Mfn-2 tethers ER and mitochondria through homo- and heterotypic interactions with Mfn-2 and Mfn-1, respectively59,101. The importance of tethering was shown in Mfn-2-knockout cells, where ER–mitochondrial Ca2+ transfer was severely reduced101. Yet, the role of Mfn-2 as a tether has been debated, since another study showed that ablation of Mfn-2 does not impair the ER–mitochondrial connection but contrarily, tightens it58,101. Here, Mfn-2 was proposed as a spacer that increases the distance at the MAMs and reduces ER–mitochondrial signaling. A typical spacer is FATE1, a cancer testis antigen, which is normally only expressed in the testis61. However, in certain cancers, FATE1 becomes upregulated and causes MAMs alterations61.
Rewiring Ca2+ signaling in cancer cells
The different possible layers of regulation of Ca2+ signaling impart an enormous flexibility to the cell concerning fine-tuning cellular processes in response to internal and external stimuli. However, this sensitive system can be hijacked to drive malignant transformations in the cell6. It is known that several types of cancer cells undergo an extensive rewiring of their Ca2+-signaling machinery, favoring oncogenesis6,102,103. At the level of the ER and the mitochondria, expression levels of Ca2+-signaling proteins, including VDAC1, IP3R, and SERCA, are often altered in cancer cells. For instance, VDAC1 expression levels are correlated with tumor growth and invasion in several types of cancer, e.g. non-small cell lung cancer and cervical cancer104,105. In this regard, recently, genetic disruption of VDAC1 in cells from cancer xenograft models displayed decreased mitochondrial membrane potential and ATP content with a consequent low migration rate and tumor regression106,107. Another example includes IP3R1 downregulation in bladder cancer cells, which attenuates cisplatin-mediated apoptosis through a decrease in ER–mitochondrial Ca2+ uptake, preventing mitochondrial Ca2+ overload108,109. Remodeling of Ca2+ signaling in tumorigenesis is also documented by the significant reduction or loss of the SERCA3 isoform in transformed colonic epithelial cells110. Different mechanisms may be responsible for the change in expression levels. Recently, two novel mechanisms dysregulated in several cancer types have been discovered to impact the proteasomal turnover and thus stead-state expression levels of IP3R3 and the apoptotic sensitivity of cells111. (i) The tumor suppressor PTEN competes with F-box/LRR-repeat protein 2 (FBXL-2), an E3 ubiquitin ligase component belonging to the Skip-Cullin1-F-box protein family112, for binding to IP3R3, thereby slowing down FBXL-2-mediated proteasomal degradation of IP3R3113. This represents a novel mechanism by which loss of PTEN allows cancer cells to evade apoptosis, since pro-apoptotic mitochondrial Ca2+ transfer becomes impaired due to downregulation of the IP3R3. (ii) The tumor suppressor BRCA1-associated protein 1 (BAP1), a deubiquitylating enzyme, promotes ER–mitochondrial Ca2+ transfer by stabilizing the IP3R3114. BAP1 function is particularly impaired during prolonged environmental stress, associated with acquired inactivating mutations in BAP1 genes. Loss of BAP1 results in IP3R3 downregulation, hampering the effective apoptotic clearance of damaged cells and favoring oncogenesis and malignant cell survival.
While MCU is not residing at the MAMs, its expression can be controlled in a tumor-specific manner, e.g. via microRNAs (miR)115. As such, miR-25, targeting MCU, was overexpressed in colon cancer cell lines and tumor samples, decreasing MCU expression compared to non-tumorigenic cells. Moreover, miR-25 overexpression in HeLa cells reduced mitochondrial Ca2+ accumulation, resulting in apoptosis resistance. In contrast, antagonizing miR-25 expression using antagomirs re-sensitized colon cancer cells to Ca2+-dependent apoptotic stimuli, like H2O2 and ceramide. Interestingly, MCU may prevent tumor cell survival in an early stage, but can become a pro-malignant factor in late-stage tumors, like triple negative breast cancer cells116. These cells express high levels of MCU, correlating with tumor size and lymph node infiltration, which negatively impact survival outcome. The mechanisms involved MCU-dependent uptake of Ca2+ into the mitochondrial matrix and subsequent generation of ROS that stabilized hypoxia-inducible factor-1α, a transcription factor driving the expression of genes involved in cancer migration and invasion116,117.
Furthermore, oncogenes like Akt/PKB and FATE1, and tumor suppressors like PML and PTEN, can play additional roles in the development of cancer via Ca2+-signaling modulation108. A striking example of this is apoptotic resistance. Since mitochondrial Ca2+ overload is involved in apoptotic cell death, modifying ER–mitochondrial Ca2+ transfer at the MAMs alters apoptotic sensitivity48. Cancer cells can gain resistance against cell death, e.g. by overexpressing proteins that suppress IP3R-mediated Ca2+ signaling, like Akt78, or by increasing the intermembrane distance at the MAMs (e.g. FATE1), thereby rendering ER–mitochondrial Ca2+ transfer less efficient61. These mechanisms are not only supporting basic cancer cell characteristics, but also underlie resistance against chemotherapy. Cell death induction strategies still play a central role in the fight against cancer. While selective oncogene inhibitors, like venetoclax/ABT-199 for Bcl-2 inhibition, emerged and entered the clinic118, chemotherapy remains a very effective way to eradicate tumor cells by triggering cell death119.
Besides ER–mitochondrial Ca2+ signaling, Ca2+ fluxes across the plasma membrane can affect cancer properties as well. We would like to refer to other excellent reviews regarding this topic120,121,122, as in this section, we would like to focus on chemotherapeutic drugs that act on Ca2+ signaling at the MAMs.
Chemotherapy and Ca2+ signaling in cancer cells
Ca2+ signaling appears to be a major contributor to the cytotoxic effects of chemotherapy. Many chemotherapeutic agents trigger a rapid onset of cytosolic [Ca2+] rises123. Furthermore, shifts in cytosolic [Ca2+] have been proposed as early markers for cytotoxicity in cells in response not only to H2O2 or staurosporine but also to chemotherapeutics like gossypol or arsenic trioxide (ATO)123. The mechanism of these early cytosolic [Ca2+] elevations is not always fully understood, but may in part depend on the presence of p53. Upon chemotherapeutic treatment, extra-nuclear p53 can accumulate at the ER membranes where it binds SERCA and activates its ER Ca2+-uptake activity66,79, 80,124,125. Thus, SERCA activation will augment the ER Ca2+-store content, overfilling the ER with Ca2+ and thus increasing the likelihood for spontaneous ER Ca2+-release events126. However, the occurrence of shifts in cytosolic [Ca2+] may not be a general phenomenon, as on-target Bcl-2 inhibitors, like ABT-737127 and venetoclax/ABT-199128, do not trigger these early cytosolic [Ca2+] elevations, even not in cancer cells that are dependent on Bcl-2 for their survival. The reason for these varying responses are not fully understood, but may actually relate to the mechanism of action of the drug applied and in particular whether p53 is involved.
Here, we will discuss the chemotherapeutic agents that act via ER–mitochondrial Ca2+ signaling. These chemotherapeutics are summarized in Table 1, whereas a schematic representation of their function at the MAMs is provided in Fig. 3.

Arrow-headed full lines indicate a stimulatory effect, while bar-headed full lines indicate an inhibitory effect. Dashed lines indicate an indirect effect. Dot-headed lines indicate a tethering or a spacing effect, depending on the inward- or outward-facing dots, respectively. This figure constitutes an overview of the chemotherapeutics discussed in this review and how they act on the MAMs. Resveratrol is an inhibitor of the ATP synthase. This inhibitory effect leads to less ATP being available for SERCA pumps to ensure rapid reuptake of Ca2+ in the ER, creating a high local Ca2+ concentration. This, together with an increased ER–mitochondrial tethering, underlies the cancer cell-specific killing by resveratrol. ATO, another chemotherapeutic agent, increases PML levels at the MAMs. This restores ER–mitochondrial Ca2+ transfer in cancer cells that have a decreased or impaired PML activity and consequently suppresses pro-survival autophagic flux. A third chemotherapeutic drug, cisplatin, covalently binds to DNA, inducing DNA damage and causing apoptotic cell death, involving ER–mitochondrial Ca2+ signaling. However, cancer cells overexpressing Bcl-2 are more resistant to cisplatin-induced cell death, seemingly via a dual mechanism: the inhibition of Ca2+ release from the ER and the inhibition of an increase in ER–mitochondrial contact points resulting from cisplatin treatment. In this sense, administering ABT-737, a Bcl-2-inhibiting BH3 mimetic, to cancer cells, restored sensitivity to cisplatin. Furthermore, there is adriamycin, which renders ER–mitochondrial Ca2+ transfer more efficient by enriching p53 at the SERCA pumps. This leads to an increased activity of SERCA, increasing the ER Ca2+ levels and sensitizing cells towards apoptosis. Lastly, mitotane is an inhibitor of SOAT1, resulting in increased free cholesterol and lipid-induced ER stress. However, sensitivity towards mitotane is dependent on the expression levels of FATE1, a spacer protein at the MAMs. Increased levels of FATE1 are responsible for decreased mitochondrial Ca2+ uptake and in this way render cancer cells less sensitive towards apoptosis
Arsenic trioxide
Acute promyelocytic leukemia (APL) is almost always characterized by a t(15;17) chromosomal translocation, resulting in a PML/retinoic acid receptor (RAR) α fusion protein that hinders the differentiation of hematopoietic cells by inhibiting gene transcription129,130. APL patients are mostly treated by a combination of all-trans retinoic acid and ATO therapy, which stimulates APL cell differentiation by triggering proteasomal degradation of the PML/RARα fusion protein129,131,132,133. This treatment approach results in high APL cure rates, reflected by high complete remission and overall survival percentages129,132.
ATO also influences ER–mitochondrial Ca2+ signaling, thereby repressing autophagy in cancer cells17. Autophagy is an important pro-survival pathway in malignant cells that experience oncogenic stress, as missing nutrients can be delivered to the cells via this process17,134. The tumor suppressor PML, which is localized at the MAMs, represses autophagy by promoting ER–mitochondrial Ca2+ transfer and mitochondrial respiration17. Hence, PML is often downregulated in cancer cells135. Loss of PML results in reduced IP3R-mediated Ca2+ transfer from the ER to the mitochondria, leading to decreased mitochondrial Ca2+ levels, thereby diminishing mitochondrial respiration and ATP production17. This triggers activation of AMP-activated protein kinase, which stimulates pro-survival autophagy by a mechanism involving mechanistic target of rapamycin and unc-51-like kinase 1 pathways136,137. Interestingly, this autophagic process is repressed in APL cells treated with ATO17. Short-term treatment of these cells with ATO promotes the selective degradation of the PML/RARα fusion protein, but not of PML. Furthermore, exposure to ATO increases the PML levels at ER–mitochondria contact sites in a p53-dependent manner (Fig. 3). Besides a block in autophagy, ATO-treated cells also displayed reduced resistance to metabolic stress17. Furthermore, this study implicates that the response of tumor cells characterized by loss of PML to chemotherapeutic agents can be improved by inhibiting autophagy.
Cisplatin
Cisplatin is a chemotherapeutic agent used to treat numerous human cancers, including lung, ovarian, head and neck, bladder, and testicular cancer138. The anticancer activity of this platinum-based drug has been linked to its ability to covalently bind purine residues on DNA138,139. This interaction causes DNA damage, interferes with DNA repair mechanisms, and blocks cell division, ultimately leading to apoptotic cell death. Unfortunately, cisplatin treatment has been associated with considerable side effects, like cardiotoxicity, hepatotoxicity, nephrotoxicity, and toxicity of other organs, and drug resistance is often acquired during therapy as well138. To overcome these obstacles, combination therapies of cisplatin with other anti-cancer drugs, including paclitaxel, doxorubicin, and gemcitabine, form the basis for treatment of many human cancers138.
Interestingly, the ER–mitochondrial Ca2+ signaling pathway contributes to cisplatin-induced cell death. Treatment of SKOV3 human ovarian cancer cells with cisplatin increased the number of ER–mitochondria contact sites, causing a Ca2+ flow from the ER to the mitochondria (Fig. 3)140. This resulted in high mitochondrial Ca2+ levels, which triggered apoptosis in the cisplatin-treated ovarian cancer cells. Furthermore, the expression level of the anti-apoptotic protein Bcl-2, which is overexpressed in many tumors and drives tumorigenesis and chemoresistance, seems to be a determinant for the sensitivity of cancer cells to cisplatin (Fig. 3). In non-small cell lung cancer and bladder cancer, cisplatin sensitivity could be enhanced by downregulating Bcl-2141,142. In addition, downregulation of Bcl-2 in SKOV3 cells with siRNA increased the cytoplasmic and mitochondrial Ca2+ levels as well as the number of ER–mitochondria contact points after cisplatin treatment, thereby increasing the sensitivity to the chemotherapeutic agent143. Hence, Bcl-2 seems to form a potential therapeutic target to improve cisplatin therapy of ovarian cancer cells. Additionally, in neuroblastoma cells, cisplatin-induced cell death was preceded by a rise in cytosolic [Ca2+]144. Cisplatin treatment also increased the expression levels of several Ca2+-transport systems, including IP3R3, RyR3, and the S100 Ca2+-binding protein A6. Therefore, cisplatin-induced cell death can be enhanced by pharmacological modulators of Ca2+-regulatory proteins, like the SERCA inhibitor thapsigargin144.
BH3 mimetics
BH3 mimetics are a class of anti-cancer drugs inhibiting the function of anti-apoptotic Bcl-2-protein family members like Bcl-2, Bcl-Xl, and Mcl-1145,146,147,148. This causes pro-apoptotic Bcl-2-family members, which are sequestered by their anti-apoptotic counterparts through a hydrophobic cleft, consisting of the BH1, BH2, and BH3 domain, to be released, resulting in apoptosis145,146. This is an effective anti-cancer therapy in cancers that rely on an upregulation of the anti-apoptotic Bcl-2-family proteins for their survival. Several molecules have been developed as BH3 mimetic drug, notably ABT-737, which targets the hydrophobic cleft of both Bcl-2 and Bcl-Xl, its orally available analog ABT-236 (navitoclax) and ABT-199 (venetoclax), which solely targets the hydrophobic cleft of Bcl-2145. While these drugs are specifically developed to suppress the canonical, anti-apoptotic function of the Bcl-2-protein family members at the mitochondria, it seems that their intracellular effects are more complex.
Thrombocytopenia is an important side effect caused by ABT-737 and ABT-263, since these BH3 mimetic drugs inhibit the function of Bcl-Xl, which is essential for platelet formation and survival149,150,151. Dysregulation of intracellular Ca2+ homeostasis might underlie ABT-737- and ABT-263-induced thrombocytopenia, as addition of ABT-263 to platelets triggered an acute rise in cytosolic Ca2+ levels149. However, a direct link between deranged Ca2+ signaling and platelet dysfunction was not provided since this effect was only observed upon addition of relatively high ABT-263 concentrations (10 µM), whereas platelet function was already decreased at much lower concentrations (100 nM–1 µM)149. Furthermore, prolonged treatment of platelets with ABT-263 and ABT-737 depleted the intracellular Ca2+-storage organelles149,152. However, it was not clear whether the effects on Ca2+ signaling were the cause of platelet apoptosis or whether they were the consequence of platelets being in late-stage apoptosis due to Bcl-Xl inhibition. Our lab excluded a direct impact of ABT-737 on intracellular Ca2+ signaling, since ABT-737 application did not affect thrombin-induced Ca2+ signaling in platelets nor ATP-induced Ca2+ signaling in HeLa cells127. Moreover, SERCA activity and IP3R-mediated Ca2+ release were unaffected by ABT-737. These results argue against a proximal role of Ca2+-signaling dysregulation in platelet dysfunction and apoptosis induced by ABT-737.
On the other hand, for ABT-199/venetoclax, which only targets the hydrophobic cleft of Bcl-2, no evidence was found for a perturbation of Ca2+ homeostasis128,148,153. In several human and mouse cell models, ABT-199 did not trigger cytosolic Ca2+ release events by itself nor did it affect agonist-induced IP3R-mediated Ca2+ signaling128. Also, clearance of Ca2+ from the cytosol after agonist application was not affected. Furthermore, ABT-199 did not interfere with the inhibition of IP3R caused by overexpression of Bcl-2128. Nevertheless, it seems that there is an interplay between ABT-199-induced cell death in Bcl-2-dependent cancer cells and basal Ca2+ signaling, since chelating intracellular Ca2+ using BAPTA-AM enhanced ABT-199-induced cell death128. The mechanisms underlying this phenomenon remain unclear, but might be due to downregulation of anti-apoptotic Bcl-2-family members or upregulation of pro-apoptotic Bcl-2-family members. Furthermore, Bcl-2-dependent cancer cells can be sensitized towards ABT-199 by the application of BIRD-2154, a BH4-domain inhibitor of Bcl-2 that triggers toxic Ca2+-release events and apoptosis in various cancer cells, including chronic lymphatic leukemia, diffuse large B-cell lymphoma, multiple myeloma, follicular lymphoma, and lung cancer cells86,154,155,156. Further, it appears that BIRD-2 upregulates Bim in a Ca2+-dependent manner, thereby likely accounting for an increased sensitivity of the cells towards BH3 mimetics like ABT-199 application154,157. As such, BIRD-2 and ABT-199 can act synergistically to trigger cell death in Bcl-2-dependent cancers. Finally, it appears that cancer cells that are less sensitive to BH3 mimetics are more sensitive to BIRD-2 and vice versa154,156,157.
Interestingly, ABT-737 can enhance the chemotherapeutic effectivity of cisplatin in cholangiocarcinoma (CC) as well as in ovarian cancer cells143,158. In the latter, ABT-737 treatment increased the cisplatin-induced growth inhibition and apoptosis in cisplatin-resistant cells, hence restoring their sensitivity to the chemotherapeutic agent143. Combination therapy with cisplatin and ABT-737 of the cisplatin-resistant ovarian cancer cells increased the number of ER–mitochondria contact sites induced by cisplatin, stimulating Ca2+ transfer from the ER to the cytosol and the mitochondria (Fig. 3). Hence, ABT-737 can reverse cisplatin resistance of ovarian cancer cells by enhancing ER-associated and mitochondria-mediated apoptosis143. Furthermore, ABT-737 sensitized CC cells to cisplatin therapy by regulating mitochondrial dynamics158. ABT-737 stimulated CC cells to undergo apoptosis after cisplatin treatment by promoting mitochondrial fission and inducing mitophagy. Therefore, ABT-737 combined with cisplatin might be an effective strategy to treat CC patients158. This also provokes the question in what manner Bcl-2-protein family members regulate the dynamics of the MAMs in cancer.
Of note, earlier versions of Bcl-2-inhibiting molecules can dysregulate intracellular Ca2+ homeostasis. HA14-1 and stabilized HA14-1s can deplete ER Ca2+ stores by inhibiting the SERCA Ca2+ pump127,159. In a separate study, both HA14-1 and BH3I-2′ trigger Ca2+ release from the ER of pancreatic acinar cells through IP3R- and RyR-mediated mechanisms, elevating cytosolic Ca2+ levels, a feature that contributes to their cell death properties160. Here, it was proposed that dissociation of Bax from Bcl-2 using these drugs sensitize IP3Rs and RyRs to cytosolic Ca2+. Excitingly, the ability of HA14-1 and BH3I-2′ to increase cytosolic Ca2+ levels in pancreatic acinar cells was strictly dependent on the presence of Bax, while the presence of Bcl-2 and Bak was not critical for this process. It was proposed that Bax, released from Bcl-2, can induce Ca2+ leak from the ER, either by itself or by acting on ER Ca2+-leak channels like IP3Rs161. Yet, further work using selective Bcl-2 inhibitors is needed to validate this model. A detailed discussion on the role of BH3 mimetics and Ca2+ signaling is provided elsewhere148,162.
Resveratrol
Resveratrol is a natural polyphenol produced in response to stressful conditions by various plant species. It is found in several foodstuffs, including grapes, mulberries, and peanuts, and has been attributed beneficial health effects since the early 1990s163,164,165. It is known as a multi-target agent exhibiting antioxidant, anti-inflammatory, and immunomodulatory activities. This pleiotropic compound affects cell proliferation, differentiation, apoptosis, and autophagy and attenuates many age-related chronic complications, such as metabolic, cardiovascular, and neurodegenerative diseases164,165. Furthermore, resveratrol has been used as a chemopreventive and chemotherapeutic agent in many types of cancer163,164,165. For instance, it has been used in clinical trials conducted in patients with colon, colorectal, and gastrointestinal cancers as well as in a trial examining the effects of resveratrol in the prevention of cancer in healthy participants163. Remarkably, resveratrol functions as a specific anticancer agent with limited toxicity in normal cells163. However, the exact mechanism by which resveratrol specifically kills cancer cells remains unclear.
Recently, it was suggested that the difference in ATP demand between cancer and somatic cells underlies the cancer cell-specific toxicity of resveratrol166. Cancer cells are characterized by a very high ATP demand at the ER because of immense protein folding activities going on at this organelle167,168. Therefore, tethering of the ER and the mitochondria, which produce ATP via the ATP synthase localized in the IMM, is strongly enriched in cancer cells, warranting high ATP levels in the proximity of the ER169,170. However, resveratrol, which acts as an inhibitor of the F1 subunit of the ATP synthase171,172,173,174 exploits the enhanced ER–mitochondria coupling in malign cells to kill these cells exclusively (Fig. 3). As a result of ATP synthase inhibition in resveratrol-treated cells, ATP formation is reduced, by which the high energy demand of cancer cells is not met anymore166. As a consequence of the reduced ATP content at the mitochondria, SERCA activity within the MAMs is decreased, hampering Ca2+ reuptake into the ER, provoking not only an accumulation of Ca2+ in the micro-domain between ER and mitochondria166, but also leading to a depletion of the ER via the Ca2+-leak channels175. Both phenomena result in a high local Ca2+ concentration and because of the enforced ER–mitochondria coupling in cancer cells, mitochondrial Ca2+ accumulation is consequently enhanced upon treatment with resveratrol. Due to the fact that cancer cells are more sensitive to ATP synthase inhibition than healthy cells, resveratrol will especially trigger apoptotic cell death via mitochondrial Ca2+ overload in those cells166.
Adriamycin
Adriamycin, also known as doxorubicin, is an anthracycline-type drug that has been used in cancer therapy for many years176,177. It is characterized by a broad-spectrum antineoplastic activity, although its mechanism of action is complex. Adriamycin inhibits topoisomerase II, intercalates in DNA, and generates free radicals, hence inhibiting biosynthesis of macromolecules and leading to oxidative stress177,178,179. Ultimately, adriamycin treatment results in apoptotic cell death. This chemotherapeutic agent is commonly used to treat various types of cancer, including breast, ovarian, bladder, stomach, and lung cancer177. However, the applicability of adriamycin as anticancer therapy is restricted due to its severe toxic effects in healthy tissues176,177,179. Especially cardiotoxicity forms a major concern during adriamycin therapy, limiting the dose that can be administered to patients177,180. To circumvent the adverse effects of adriamycin, several drug delivery systems have been used176,177,179. For instance, adriamycin-induced toxicity is decreased when using liposomal, nanoparticle, or hydrogel drug formulations.
Adriamycin also renders cells more prone to programmed cell death by influencing the ER–mitochondrial Ca2+ signaling axis66,79,181. The tumor suppressor p53, an important transcription factor regulating DNA repair, cell-cycle arrest, and apoptosis, modulates Ca2+ homeostasis by stimulating the SERCA pump located at the ER and the MAMs66. As a consequence, Ca2+ accumulation in the ER is enhanced. Interestingly, treatment of cancer cells with adriamycin caused an enrichment of p53 at the ER and the MAMs (Fig. 3)66,79. This induction of p53 by adriamycin led to higher ER Ca2+ levels and higher cytosolic and mitochondrial [Ca2+] increases evoked by agonist stimulation66. Moreover, Ca2+ transport from the ER to the mitochondria was increased by adriamycin, allowing apoptotic stimuli to rapidly overload the mitochondria with Ca2+, resulting in apoptotic cell death. Hence, chemotherapeutic agents like adriamycin boost toxic ER–mitochondrial Ca2+ signaling through modulation of the Ca2+ homeostasis at the MAMs, thereby triggering apoptotic cell death in cancer cells66,79.
Another way in which Ca2+ signaling is able to contribute to the sensitivity of cancer cells to chemotherapeutics, is the induction of autophagy182. Recently, valproic acid was found to reduce the intracellular availability of IP3, hence blocking Ca2+ transfer to the mitochondria and altering the AMP-activated protein kinase 1/2–mechanistic target of rapamycin pathway. This lack of ER–mitochondrial signaling induced autophagy, thereby sensitizing cancer cells to adriamycin182.
Lipid-interfering strategies
A recent insight in tumor biology is the occurrence of dysregulation of lipid metabolism in cancer cells and its importance to several aspects of cancer cell function and survival. Consequently, disrupting or altering lipid homeostasis in cancer cells might be an efficient way of inducing cell death183. One of the ways to induce apoptosis in this manner in cancer cells is the use of chemotherapeutics that increase intracellular, free cholesterol183. Alkyl phospholipids were observed to hinder cholesterol transport from the mitochondria to the ER, avoiding its esterification, whereas cholesterol synthesis and incorporation was increased at the same time in glioblastoma cells184. In macrophages it was found that this free cholesterol accumulates at the ER, inhibiting SERCA pumps and causing depletion of the ER. This results in an increased transfer of Ca2+ to the mitochondria and subsequent apoptosis185.
Another chemotherapeutic drug that acts on lipid metabolism is mitotane, a derivative of the insecticide dichlorodiphenyl-trichloroethane. Mitotane is the only chemotherapeutic drug approved for the treatment of adrenocortical carcinoma (ACC), one of the deadliest endocrine malignancies186,187,188,189. In ACC cells, but not in non-adrenal cancer tissues, mitotane counteracts tumor growth and steroid hormone production186,190. These effects of mitotane are believed to be the result of the inhibition of the sterol-O-acyl-transferase 1 (SOAT1), an enzyme that protects cells against the harmful effects of free cholesterol by transforming it into cholesterol esters. Because of SOAT1 inhibition, mitotane therapy leads to the accumulation of toxic lipids, including free cholesterol and oxysterols, inside ACC cells, which triggers lipid-induced ER stress190. Moreover, in ACC tissue samples, SOAT1 expression correlated with the response to mitotane treatment.
Interestingly, mitotane-induced apoptosis in ACC cells also depends on FATE1 expression61. Under physiological conditions, FATE1 expression is restricted to the testis and adrenal gland, while FATE1 overexpression is observed in a variety of cancers191,192. In ACC cells, FATE1 expression is controlled by the steroidogenic factor-1 (SF-1), a transcription factor that is important for adrenal development and plays a role in the formation of adrenocortical tumors61,191. FATE1, localized at the MAMs where it uncouples the ER and mitochondria, decreases mitochondrial Ca2+ uptake in ACC cells61. In this way, FATE1 protects cancer cells from Ca2+-dependent apoptotic stimuli. Furthermore, FATE1 expression conferred mitotane resistance to ACC cells, when this chemotherapeutic drug was used in a dose that falls inside the therapeutic window for ACC patients, whereas knockdown of FATE1 in these cells increased the sensitivity to the drug61. FATE1 probably protects against mitotane-induced apoptosis because of its localization in the MAMs61. Mitotane inhibits the SOAT1 enzyme190, which is also localized in the MAMs, resulting in the accumulation of toxic cholesterol lipids. Hence, in the presence of FATE1 mitotane-mediated SOAT1 inhibition may be less efficient (Fig. 3)61. Interestingly, FATE1 expression in ACC tumor cells can even be used as a prognosis indicator since FATE1 expression is inversely correlated with the overall survival of ACC patients61.
Conclusion
In conclusion, MAMs form important intracellular signaling platforms, allowing for Ca2+-encoded messages between the ER and the mitochondria. This ER–mitochondrial Ca2+ exchange can be altered during cancer development to promote cancer hallmarks like evasion of apoptosis, excessive cell proliferation, and a metabolic rewiring. In addition, many chemotherapeutics act via Ca2+ signaling at the MAMs (Fig. 3). Moreover, chemotherapeutics can interfere with the function of oncogenes and tumor suppressors, thereby altering ER–mitochondrial Ca2+ transfer. In this sense, chemotherapeutics that modify ER–mitochondrial Ca2+ signaling can be used to increase the response of cancer cells towards therapeutics that harbor a Ca2+ component in their working mechanism.
References
Rizzuto, R., De Stefani, D., Raffaello, A. & Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell. Biol. 13, 566–578 (2012).
Mesmin B. Mitochondrial lipid transport and biosynthesis: a complex balance. J. Cell Biol. 214, 9–11 (2016).
Wang, C. & Youle, R. J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118 (2009).
Antico Arciuch, V. G., Elguero, M. E., Poderoso, J. J. & Carreras, M. C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 16, 1150–1180 (2012).
Okamoto, K. & Kondo-Okamoto, N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochim. Biophys. Acta 1820, 595–600 (2012).
Ivanova, H., Kerkhofs, M., La Rovere, R. M. & Bultynck, G. Endoplasmic reticulum-mitochondrial Ca2+ fluxes underlying cancer cell survival. Front. Oncol. 7, 70 (2017).
Schrepfer, E. & Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 61, 683–694 (2016).
Vance, J. E. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609 (2014).
Demarquoy, J. & Le Borgne, F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 6, 301–309 (2015).
Stefan, C. J., Manford, A. G. & Emr, S. D. ER-PM connections: sites of information transfer and inter-organelle communication. Curr. Opin. Cell. Biol. 25, 434–442 (2013).
Giacomello, M. & Pellegrini, L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death. Differ. 23, 1417–1427 (2016).
Vance, J. E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 (1990).
van Vliet, A. R., Verfaillie, T. & Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 1843, 2253–2262 (2014).
Giorgi, C., Wieckowski, M. R., Pandolfi, P. P. & Pinton, P. Mitochondria associated membranes (MAMs) as critical hubs for apoptosis. Commun. Integr. Biol. 4, 334–335 (2011).
Bononi, A. et al. Mitochondria-associated membranes (MAMs) as hotspot Ca2+ signaling units. Adv. Exp. Med. Biol. 740, 411–437 (2012).
Hamasaki, M. et al. Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 (2013).
Missiroli, S. et al. PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep. 16, 2415–2427 (2016).
Giorgi, C. et al. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 22, 995–1019 (2015).
Wang, H. J., Guay, G., Pogan, L., Sauve, R. & Nabi, I. R. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell. Biol. 150, 1489–1498 (2000).
Csordas, G. et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 (2006).
Sood, A. et al. A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 111, 16017–16022 (2014).
Clapham, D. E. Calcium signaling. Cell 131, 1047–1058 (2007).
Bootman, M. D. Calcium signaling. Cold Spring Harb. Perspect. Biol. 4, a011171 (2012).
Parys, J. B. & De Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 740, 255–279 (2012).
Lanner, J. T., Georgiou, D. K., Joshi, A. D., Hamilton, S. L. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2, a003996 (2010).
Gincel, D., Zaid, H. & Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358(Pt. 1), 147–155 (2001).
Rapizzi, E. et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 (2002).
Marchi, S. & Pinton, P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J. Physiol. 592, 829–839 (2014).
Foskett, J. K. & Philipson, B. The mitochondrial Ca2+ uniporter complex. J. Mol. Cell Cardiol. 78, 3–8 (2015).
De Stefani, D., Rizzuto, R. & Pozzan, T. Enjoy the trip: calcium in mitochondria back and forth. Annu. Rev. Biochem. 85, 161–192 (2016).
Csordas, G., Thomas, A. P. & Hajnoczky, G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 18, 96–108 (1999).
Denton, R. M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316 (2009).
Shanmughapriya, S. et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal. 8, ra23 (2015).
Booth, D. M., Enyedi, B., Geiszt, M., Varnai, P. & Hajnoczky, G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol. Cell 63, 240–248 (2016).
Glancy, B. & Balaban, R. S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973 (2012).
Marmol, P. et al. Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells. J. Biol. Chem. 284, 515–524 (2009).
Gellerich, F. N. et al. Extramitochondrial Ca2+ in the nanomolar range regulates glutamate-dependent oxidative phosphorylation on demand. PLoS ONE 4, e8181 (2009).
Gellerich, F. N. et al. The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta 1797, 1018–1027 (2010).
Gellerich, F. N. et al. Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem. J. 443, 747–755 (2012).
Lasorsa, F. M. et al. Recombinant expression of the Ca2+-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J. Biol. Chem. 278, 38686–38692 (2003).
Paillard, M. et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 18, 2291–2300 (2017).
Csordas, G. et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987 (2013).
Patron, M. et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737 (2014).
Kamer, K. J., Grabarek, Z. & Mootha, V. K. High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 18, 1397–1411 (2017).
Liu, J. C. et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16, 1561–1573 (2016).
Morciano, G. et al. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 78, 142–153 (2015).
Bonora, M. et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO. Rep. 18, 1077–1089 (2017).
Pinton, P., Giorgi, C., Siviero, R., Zecchini, E. & Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27, 6407–6418 (2008).
Decuypere, J. P. et al. The IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 1813, 1003–1013 (2011).
Halestrap, A. P. The C ring of the F1Fo ATP synthase forms the mitochondrial permeability transition pore: a critical Appraisal. Front. Oncol. 4, 234 (2014).
Danese, A. et al. Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta 1858, 615–627 (2017).
Hwang, M. S. et al. Mitochondrial Ca(2+) influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death. Differ. 21, 1733–1745 (2014).
Giorgio, V. et al. Ca2+ binding to F-ATP synthase beta subunit triggers the mitochondrial permeability transition. EMBO Rep. 18, 1065–1076 (2017).
Rizzuto, R., Brini, M., Murgia, M. & Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747 (1993).
Szabadkai, G. et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 (2006).
Giorgi, C. et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251 (2010).
Bononi, A. et al. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death. Differ. 20, 1631–1643 (2013).
Filadi, R. et al. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 112, E2174–E2181 (2015).
de Brito, O. M. & Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 (2008).
Verfaillie, T. et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death. Differ. 19, 1880–1891 (2012).
Doghman-Bouguerra, M. et al. FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep. 17, 1264–1280 (2016).
Bultynck, G., Parys, J. B. Ca2+ signaling and cell death: focus on Ca2+-transport systems and their implication in cell death and survival. Cell Calcium 69, 1–3 (2018).
De Stefani, D. et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death. Differ. 19, 267–273 (2012).
Mendes, C. C. et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900 (2005).
Ivanova, H. et al. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 1843, 2164–2183 (2014).
Giorgi, C. et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 112, 1779–1784 (2015).
Lynes, E. M. et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J. Cell Sci. 126(Pt. 17), 3893–3903 (2013).
Simmen, T., Lynes, E. M., Gesson, K. & Thomas, G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 1798, 1465–1473 (2010).
Brini, M. & Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 89, 1341–1378 (2009).
Pinton, P. et al. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol. 148, 857–862 (2000).
Foyouzi-Youssefi, R. et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 97, 5723–5728 (2000).
Pinton, P. et al. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20, 2690–2701 (2001).
Chami, M. et al. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 32, 641–651 (2008).
Chami, M. et al. SERCA1 truncated proteins unable to pump calcium reduce the endoplasmic reticulum calcium concentration and induce apoptosis. J. Cell Biol. 153, 1301–1314 (2001).
Herrera-Cruz, M. S. & Simmen, T. Cancer: untethering mitochondria from the endoplasmic reticulum? Front. Oncol. 7, 105 (2017).
Bittremieux, M., Parys, J. B., Pinton, P. & Bultynck, G. ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca2+ signaling. Biochim. Biophys. Acta 1863, 1364–1378 (2016).
Akl, H. & Bultynck, G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim. Biophys. Acta 1835, 180–193 (2013).
Szado, T. et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 105, 2427–2432 (2008).
Bittremieux, M. & Bultynck, G. p53 and Ca2+ signaling from the endoplasmic reticulum: partners in anti-cancer therapies. Oncoscience 2, 233–238 (2015).
Kroemer, G., Bravo-San Pedro, J. M. & Galluzzi, L. Novel function of cytoplasmic p53 at the interface between mitochondria and the endoplasmic reticulum. Cell Death Dis. 6, e1698 (2015).
Monaco, G. et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 290, 9150–9161 (2015).
Vervliet, T. et al. Modulation of Ca2+ signaling by anti-apoptotic B-cell lymphoma 2 proteins at the endoplasmic reticulum-mitochondrial interface. Front. Oncol. 7, 75 (2017).
Rong, Y. & Distelhorst, C. W. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 70, 73–91 (2008).
Vervliet, T., Parys, J. B. & Bultynck, G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene 35, 5079–5092 (2016).
Rong, Y. P. et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 106, 14397–14402 (2009).
Zhong, F. et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood 117, 2924–2934 (2011).
Monaco, G. et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell. Death Differ. 19, 295–309 (2012).
Monaco, G. et al. A double point mutation at residues Ile14 and Val15 of Bcl-2 uncovers a role for BH4 domain in both protein stability and function. FEBS J 285, 127–145 (2018).
Ivanova, H. et al. The trans-membrane domain of Bcl-2alpha, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget 7, 55704–55720 (2016).
Xu, L. et al. Suppression of IP3-mediated calcium release and apoptosis by Bcl-2 involves the participation of protein phosphatase 1. Mol. Cell. Biochem. 295, 153–165 (2007).
Chang, M. J. et al. Feedback regulation mediated by Bcl-2 and DARPP-32 regulates inositol 1,4,5-trisphosphate receptor phosphorylation and promotes cell survival. Proc. Natl. Acad. Sci. USA 111, 1186–1191 (2014).
Eckenrode, E. F., Yang, J., Velmurugan, G. V., Foskett, J. K. & White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 285, 13678–13684 (2010).
Monaco, G. et al. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem. Biophys. Res. Commun. 428, 31–35 (2012).
White, C. et al. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat. Cell Biol. 7, 1021–1028 (2005).
Akl, H. et al. A dual role for the anti-apoptotic Bcl-2 protein in cancer: mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 1843, 2240–2252 (2014).
Abu-Hamad, S. et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 122, 1906–1916 (2009).
Arbel, N. & Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 285, 6053–6062 (2010).
Shimizu, S., Konishi, A., Kodama, T. & Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA 97, 3100–3105 (2000).
Naon, D. & Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta 1843, 2184–2194 (2014).
Marchi, S. et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72 (2018).
Naon, D. et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 113, 11249–11254 (2016).
Monteith, G. R., Davis, F. M. & Roberts-Thomson, S. J. Calcium channels and pumps in cancer: changes and consequences. J. Biol. Chem. 287, 31666–31673 (2012).
Monteith, G. R., Prevarskaya, N. & Roberts-Thomson, S. J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 17, 367–380 (2017).
Zhang, G. et al. Decreased expression of microRNA-320a promotes proliferation and invasion of non-small cell lung cancer cells by increasing VDAC1 expression. Oncotarget 7, 49470–49480 (2016).
Wu, C. H., Lin, Y. W., Wu, T. F., Ko, J. L. & Wang, P. H. Clinical implication of voltage-dependent anion channel 1 in uterine cervical cancer and its action on cervical cancer cells. Oncotarget 7, 4210–4225 (2016).
Arif, T., Vasilkovsky, L., Refaely, Y., Konson, A. & Shoshan-Barmatz, V. Silencing VDAC1 expression by siRNA inhibits cancer cell proliferation and tumor growth in vivo. Mol. Ther. Nucleic Acids 8, 493 (2017).
Shoshan-Barmatz, V., Krelin, Y., Shteinfer-Kuzmine, A. & Arif, T. Voltage-dependent anion channel 1 as an emerging drug target for novel anti-cancer therapeutics. Front Oncol. 7, 154 (2017).
Prevarskaya, N., Ouadid-Ahidouch, H., Skryma, R. & Shuba, Y. Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130097 (2014).
Tsunoda, T. et al. Inositol 1,4,5-trisphosphate IP3 receptortype1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 24, 1396–1402 (2005).
Gelebart, P. et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J. Biol. Chem. 277, 26310–26320 (2002).
Bultynck, G. & Campanella, M. Tumor suppressive Ca2+ signaling is driven by IP3 receptors’ fitness. Cell Stress 1, 73–78 (2017).
Chen, B. B., Glasser, J. R., Coon, T. A. & Mallampalli, R. K. FBXL2 is a ubiquitin E3 ligase subunit that triggers mitotic arrest. Cell Cycle 10, 3487–3494 (2011).
Kuchay, S. et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558 (2017).
Bononi, A. et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546, 549–553 (2017).
Marchi, S. et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 23, 58–63 (2013).
Tosatto, A. et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 8, 569–585 (2016).
Kania, E., Roest, G., Vervliet, T., Parys, J. B. & Bultynck, G. IP3 receptor-mediated calcium signaling and its role in autophagy in cancer. Front Oncol. 7, 140 (2017).
Green, D. R. A BH3 mimetic for killing cancer cells. Cell 165, 1560 (2016).
Anderson, M. A. et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 127, 3215–3224 (2016).
Mignen, O. et al. Constitutive calcium entry and cancer: updated views and insights. Eur. Biophys. J. 46, 395–413 (2017).
Chen, Y. F., Hsu, K. F. & Shen, M. R. The store-operated Ca2+ entry-mediated signaling is important for cancer spread. Biochim. Biophys. Acta 1863, 1427–1435 (2016).
Deliot, N. & Constantin, B. Plasma membrane calcium channels in cancer: alterations and consequences for cell proliferation and migration. Biochim. Biophys. Acta 1848(10 Pt. B), 2512–2522 (2015).
Wyrsch, P., Blenn, C., Pesch, T., Beneke, S. & Althaus, F. R. Cytosolic Ca2+ shifts as early markers of cytotoxicity. Cell Commun. Signal. 11, 11 (2013).
Giorgi, C. et al. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 6, 1435–1445 (2015).
Bonora, M., Giorgi, C. & Pinton, P. Novel frontiers in calcium signaling: a possible target for chemotherapy. Pharmacol. Res. 99, 82–85 (2015).
Wang, T. F., Zhou, C., Tang, A. H., Wang, S. Q. & Chai, Z. Cellular mechanism for spontaneous calcium oscillations in astrocytes. Acta Pharmacol. Sin. 27, 861–868 (2006).
Akl, H. et al. HA14-1, but not the BH3 mimetic ABT-737, causes Ca2+ dysregulation in platelets and human cell lines. Haematologica 98, e49–e51 (2013).
Vervloessem, T., Ivanova, H., Luyten, T., Parys, J. B. & Bultynck, G. The selective Bcl-2 inhibitor venetoclax, a BH3 mimetic, does not dysregulate intracellular Ca2+ signaling. Biochim. Biophys. Acta 1864, 968–976 (2017).
Cull, E. H. & Altman, J. K. Contemporary treatment of APL. Curr. Hematol. Malig. Rep. 9, 193–201 (2014).
Salomoni, P., Ferguson, B. J., Wyllie, A. H. & Rich, T. New insights into the role of PML in tumour suppression. Cell. Res. 18, 622–640 (2008).
Alimoghaddam, K. A review of arsenic trioxide and acute promyelocytic leukemia. Int. J. Hematol. Oncol. Stem Cell Res. 8, 44–54 (2014).
McCulloch, D., Brown, C. & Iland, H. Retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia: current perspectives. OncoTargets Ther. 10, 1585–1601 (2017).
Lallemand-Breitenbach, V. et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 193, 1361–1371 (2001).
Maes, H., Rubio, N., Garg, A. D. & Agostinis, P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 19, 428–446 (2013).
Gurrieri, C. et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 96, 269–279 (2004).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Nazio, F. et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15, 406–416 (2013).
Dasari, S. & Tchounwou, P. B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378 (2014).
Wozniak, K. & Blasiak, J. Recognition and repair of DNA-cisplatin adducts. Acta Biochim. Pol. 49, 583–596 (2002).
Xu, Y. et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol. Rep. 34, 3051–3060 (2015).
Huang, Z. et al. Bcl-2 small interfering RNA sensitizes cisplatin-resistant human lung adenocarcinoma A549/DDP cell to cisplatin and diallyl disulfide. Acta Biochim. Biophys. Sin. (Shanghai). 39, 835–843 (2007).
Schaaf, A. et al. Cytotoxicity of cisplatin in bladder cancer is significantly enhanced by application of bcl-2 antisense oligonucleotides. Urol. Oncol. 22, 188–192 (2004).
Xie, Q. et al. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int. J. Oncol. 49, 2507–2519 (2016).
Florea, A. M. et al. Calcium-regulatory proteins as modulators of chemotherapy in human neuroblastoma. Oncotarget 8, 22876–22893 (2017).
Delbridge, A. R. & Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell. Death Differ. 22, 1071–1080 (2015).
Billard, C. BH3 mimetics: status of the field and new developments. Mol. Cancer Ther. 12, 1691–1700 (2013).
Lee, E. F. et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J. Cell. Biol. 180, 341–355 (2008).
Vervloessem, T. et al. Bcl-2 inhibitors as anti-cancer therapeutics: the impact of and on calcium signaling. Cell Calcium (2017). pii: S0143-4160(17)30103-3. https://doi.org/10.1016/j.ceca.2017.05.014 (in press).
Vogler, M. et al. BCL2/BCL-XL inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 117, 7145–7154 (2011).
Mason, K. D. et al. Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 (2007).
Schoenwaelder, S. M. & Jackson, S. P. Bcl-xL-inhibitory BH3 mimetics (ABT-737 or ABT-263) and the modulation of cytosolic calcium flux and platelet function. Blood 119, 1320–1321 (2012). author reply 1321–1322.
Harper, M. T. & Poole, A. W. Bcl-xL-inhibitory BH3 mimetic ABT-737 depletes platelet calcium stores. Blood 119, 4337–4338 (2012).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
Lavik, A. R. et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 6, 27388–27402 (2015).
Akl, H. et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3 R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 4, e632 (2013).
Greenberg, E. F. et al. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 6, e2034 (2015).
Vervloessem, T. et al. Reciprocal sensitivity of diffuse large B-cell lymphoma cells to Bcl-2 inhibitors BIRD-2 versus venetoclax. Oncotarget 8, 111656–111671 (2017).
Fan, Z. et al. ABT737 enhances cholangiocarcinoma sensitivity to cisplatin through regulation of mitochondrial dynamics. Exp. Cell. Res. 335, 68–81 (2015).
Hermanson, D. et al. Dual mechanisms of sHA 14-1 in inducing cell death through endoplasmic reticulum and mitochondria. Mol. Pharmacol. 76, 667–678 (2009).
Gerasimenko, J., Ferdek, P., Fischer, L., Gukovskaya, A. S. & Pandol, S. J. Inhibitors of Bcl-2 protein family deplete ER Ca2+ stores in pancreatic acinar cells. Pflug. Arch. 460, 891–900 (2010).
Ferdek, P. E. et al. BH3 mimetic-elicited Ca2+ signals in pancreatic acinar cells are dependent on Bax and can be reduced by Ca2+-like peptides. Cell Death Dis. 8, e2640 (2017).
Ferdek, P. E. & Jakubowska, M. A. On BH3 mimetics and Ca2+ signaling. Drug Dev. Res. 78, 313–318 (2017).
Varoni, E. M., Lo Faro, A. F., Sharifi-Rad, J. & Iriti, M. Anticancer molecular mechanisms of Resveratrol. Front. Nutr. 3, 8 (2016).
Aluyen, J. K. et al. Resveratrol: potential as anticancer agent. J. Diet. Suppl. 9, 45–56 (2012).
Kulkarni, S. S. & Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 1852, 1114–1123 (2015).
Madreiter-Sokolowski, C. T. et al. Resveratrol specifically kills cancer cells by a devastating increase in the Ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell. Physiol. Biochem. 39, 1404–1420 (2016).
Wang, W. A., Groenendyk, J. & Michalak, M. Endoplasmic reticulum stress associated responses in cancer. Biochim. Biophys. Acta 1843, 2143–2149 (2014).
Dolfi, S. C. et al. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 1, 20 (2013).
Cardenas, C. et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep. 14, 2313–2324 (2016).
Bultynck, G. Onco-IP3Rs feed cancerous cravings for mitochondrial Ca2+. Trends Biochem. Sci. 41, 390–393 (2016).
Dadi, P. K., Ahmad, M. & Ahmad, Z. Inhibition of ATPase activity of Escherichia coli ATP synthase by polyphenols. Int. J. Biol. Macromol. 45, 72–79 (2009).
Gledhill, J. R. & Walker, J. E. Inhibition sites in F1-ATPase from bovine heart mitochondria. Biochem. J. 386, 591–598 (2005).
Zheng, J. & Ramirez, V. D. Piceatannol, a stilbene phytochemical, inhibits mitochondrial F0F1-ATPase activity by targeting the F1 complex. Biochem. Biophys. Res. Commun. 261, 499–503 (1999).
Zheng, J. & Ramirez, V. D. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br. J. Pharmacol. 130, 1115–1123 (2000).
Luyten, T. et al. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2+. Biochim. Biophys. Acta 1864, 947–956 (2017).
Tacar, O., Sriamornsak, P. & Dass, C. R. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 65, 157–170 (2013).
Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 10, 853–858 (2014).
Gewirtz, D. A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 57, 727–741 (1999).
Hanusova, V., Bousova, I. & Skalova, L. Possibilities to increase the effectiveness of doxorubicin in cancer cells killing. Drug. Metab. Rev. 43, 540–557 (2011).
Volkova, M. & Russell, R. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 7, 214–220 (2011).
Giorgi, C. et al. Alterations in mitochondrial and endoplasmic reticulum signaling by p53 mutants. Front. Oncol. 6, 42 (2016).
Ji, M. M. et al. Induction of autophagy by valproic acid enhanced lymphoma cell chemosensitivity through HDAC-independent and IP3-mediated PRKAA activation. Autophagy 11, 2160–2171 (2015).
Beloribi-Djefaflia, S., Vasseur, S. & Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 5, e189 (2016).
Rios-Marco, P. et al. Alkylphospholipids deregulate cholesterol metabolism and induce cell-cycle arrest and autophagy in U-87 MG glioblastoma cells. Biochim. Biophys. Acta 1831, 1322–1334 (2013).
Li, Y. et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 279, 37030–37039 (2004).
Lalli, E. Mitotane revisited: a new target for an old drug. Endocrinology 156, 3873–3875 (2015).
Berruti, A. et al Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 23(Suppl 7), vii131–vii138 (2012).
Berruti, A. et al. Long-term outcomes of adjuvant mitotane therapy in patients with radically resected adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 102, 1358–1365 (2017).
Fassnacht, M. et al. Combination chemotherapy in advanced adrenocortical carcinoma. N. Engl. J. Med. 366, 2189–2197 (2012).
Sbiera, S. et al. Mitotane inhibits sterol-O-acyl transferase 1 triggering lipid-mediated endoplasmic reticulum stress and apoptosis in adrenocortical carcinoma cells. Endocrinology 156, 3895–3908 (2015).
Doghman, M. et al. Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol. Endocrinol. 21, 2968–2987 (2007).
Yang, X. A. et al. Immunohistochemical analysis of the expression of FATE/BJ-HCC-2 antigen in normal and malignant tissues. Lab. Invest. 85, 205–213 (2005).
Acknowledgements
G.B. and J.B.P. are supported by grants from the Research Foundation—Flanders (FWO) (grants G.0571.12N, G.0819.13N, G.0634.13N, G.0C91.14N, G.0927.15N, and G.0A34.16N) and the Research Council—KU Leuven (OT14/101). M.B. and M.K. are holders of a Ph.D. Fellowship from the FWO. P.P. is grateful to Camilla degli Scrovegni for continuous support. P.P. is supported by the Italian Ministry of Education, University and Research, the Italian Ministry of Health, Telethon (GGP15219/B), the Italian Association for Cancer Research (IG-18624), and by local funds from the University of Ferrara. C.G. is supported by local funds from the University of Ferrara, the Italian Association for Cancer Research, the Italian Ministry of Health, and by Cariplo grant. G.B., J.B.P., and P.P. are part of the FWO—Scientific Research Community Casign (W0.019.17N).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kerkhofs, M., Bittremieux, M., Morciano, G. et al. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis 9, 334 (2018). https://doi.org/10.1038/s41419-017-0179-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-017-0179-0
This article is cited by
-
Nano-modulators with the function of disrupting mitochondrial Ca2+ homeostasis and photothermal conversion for synergistic breast cancer therapy
Journal of Nanobiotechnology (2023)
-
Calcium signalling pathways in prostate cancer initiation and progression
Nature Reviews Urology (2023)
-
Lon upregulation contributes to cisplatin resistance by triggering NCLX-mediated mitochondrial Ca2+ release in cancer cells
Cell Death & Disease (2022)
-
Paclitaxel Promotes Oxidative Stress–Mediated Human Laryngeal Squamous Tumor Cell Death through the Stimulation of Calcium and Zinc Signaling Pathways: No Synergic Action of Melatonin
Biological Trace Element Research (2022)
-
CD137 Signal Mediates Cardiac Ischemia–Reperfusion Injury by Regulating the Necrosis of Cardiomyocytes
Journal of Cardiovascular Translational Research (2022)
