
Abstract
The endoplasmic reticulum (ER) and mitochondria are tightly associated with very dynamic platforms termed mitochondria-associated membranes (MAMs). MAMs provide an excellent scaffold for crosstalk between the ER and mitochondria and play a pivotal role in different signaling pathways that allow rapid exchange of biological molecules to maintain cellular health. However, dysfunctions in the ER–mitochondria architecture are associated with pathological conditions and human diseases. Inflammation has emerged as one of the various pathways that MAMs control. Inflammasome components and other inflammatory factors promote the release of pro-inflammatory cytokines that sustain pathological conditions. In this review, we summarize the critical role of MAMs in initiating inflammation in the cellular defense against pathogenic infections and the association of MAMs with inflammation-mediated diseases.
Similar content being viewed by others
Facts
-
MAMs are a molecular platform essential for NLRP3 inflammasome formation.
-
The adaptor MAVS is required for NLRP3 mitochondrial localization and promotes its activation.
-
MAMs are directly involved in DAMP generation and in the cellular antiviral response.
-
Several MAM-resident proteins such as α-synuclein and presenilins play a crucial role in the pathogenesis of Parkinson’s disease and Alzheimer’s disease, respectively.
Open questions
-
How can such a small subcellular organelle have such an important function in NLRP3 inflammasome formation?
-
Could MAMs be considered a potential target for pathogenic bacteria to disrupt inflammasome activation and signaling during infection?
-
How could MAMs be developed to target or treat diseased conditions?
-
Is it feasible to assume that the integrity and functions of MAMs are relevant for therapeutic intervention?
Introduction
Studies over the past few decades have demonstrated that the endoplasmic reticulum (ER) and mitochondria are physically connected to form junctions termed mitochondria-associated membranes (MAMs).
The MAMs fraction was first identified by J.E. Vance1 in 1990, who described the isolation of a unique membrane “fraction X” from rat liver enriched in a protein implicated in lipid synthesis and trafficking (for a detailed review, see ref. 2).
Several biochemical techniques since have been described to isolate the MAM fraction, confirming that MAMs are composed of membrane fragments from both the ER and the outer mitochondrial membrane (OMM)3.
In the past few years, different proteomics studies have identified the molecular components of the MAM fraction, demonstrating both from human fibroblast4 and from mouse brain5 that >1000 “MAM proteins” reside in this fraction. Recently, Sala-Vila et al. performed in-depth mass spectrometric analysis of the proteins composing MAM-enriched fractions and identified 1052 proteins including caveolin-1 (CAV1), which is an integral component of hepatic MAMs that determines the relative cholesterol content of these ER subdomains6.
MAMs provide a platform that is fundamental for several cellular functions, such as calcium (Ca2+) homeostasis, autophagy, lipid metabolism, apoptosis, and tumor growth7,8, that allow rapid exchange of biological molecules to maintain cellular health. Therefore, the accurate connections and crosstalk between the ER and mitochondria are events that coordinate important functions of the two organelles and thus determine key aspects of cell fate. In fact, alterations in the composition of MAMs and the abnormal induction of this ER–mitochondria association lead to different pathological conditions9.
For these reasons, several regulatory proteins, oncogenes, and tumor suppressors reside at MAMs in order to maintain normal cellular function and thereby preserve the intracellular equilibrium10,11.
Considering that the ER–mitochondria interface is involved in several molecular pathways, it is not surprising that MAMs play an emerging role also in inflammatory signaling pathways. In fact, MAMs provide a critical site for inflammasome formation, are attractive targets for pathogenic bacteria and are relevant for the antiviral response (see the next paragraphs).
Additionally, increasing evidence strongly implicates the involvement of MAMs in the initiation or progression of diseases associated with high inflammation, such as metabolic disorders and neurodegenerative diseases12.
Finally, recent developments of promising therapeutics that target inflammasome activities at MAMs in inflammatory diseases have been reported. In this review, we discuss how MAMs represent a primary platform for initiating inflammatory mechanisms and how these networks might be manipulated to provide novel therapies for inflammatory diseases.
MAMs as a critical site for inflammasome formation
A link between the ER–mitochondria interface and inflammation was first recognized a few years ago with the observation that reactive oxygen species (ROS) promote the activation of NOD-like receptor protein 3 (NLRP3) inflammasomes13.
The inflammasome is a multiprotein complex composed of a sensor protein, an adaptor protein called apoptosis-associated speck-like protein, containing a caspase-recruitment domain (ASC) and pro-caspase 1, a cysteine protease. There are four subfamilies of inflammasomes depending on the sensor molecule: NLRP3, NLRP1, NLRC4 (NLR family, CARD domain containing 4), and AIM2 (absent in melanoma 2)14.
These sensor molecules can sense diverse stimulators ranging from microbial products (pathogen-associated molecular patterns, PAMPs) to host-derived damage signals (damage-associated molecular patterns, DAMPs) or any other insults that may occur within the cytosol. Once assembled, pro-caspase 1 is autoprocessed by proximity to active caspase 1, which induces the maturation of pro-interleukin-1β (IL-1β) or pro-IL-18 to their activated form. Among the inflammasomes, NLRP3 is the most studied and characterized due to its implication in the pathogenesis of different human diseases15. To date, the NLRP3 complex is the only inflammasome complex to be described as associated with MAMs13.
Despite receiving much attention and investigation16,17, the activation of the NLRP3 inflammasome remains an enigmatic mechanism. So far, a two-step process has been proposed for the activation of NLRP3: (i) a priming step triggered by the interaction of Toll-like endosomal receptors (TLRs) with their ligands, leading to the transcription of NLRP3 and pro-IL-1β by nuclear factor kB (NF-κB), and (ii) an activation step promoted by several stimulators that initiates complex assembly in a manner dependent on K+ efflux through P2×7 activation18, lysosomal rupture, or mitochondrial ROS19. Moreover, different models have been proposed to explain the role of Ca2+-signaling in NLRP3 activation (for a detailed review, see ref. 20). Lee et al. proposed a critical role for calcium-sensing receptor (CASR) in activating the NLRP3 inflammasome that is mediated by increased intracellular Ca2+ and decreased cellular cyclic AMP (cAMP)21. An alternative model proposed that sustained Ca2+ influx (via ER Ca2+ release channels) triggers mitochondrial Ca2+ overload and associated mitochondrial destabilization22 and is characterized by high levels of ROS production and the induction of mitochondrial permeability transition pore (mPTP)23. Supporting this model, Misawa et al. observed that during inflammasome formation microtubules drive the perinuclear migration of mitochondria that results in subsequent apposition of ASC on mitochondria to NLRP3 on the ER24.
NLRP3 is expressed in most tissues but predominantly in macrophages. In the inactive state, NLRP3 localizes to the ER membrane and cytosol, but when activated both NLRP3 and its adaptor ASC relocate to the MAM fraction (Fig. 1) where they detect increased ROS production from damaged mitochondria13. In particular, respiratory chain inhibitors (like complex I inhibitor rotenone) activate the inflammasome implicating a loss of the ΔΨm and ROS accumulation. Therefore, by virtue of its ER–mitochondria localization upon activation, the NLRP3 inflammasome is strategically located to sense signals emanating from mitochondria.

Under non-stimulatory conditions, most NLRP3 protein localizes to cytoplasmic granular structures. Stimulation with extracellular ATP and particulate/crystalline activators, which lead to lysosomal rupture, DAMPs, and PAMPs, triggers the generation of ROS that activate NLRP3. Once activated, NLRP3 recruits an adaptor protein called apoptosis-associated speck-like protein containing a CARD (ASC) and redistributes to the MAM fraction. Thus, upon pro-inflammatory stimuli, NLRP3 oligomerizes and exposes its effector domain to interact with ASC, which in turn recruits pro-caspase-1. Finally, activated caspase-1 cleaves pro-IL-1β to generate mature IL-1β. ASC apoptosis-associated speck-like protein containing a CARD, IL-1β interleukin-1 beta, NLRP3 NOD-like receptor protein 3
Consistent with these observations, defective mitophagy has been shown to enhance inflammasome activation. In fact, inhibition of mitophagy/autophagy by 3-methyladenine (3MA) treatment or silencing the autophagy regulator Beclin 1 and autophagy protein 5 (ATG5) in macrophages resulted in increased NLRP3 activation and IL-1β release upon stimulation with monosodium urate (MSU) crystals and nigericin due to the accumulation of damaged mitochondria and increased ROS generation25. Moreover, treatment with the antioxidant 4-amino-2,4-pyrrolidinedicarboxylic acid (APDC) blocked NLRP3 inflammasome activation and IL-1β secretion13.
Interestingly, a study by the group of M. Karin demonstrated that NF-κB could exert its anti-inflammatory activity and restrain NLRP3 inflammasome activation, by inducing delayed accumulation of the autophagy receptor p62/SQSTM126. p62, induced on NF-κB activation, suppresses NLRP3-inflammasome-dependent IL-1β production.
Therefore, the “NF-κB–p62/SQSTM1–mitophagy” pathway provides a crucial regulatory loop through which NF-κB orchestrates NLRP3 inflammasome activation and cytokine release thereby focusing on a reparative inflammatory response and preventing excessive collateral damage26.
However, the exact role of autophagy on the secretion of IL-1β is still a matter of intense study and debate27. Indeed increasing data from the literature support the idea that induction of autophagy can directly promote IL-1β secretion after inflammasome activation by incorporating it into the autophagosomal carrier28. Conversely, autophagy inhibition induces interleukin-1β accumulation inside the cells blocking its release28.
Therefore, a better understanding of the NLRP3 inflammasome is required for the development of effective therapeutic treatments for NLRP3-related inflammatory diseases.
Additionally, mitochondrial antiviral-signaling protein (MAVS), best known for its role in the innate immune system (see next section for further details), has been suggested to recruit NLRP3 to mitochondria in response to viral infection29. This study by Subramanian N. demonstrated that MAVS is required for optimal NLRP3 inflammasome activity and identified the N-terminal amino acid sequence in NLRP3 that is fundamental for its association with MAVS at the mitochondria29.
Another NLRP3-binding partner thioredoxin-interacting protein (TXNIP) redistributes to MAMs/mitochondria in response to oxidative stress30 or NLRP3 inflammasome activation31. In resting cells, TXNIP interacts with thioredoxin (TRX, a cellular antioxidant protein) and is therefore unavailable for interaction with NLRP3. Inflammasome activators, such as uric acid crystals, induce the dissociation of TXNIP from oxidized TRX in a ROS-sensitive manner and allow it to bind NLRP3 and translocate to MAMs/mitochondria. This raises the possibility that TXNIP is involved in IL-1β production through NLRP3 under ER stress conditions31.
Together with ROS, mitochondrial DNA (mtDNA) is one of the mitochondrial damage signals that interacts directly with NLRP3 and AIM2, and oxidized mtDNA (ox-mtDNA) interacts specifically with NLRP3 in cells with ATP and nigericin treatment32. Moreover, IL-1β release is drastically reduced in AIM2 KO macrophages in response to mtDNA. Importantly, IL-1β release is higher after ox-mtDNA stimulation than mtDNA, and AIM2 does not seem to be involved in ox-mtDNA–mediated inflammasome activation32.
Although these numerous studies support the idea that mitochondrial dysfunctions and ROS are closely associated with inflammasome assembly and activation, there is a lack of understanding how these factors trigger NLRP3 activation.
Recently, multiple works have focused on studying the possible pathway and activation mechanisms of the other inflammasomes. For instance, D’Osualdo et al. showed that expression of NLRP1, a core inflammasome component, is specifically upregulated during severe ER stress conditions in human cell lines33, which suggests a plausible involvement of MAMs.
All these data highlight the importance of the MAM interface as a platform for inflammasome formation and require additional studies to identify the mechanism that induces inflammasome activation at this critical cellular site.
Antiviral response at the MAM interface
In addition to their established role, MAMs can exert a special role in the immune-viral response34,35.
The innate immune response is regulated by several germline-encoded receptors, called pattern recognition receptors (PRRs), which are able to detect PAMPs and DAMPs. In particular, TLRs and cytosolic sensors of the RIG-I-like receptor family (RLRs) can also sense the cell’s double-stranded DNA and the DNA derived from viral infection, respectively, when present in the cytosol, or single-stranded viral RNA34,36.
The RIG-I-like receptor family includes the retinoic acid-inducible gene-I (RIG-I) protein, the melanoma differentiation-associated gene 5 (MDA5) protein, and the RIG-I-like receptor LGP2, and this receptor family triggers damage signaling by sensing viral RNA. These proteins share several homologous domains, including the presence of caspase activation and recruitment domains (CARDs) and a DEAD box helicase/ATPase domain37. In particular, the CARD allows RIG-I and MDA5 to interact with MAVS. MAVS is localized at the mitochondrial membrane through its C-terminal transmembrane domain. The MAVS mitochondrial localization is required to trigger the downstream antiviral signaling pathways38. In fact, MAVS activation induces the recruitment of several members of the TRAF family, followed by phosphorylation and nuclear translocation of interferon regulatory factor 3 (IRF3), as well as activation of NF-κB; this induces transcription of interferon I (I IFN) and III genes and other inflammatory cytokines39 (Fig. 2).

MAVS is located on the outer mitochondrial membrane (OMM) and mediates antiviral signaling by inducing the recruitment of several members of the TRAF family. Moreover, MAVS interacts with the helicases RIG-1 or MDA5 (melanoma differentiation-associated gene 5). Upon virus infection, MAVS and RIG-I create a complex with STING that increases the interferon response. Gp78 can be considered another MAVS interactor that regulates MAVS-mediated antiviral signaling. The UL37 protein from HCMV traffics into the MAMs during permissive infection and induces alteration of the Ca2+ signaling. Infection with HIV-1 directly targets MAMs leading to mitochondrial polarization and apoptosis. Infection with DENV results in the compromised integrity of MAMs and reduced RIG-1-dependent IFN response. DENV dengue virus, ER endoplasmic reticulum, HCMV human cytomegalovirus of the large family of DNA viruses Herpesviridae, HCV hepatitis C virus, HIV-1 immunodeficiency virus 1, I IFN interferon I, IRF3 interferon regulatory factor 3, MAVS mitochondrial antiviral-signaling protein, MCU mitochondrial Ca2+ uniporter, NF-kB nuclear factor-κB, NS3 nonstructural protein 3, NS4 nonstructural protein 4, RIG-I retinoic acid-inducible gene-I protein, ROS reactive oxygen species, Sig1-R sigma-1 receptor, STING Stimulator of interferon genes, Vpr viral protein R
Stimulator of interferon genes (STING) has a central role in controlling immune responses to cytoplasmic DNA. STING activation depends on the binding of a type of cyclic dinucleotide (CDN) termed cGAMP (cyclic GMP–AMP) by GMP-AMP synthase (cGAS)40. This leads to the phosphorylation of IRF3 and NF-κB and the subsequent induction of cytokines and proteins, such as the type I IFN, that exerts its antipathogenic activities41,42. Although these pathways, in principle, detect different signals, crosstalk between these distinct signaling pathways has been reported to be important. In several studies, STING appeared to interact with RIG-I and MAVS in a complex that was stabilized upon virus infection43. STING can bind MAVS at MAMs thus increasing the interferon response to viral infection43,44, whereas genetic ablation of STING inhibited the activation of the IRF/IFN pathway in the presence of the ssRNA genome of the Japanese encephalitis virus45. The physical interaction between STING and MAVS for microbial DNA and RNA recognition is of particular interest for several reasons: (i) converges the two signaling pathways at the mitochondrial level, where MAVS is anchored; (ii) incorporates MAMs into antiviral signaling, and (iii) orients viral strategies to dampen or prevent the activation of the IFN response and also toward MAM dysregulation. In fact, the alteration of the contacts between the ER and mitochondria and destruction of the MAMs/mitochondria-associated MAVS is a common strategy for many viruses.
Lastly, Gp78, an E3 ubiquitin ligase which is active in the ER-associated degradation (ERAD) pathway, has been considered a novel regulator of RLR signaling that localize to the ER–mitochondria interface46. Gp78 physically interacts with MAVS and regulates MAVS-mediated antiviral signaling through two possible mechanisms. The first requires its E3 ubiquitin ligase and ERAD activity to directly degrade MAVS, while the second occurs independent of these activities, requires the Gp78 RING domain, and occurs via a direct association between this region and MAVS46.
Taken together, these data suggest that other MAM-localized components might also serve to specifically target MAVS as a means to regulate inflammatory signaling within the cell. Defining the detailed components of the MAVS regulome specifically within the MAMs will certainly provide exciting new insights into the regulation of antiviral signaling.
Given the important role of the MAMs in the antiviral response, it is not surprising that numerous viral proteins target this structure (Fig. 2). One well-characterized example is vMIA (also named pUL37×1), which is an immediate-early protein synthesized by HCMV, the human cytomegalovirus of the large family of DNA viruses Herpesviridae. This protein exerts a potent antiapoptotic function in infected cells47. vMIA is initially synthesized in the ER and localizes on the ER membrane. However, the presence of a moderately hydrophobic leader peptide retargets all isoforms of vMIA to the MAMs and OMM4,48,49. This association of vMIA to MAMs allows the virus to exploit cellular level cytotoxic effects, such as the prevention of the antiviral response. Specifically, the additional presence of a consensus cholesterol-binding domain allows vMIA to associate with detergent-resistant membranes at the MAMs, without affecting the OMM localization49. The cholesterol-dependent association of vMIA with MAMs is responsible for at least part of the toxic action of the virus because it allows vMIA to interact with the sigma-1 receptor (Sig-1R); this contributes to the regulation of inositol 1,4,5-triphosphate receptor (IP3R) at the ER/MAM interface, thus influencing their Ca2+ transfer and degradation50,51,52. In addition to the regulation of IP3Rs, vMIA affects Ca2+ transfer increasing the expression of mitochondrial Ca2+ uniporter (MCU), MICU1, ER pump SERCA, and VDAC4,53,54, contributing to overload of Ca2+ entry in the mitochondria and inducing a condition of programmed cell death.
To prevent the cellular antiviral response, vMIA deeply affects mitochondrial morphology by both increasing mitochondrial biogenesis55 and by causing mitochondrial fragmentation in transfected cells and during viral infection56,57,58,59,60. Consequently, this results in the reduction of the number of MAMs and of the interactions between MAVS and STING. These alterations thereby reduce the downstream signaling against HCMV infection60.
Furthermore, HMCV re-localizes various cellular proteins, including calreticulin, calnexin, PACS-2, and glycolytic enzymes, to the MAMs platform. The whole effect is to induce a metabolic reprogramming that reduces pyruvate utilization at mitochondria level but increase the metabolic intermediates for biosynthetic reactions61,62.
MAVS activity is a not only a target of HCMV but also of hepatitis C virus (HCV), a small enveloped single-stranded, positive-sense RNA virus. Upon interaction with HCV viral RNA, RIG-1 tetramerizes and recruits MAVS, which in turn elicits an IFN-mediated antiviral response. To avoid this, the multifunction HCV serine proteases residing in nonstructural protein 3 (NS3) and 4 (NS4) can cleave MAVS. NS3/4A can associate to intracellular membranes through their membrane-targeting domains within NS4A and the amphipathic α helix of NS3 and thus target MAVS specifically localized at MAMs39,63. So, an HCV viral strategy, based on protease inactivation of MAVS, leads to the suppression of type I and III IFN production39. Other HCV proteins have been found to localize to MAMs64, where they could be responsible for the documented elevation of mitochondrial ROS by manipulating Ca2+ entry, in particular at the level of MCU65,66,67.
Another example of a virus that directly targets MAMs is the human immunodeficiency virus 1 (HIV-1) lentivirus, a single-stranded positive-sense RNA virus that can have a strong cytotoxic effect on host cells, in particular on human primary CD4+T cells68. This event occurs after insertion of viral protein R (Vpr) into the membranes of the ER, OMM, and MAMs. This insertion can dissipate the mitochondrial transmembrane potential and lead to apoptosis. Membrane insertion of Vpr is obtained through its C-terminal transmembrane hydrophobic segment, which shares homology with proteins of Myxoma M11L, vaccinia F1L, Epstein-Barr BHRF-1, and HCVs, and contains mitochondrial targeting sequence69,70,71. In particular, it has been found that Vpr transport occurs from the ER to OMM through MAMs and suggests the presence of dynamin-related protein 1 (Drp1), Mitofusin 2 (Mfn2), and ATPase family, AAA domain containing 3A72. As a consequence of Vpr interaction, DRP-1 and Mfn2 are downregulated via the VprBP-DDB1-CUL4A ubiquitin ligase complex, and mitochondria showed fragmentation and disruption of OMM73.
Additionally, dengue virus (DENV) carries out a strategy similar to HCMV and HCV. The mosquito genus Aedes is the host for this RNA virus of the Flaviviridae family that infects 10 million cases per year worldwide. DENV promotes infection by altering mitochondrial morphology and weakening the IFN response at the MAMs. DENV replicates at ER-derived cytoplasmic structures, such as the convoluted membranes (CM)74,75. The DENV nonstructural protein NS4B induces elongation of mitochondria, which is associated with the downregulation of Drp1. Elongated mitochondria physically contact CMs and exhibit compromised MAMs integrity and reduced RIG-1-dependent IFN response74. In particular, NS4B co-localizes with MAVS in the MAMs by interacting with the N-terminal CARD-like domain and the C-terminal transmembrane domain of MAVS. This association prevents the binding of MAVS to RIG-I and results in the suppression of RIG-I-induced IRF3 activation and, consequently, the abrogation of IFN production74. Similar to HCV, alternative strategies of DENV include the proteolytic inactivation of MAM-resident proteins. DENV NS2B-3 protease, which is highly enriched in CMs, proteolytically inactivates the MAM-resident signaling adaptor STING76 and prevents RIG-I translocation to mitochondria by targeting the adaptor protein 14-3-3ε using a highly conserved phosphomimetic motif77. Taken together, these studies highlight the importance of MAMs in the antiviral response.
MAMs are attractive targets for pathogenic bacteria
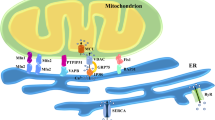
Among the numerous processes coordinated by MAMs, inflammatory signaling pathways associated with MAMs play critical roles in the cellular defense against pathogenic infections (Fig. 3). MAMs provide an excellent platform for the coordination of lipid synthesis and trafficking, mitochondrial morphology, and autophagosome formation78. Additionally, all these important cellular processes are modulated by bacterial pathogens to suppress host functions and promote infections; in fact, pathogenic bacteria frequently target the ER and mitochondria to carry out host functions. Among these bacterial pathogens, intracellular bacteria, such as Salmonella typhimurium, Chlamydia trachomatis, Mycobacterium tuberculosis, Listeria monocytogenes, Legionella pneumophila, or Yersinia pestis, have the ability to grow and replicate inside host cells. MAMs could be targeted by pathogenic bacteria to subvert key host cellular processes79. However, why are MAMs such attractive targets for pathogenic bacteria?

MAMs are a molecular platform involved in several cellular processes. As such, different pathogenic bacteria could alter these signaling pathways. Interestingly, once released into the extracellular space, mitochondrial DAMPs of bacterial origin, such as cardiolipin and NFPs, can stimulate the innate and adaptive immune responses. ACAT enzyme acyl-CoA:cholesterol acyltransferase, CASP2 caspase 2, Ca2+ calcium, IP3R3 inositol 1,4,5-triphosphate receptor type 3, MCU mitochondrial Ca2+ uniporter, NFPs N-formyl peptides, NLRP3 NOD-like receptor protein 3, PE phosphatidylethanolamine, PS phosphatidylserine, PSD phosphatidylserine decarboxylase, PSS1 phosphatidylserine synthase-1, PSS2 phosphatidylserine synthase-2, Sig1-R sigma-1 receptor, TXNIP thioredoxin-interacting protein
MAMs coordinate the synthesis and transformation of phosphatidylserine (PS) into phosphatidylethanolamine (PE), which is one of the most abundant phospholipids in bacterial cell membranes. The two enzymes phosphatidylserine synthase-1 (PSS1) and 2 (PSS2) are highly enriched in MAMs80 and are responsible for PS synthesis. After being transported to the mitochondria, PS is decarboxylated into PE by the enzyme phosphatidylserine decarboxylase (PSD) located in the inner mitochondrial membrane (IMM).
PE can be readily broken down into glycerol and ethanolamine by phosphodiesterases, and bacteria take advantage of ethanolamine as a nutrient source81. Interestingly, S. typhimurium in the lumen of the inflamed intestine use PE-derived ethanolamine as a carbon source to produce a respiratory electron acceptor (tetrathionate). This supports anaerobic growth with ethanolamine, which is released from host tissue but is not utilizable by competing bacteria82. Interestingly, the contribution of ethanolamine utilization to L. monocytogenes pathogenesis, in an intravenous mouse infection model, suggests that ethanolamine utilization is important outside of the intestine and possibly in the intracellular environment83.
Phospholipid synthesis is one of the major functions of MAMs. Given the enzyme acyl-CoA:cholesterol acyltransferase (ACAT) is highly enriched at MAMs, it has been suggested that MAMs might also serve as a site for cholesterol synthesis and neutral lipid synthesis84. Coxiella burnetii require lipids for both normal bacterial functions as well as formation of the acidic, phagolysosomal-like parasitophorous vacuole (PV) surrounding the bacteria. C. burnetii does not have the capability to generate cholesterol and thus utilizes host cell lipids for membrane biogenesis and possibly energy. This suggests that sterols are actively diverted from the host cell85.
Since the central role of MAMs in Ca2+ homeostasis, which in turn is key for the initiation of apoptosis86, and considering that bacterial pathogens can modulate Ca2+ fluxes as a strategy for pathogenesis (for a detailed review, see ref.87), it is plausible to think that MAMs might be involved in the subversion of apoptosis and Ca2+ signaling during bacterial infections. Just to mention a few examples, bacterial toxins from pathogens such as Escherichia coli or L. monocytogenes, as well as flagellins of S. typhimurium and Pseudomonas aeruginosa, can induce an increase in free cytosolic Ca2+ in host cells, required for the toxin-mediated effects. Rimessi et al. demonstrated that P. aeruginosa affects Ca2+ signaling and mitochondrial function, in which flagellin is the inducer and MCU is a signal-integrating organelle member for NLRP3 activation and IL-1β and IL-18 processing88. Interestingly, Neisseria meningitidis mediates Ca2+ release by activating the IP3 receptors on the surface of the ER, promoting adherence and invasion into host endothelial cells89.
Notably, MAMs are the sites where autophagosome formation occurs. This is due to key factors of the initiating machinery of autophagy, such as ATG14 and ATG5, redistributing to MAMs upon autophagy induction90. Certain intracellular pathogens such as M. tuberculosis and S. typhimurium target and inhibit the autophagy response of the host cells during infection79; additionally, L. pneumophila restrains autophagy by the secretion of bacterial effectors91.
Taken together, these and other data demonstrated that bacterial pathogens can modulate some cellular processes in which MAMs are involved; however, whether pathogenic bacteria might target MAMs to disrupt these pathways has not been demonstrated yet.
Another pivotal role of MAMs is in the generation of DAMPs within host cells in response to cellular damage triggered by cellular stress. DAMPs derived from mitochondria, such as ATP, cardiolipin, cytochrome C, N-formyl peptides (NFPs), succinate, and others, play a central position as modulators of inflammation during different pathologies and have a central role in the activation of inflammation via NLRP392. Moreover, ROS derived from mitochondria can interact with and directly modify the function of DAMPs that, in turn, can regulate immune responses and contribute to the development of inflammatory diseases92.
An elegant study by Bronner et al. revealed that microbial infection with the Brucella abortus strain RB51 induced NLRP3 inflammasome activation leading to the induction of mitochondrial DAMPs93. This model of infection induced the activation of the ER stress marker IRE-1 that consequently promotes TXNIP1 translocation to mitochondria, which in turn promotes mitochondrial ROS production. This event leads to NLRP3-mediated crosstalk between ER and mitochondria, resulting in the release of mitochondrial contents through activation of the caspase-2–Bid signaling axis93.
In particular, the expression of TXNIP induced by ER stress is under the control of the IRE1α and PERK–eIF2α pathways of the UPR94. In different studies, it has been proposed that ER stress activates the NLRP3 inflammasome in a K+ efflux- and ROS-dependent manner that may also affect the mitochondria, suggesting the critical role played by the MAM as a site for signals exchange between the two organelles.
As previously described, TXNIP1 and IRE1a localize at MAMs31,95.
Under ER stress or after exposure to high concentrations of ROS, IRE1 is stabilized at the MAMs by Sig-1R95, and this enhanced cellular survival by prolonging the activation of the IRE1–XBP1 signaling pathway. Infection was often associated with ER stress, and animals deficient in components of the IRE1 signaling pathway were more susceptible to bacterial infection than controls96,97,98.
These findings indicate the direct involvement of MAMs in the generation of DAMPs and give rise to further speculations. Exploration of this platform during bacterial infection ensures promising and stimulating new research opportunities.
Role of MAMs in inflammatory diseases
Considering what has been previously outlined, it is not surprising that members of the inflammatory response are strongly implicated in the initiation or progression of different human pathologies. In particular, inflammatory response proteins have been most commonly implicated in neurodegenerative diseases, which may be attributed to different causes. For example, continuous release of IL-1β negatively modulates the integrity of the brain–blood barrier, which results in the infiltration of immune cells into the central nervous system99. The same cytokine amplifies the generation of other pro-inflammatory factors by stimulating the activation of microglia and astrocytes100. Moreover, it has been demonstrated that overexpression of IL-1β mediates neuronal injury and cell death throughout glutamate excitotoxicity101. An important link between inflammation and neurodegeneration could be found in the fact that the maintenance of the integrity of ER compartment is critical for the conservation of an appropriate MAMs network and functionality. Notably, misfolded protein aggregates and excessive accumulation of metabolites are critical determinants for the activation of ER-stress and NLRP3 inflammasome and, at the same time, are specific hallmarks of initiation and progress of neurodegeneration15.
A classic example may be found in Alzheimer’s disease (AD) that is characterized by aberrant accumulation of amyloid-β plaques in the cerebrum. Indeed, amyloid-β is a potent trigger for ER stress and ROS production102. In addition, it has been recently demonstrated that amyloid-β precursor and its catabolites also localize to the MAMs compartment, where they interact with MAM-resident proteins and modulate ER functions103. Considering all these aspects, it is easy to speculate that amyloid-β, ROS, and MAMs may be a platform sufficient to trigger the inflammatory processes (Fig. 4).

The activities of several MAM proteins linked to inflammation have been found to be disrupted during the pathogenesis of a number of human diseases. See text for further details
Intriguingly, amyloid-β was the first peptide associated with neurodegeneration to be shown to activate NLRP3 and promote the production and release of pro-inflammatory cytokines104.
These processes appear to be driven by the lysosomal protease cathepsin B. Indeed, during the phagocytosis of amyloid-β by microglial cells, lysosomes that have sequestered this protein lose their function and integrity, resulting the release of lysosomal components. Among these, cathepsin B was found to be necessary to mediate IL-1β production.
This discovery also displays important therapeutic aspects. Remarkably, it has been observed that, throughout inhibition of cathepsin B, it is possible to reduce the numbers of amyloid plaques and improve the memory in the AD mouse model104.
Other findings highlight the intimate relationship between amyloid-β and NLRP3. Recently, it has been observed that transgenic mice Nlrp3−/− or Casp1−/− with mutations associated with familial AD have reduced accumulation of amyloid-β, loss of spatial memory, and present enhanced tissue remodeling105. Accordingly, excessive levels of caspase-1 were found in the brains of AD patients.
Parkinson’s disease (PD) is a neurodegenerative disease characterized by an excessive death of neurons in the substantia nigra pars compacta (SNpc) caused by α-synuclein (αSyn) aggregates. The majority of αSyn resides in the cytoplasm. However, different investigations show the presence of this protein in the mitochondrial compartment in the striatum, SNpc, and cortex of PD brains, impairing the function of mitochondrial electron chain complexes106,107. Most recently, it has been shown that αSyn also localizes at the MAMs108. Additionally, in cells and animal models of PD with pathogenic mutations of α-syn, MAM functions are downregulated and, most importantly, that inflammasome is activated108,109.
Indeed, studies show that αSyn aggregates are sufficient to provoke IL-1β production by activating microglia and astrocytes. In addition, it has been demonstrated that there is an important difference between the fibrillar and monomeric form of this protein. Meanwhile, the monomeric αSyn only induces the expression of pro-IL-1β. The fibrillary form is able to provoke caspase-1 activation and maturation of IL-1β and thus fully activates the inflammasome. Interestingly, these findings have also been found in vivo. Indeed, transgenic mice lacking NLRP3 are resistant to developing PD following treatment with the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Despite this, the endogenous regulatory mechanisms of inflammasome activation in PD are still unclear. A role for ROS and cathepsin-B has also been suggested for this pathology. In fact, similar to AD, stimulating caspase-1 activation and the release of IL-1β is necessary to induce the production of ROS and activity of cathepsin-B110. Accordingly, through specific inhibition of cathepsin-B, it is possible to interfere with the inflammasome assembly. However, an in vivo animal model did not validate the findings of this study and experiments were only performed in monocytes. More relevant to this study, Yan et al. showed that dopamine-producing neurons and the NLRP3 inflammasome are tightly interconnected and are able to regulate each other111. Intriguingly, the authors demonstrated that the neurotransmitter dopamine (DA) has the potential to inhibit NLRP3 inflammasome activation and subsequent IL-1β production and that the mechanism underlying the inhibitory activity of DA on NLRP3 occurs via the dopamine D1 receptor (DRD1) signaling through an autophagic-dependent process. Most importantly, experiments in transgenic mice deleted for DRD1 or NLRP3 and treated with MPTP showed that DRD1 signaling counteracts MPTP-induced neuroinflammation by inhibiting the NLRP3 inflammasome. In addition, authors also found an important role for DA and DRD1 in regulating lipopolysaccharide-induced systemic inflammation and MSU-induced peritoneal inflammation111. Overall, these data highlight the possibility to consider DA and DRD1 as potential targets for counteracting inflammation in AD.
Insulin resistance and islet β-cell dysfunction in type 2 diabetes (T2D) are widely associated with disruptions of MAM compartments (Fig. 4). The group coordinated by J. Rieusset clearly demonstrated a strong relationship between MAM integrity and efficient insulin action in hepatic cells, indicating MAMs as novel actors in the mechanism of action of insulin liver112,113. Indeed, they demonstrated in vitro and in vivo that defective ER–mitochondria coupling is closely associated with impaired hepatic insulin sensitivity and restoration of MAM integrity by cyclophilin D overexpression improves insulin signaling in primary hepatocytes of diabetic mice113.
Accordingly, in the skeletal muscle of obese and diabetic humans, the expression levels of the ER–mitochondria tethering protein MFN2 are reduced. At confirmation, in vivo experiments demonstrated that feeding animals with high-glucose diet reduced MFN2 expression and attenuated insulin signaling that is reduced by promoting MFN2 overexpression. These processes appeared to be driven by a significant downregulation of mitochondrial activities. Indeed, the livers of transgenic mice deleted for MFN2 possessed a low insulin response and high number of fragmented mitochondria that reflected a reduction in mitochondrial respiration due to an atypical functioning of OXPHOS complex subunits. As a consequence, ROS production increased with subsequent accumulation of mutation at the level of mtDNA114. Increases in ROS levels appeared to be a primary contributor to inflammation in T2D. In fact, pro-inflammatory cytokines improved ER and oxidative stress events, leading to β-cell loss, recruitment of NLRP3 inflammasome, and finally, to the pathogenesis of T2D. Different molecular pathways have been proposed to work on this process. As previously described, increased ROS production triggers conformational changes in TXNIP and subsequent loss of the complex TRX–TRXNIP that binds and activates NLRP3, which produces IL-1β secretion31.
Interestingly, a recent study in human monocytes demonstrated that high glucose causes excess ROS production and TXNIP-mediated NLRP3 inflammasome activation through TRPM2-mediated Ca2+ influx and p47 phox signaling115; as such, TXNIP1 could be considered a potential therapeutic target for diabetes94.
Alternatively, a recent study suggested that ROS might activate NLRP3 via the action of intracellular AMP-activated protein kinase (AMPK) in an autophagic-dependent manner116. Notably, AMPK is a crucial mediator of metabolism of fatty acids and inhibits ROS production by regulating the expression and function of NADPH oxidase. In this elegant study, the authors demonstrated that a diet rich in saturated fatty acids promotes the inhibition of AMPK activation and reduction in autophagic activity. When the autophagic machinery is silenced, the ROS production increases and activates the NLRP3 inflammasome116.
MAMs and mitochondrial dynamics are also recognized as key factors in the pathogenesis of cardiac and vascular diseases (Fig. 4). The first support for this concept was found in the early 1990s, when it was demonstrated that the precise Ca2+ transport from the ER to the mitochondria regulates the cardiac contraction cycle117. Subsequent studies demonstrated that mitochondrial Ca2+ fluctuations and Ca2+ oscillation triggered by ER are present during cardiomyocyte beating118. Later, it was demonstrated that the precise transport from the ER to mitochondria is widely regulated by the appropriate composition of the MAMs fraction118. Among the proteins involved in the maintenance of MAMs, MFN1/2 seem to be the most relevant ones119. In fact, adult hearts deleted for both mitofusins showed compromised cardiac function, augmented left ventricular end-diastolic volume, and reduced fractional shortening120. In addition, the transgenic MFN2−/− mice exhibited reduced contact length between these organelles, a reduction in the ER-mitochondrial Ca2+ transfer, and increased ROS production.
Although only few studies identified a specific role for MAMs in cardiovascular diseases (CVD), it seems that these reactive chemical species are important contributors for different CVD. Interestingly, excessive ROS production and subsequent NLRP3 activation are frequently found in CVD, such as atherosclerosis (AS), where an excess of cholesterol is deposited in the arterial wall as cholesterol crystals leading to inflammatory injury. It has been demonstrated that, by internalizing these crystals, macrophages promote NLRP3 inflammasome activation in a process involving leakage of cathepsin B and L into the cytoplasm, excessive formation of mitochondrial ROS, and lowering in potassium concentrations121.
The important role of inflammasomes was confirmed in AS using ApoE−/− mice (transgenic mouse model of AS, which develop spontaneous AS when fed with a high-fat diet); deletion of IL-1β reduced the size of atherosclerotic lesions by up to 30%122. Moreover, the deletion of the IL-18 receptor (IL-18R−/−) decreased the size of the lesions123,124. Despite this, NLRP3 may be not the only source of pro-inflammatory cytokines in AS. In fact, transgenic mice ApoE−/− crossed with mice deleted for different components of the NLRP3 (Nlrp3−/−, Asc−/−, or caspase-1−/−) exhibited no differences in atherosclerotic lesions and plaques when compared to the double-knockout and control mice125. As such, further studies to elucidate the implication of NLRP3 inflammasome in AS are needed.
NLRP3 inflammasome recruitment and the appropriate MAM composition also have an important role during ischemia/reperfusion (I/R). Notably, IL-1β and IL-18 are primary mediators of I/R-induced human myocardial injury, and through the inhibition of caspase-1 activity it is possible to reduce the depression in contractile force after I/R in an I/R model126. Similarly, in ASC−/− mice the amount of inflammatory cytokines and resulting injuries such as the development of infarctions, myocardial fibrosis, and dysfunction in myocardial I/R injury were significantly reduced compared to wild-type controls127. Finally, it has been reported that specific proteins conserving the ER–mitochondria interface are involved in I/R. For example, OPA1 deficiency was associated with increased sensitivity to I/R, whereas the inhibition of FIS1 and DRP1 function was reported to be cardioprotective128.
To summarize, it is necessary to improve the understanding of the role that MAMs and inflammasome activation play in CVD. Nevertheless, the data suggest that both mechanisms may be considered potential therapeutic targets. Accordingly, treatments aimed to modulate ER–mitochondrial dynamics have been developed to target or treat cardiovascular diseases, and specific drugs counteracting the activity of components of inflammasome are available.
Conclusions
Accumulating evidence indicates that ER–mitochondria contact sites play important roles in promoting inflammation and the development of inflammatory diseases.
However, the new understanding of how MAMs could impact inflammatory signaling raises several questions. Can MAMs be attractive targets for bacterial proteins? Several important processes regulated at MAM level are modulated by pathogens to subvert host functions and promote infection, thus it is tempting to assume that pathogenic bacteria target MAMs to undermine key host cellular processes.
Importantly, inflammasome activity is crucial for the host response to microbial pathogens and possibly for optimal response to vaccine adjuvants.
Recent studies have shed light on MAM dynamics as critical regulators/effectors of antiviral signaling129; however, the function of MAM-mediated antiviral signaling deserves further investigation. With the current knowledge, MAMs appear to be a recruitment platform involved in the antiviral pathway. However, are other MAMs proteins, such as MAVS, required for this signaling for a proper antipathogen response and/or inflammatory response? Do the MAMs also transduce other innate immunity signaling? Future studies should provide answers to some of these interesting questions.
With regard to inflammasome activity, NLRP3 and other inflammasome members locate to ER–mitochondria sites to promptly sense the level of damage and to coordinate the appropriate response. In fact, not all inflammasome activation can be considered harmful, and the therapeutic inhibition of this pathway must be balanced with its beneficial contribution.
Importantly, a deeper understanding of the balance between beneficial and damaging inflammasome activation is also required in order to create new therapies for patients with inflammatory diseases.
Hence, increasing our knowledge regarding the molecular aspects and functions of different inflammatory players at MAMs is the only way to provide future therapeutic targets for a wide range of inflammatory diseases and to ameliorate patient outcomes.
References
Vance, J. E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 (1990).
Vance, J. E. Phospholipid synthesis and transport in mammalian cells. Traffic 16, 1–18 (2015).
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J. & Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 (2009).
Zhang, A. et al. Quantitative proteomic analyses of human cytomegalovirus-induced restructuring of endoplasmic reticulum-mitochondrial contacts at late times of infection. Mol. Cell. Proteomics 10, M111 009936 (2011).
Poston, C. N., Krishnan, S. C. & Bazemore-Walker, C. R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 79, 219–230 (2013).
Sala-Vila, A. et al. Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6, 27351 (2016).
Giorgi, C. et al. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox. Signal. 22, 995–1019 (2015).
Missiroli, S. et al. Endoplasmic reticulum-mitochondria Ca2+ crosstalk in the control of the tumor cell fate. Biochim. Biophys. Acta 1864, 858–864 (2017).
Marchi, S. & Pinton, P. Alterations of calcium homeostasis in cancer cells. Curr. Opin. Pharmacol. 29, 1–6 (2016).
Missiroli, S. et al. PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell. Rep. 16, 2415–2427 (2016).
Giorgi, C. et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 112, 1779–1784 (2015).
Marchi, S. et al. Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol. Cell. Oncol. 1, e956469 (2014).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 (2015).
Salaro, E. et al. Involvement of the P2X7-NLRP3 axis in leukemic cell proliferation and death. Sci. Rep. 6, 26280 (2016).
Baroja-Mazo, A. et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 15, 738–748 (2014).
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L. & Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 47, 15–31 (2017).
Jo, E. K., Kim, J. K., Shin, D. M. & Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 13, 148–159 (2016).
Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 35, 253–261 (2014).
Lee, G. S. et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 (2012).
Murakami, T. et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 109, 11282–11287 (2012).
Bonora, M. & Pinton, P. The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front. Oncol. 4, 302 (2014).
Misawa, T. et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 (2013).
Wu, J. et al. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp. Cell Res. 341, 42–53 (2016).
Zhong, Z. et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016).
Zhang, M., Kenny, S. J., Ge, L., Xu, K. & Schekman, R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife 4, e11205 (2015).
Dupont, N. et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 30, 4701–4711 (2011).
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z. & Germain, R. N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013).
Saxena, G., Chen, J. & Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 285, 3997–4005 (2010).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
D’Osualdo, A. et al. Transcription factor ATF4 induces NLRP1 inflammasome expression during endoplasmic reticulum stress. PLoS ONE 10, e0130635 (2015).
Paludan, S. R. & Bowie, A. G. Immune sensing of DNA. Immunity 38, 870–880 (2013).
Galluzzi, L., Kepp, O. & Kroemer, G. Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788 (2012).
Jacobs, J. L. & Coyne, C. B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 425, 5009–5019 (2013).
Yoneyama, M. et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858 (2005).
Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682 (2005).
Bender, S. et al. Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog. 11, e1005264 (2015).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Abe, T. & Barber, G. N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 88, 5328–5341 (2014).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Ishikawa, H. & Barber, G. N. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell. Mol. Life. Sci. 68, 1157–1165 (2011).
Nazmi, A., Mukhopadhyay, R., Dutta, K. & Basu, A. STING mediates neuronal innate immune response following Japanese encephalitis virus infection. Sci. Rep. 2, 347 (2012).
Jacobs, J. L., Zhu, J., Sarkar, S. N. & Coyne, C. B. Regulation of mitochondrial antiviral signaling (MAVS) expression and signaling by the mitochondria-associated endoplasmic reticulum membrane (MAM) protein Gp78. J. Biol. Chem. 289, 1604–1616 (2014).
Goldmacher, V. S. et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 96, 12536–12541 (1999).
Williamson, C. D. & Colberg-Poley, A. M. Intracellular sorting signals for sequential trafficking of human cytomegalovirus UL37 proteins to the endoplasmic reticulum and mitochondria. J. Virol. 84, 6400–6409 (2010).
Williamson, C. D., Zhang, A. & Colberg-Poley, A. M. The human cytomegalovirus protein UL37 exon 1 associates with internal lipid rafts. J. Virol. 85, 2100–2111 (2011).
Pearce, M. M., Wang, Y., Kelley, G. G. & Wojcikiewicz, R. J. SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J. Biol. Chem. 282, 20104–20115 (2007).
Browman, D. T., Resek, M. E., Zajchowski, L. D. & Robbins, S. M. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J. Cell Sci. 119(Pt 15), 3149–3160 (2006).
Hayashi, T. & Fujimoto, M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 77, 517–528 (2010).
Mavinakere, M. S. & Colberg-Poley, A. M. Dual targeting of the human cytomegalovirus UL37 exon 1 protein during permissive infection. J. Gen. Virol. 85(Pt 2), 323–329 (2004).
Bozidis, P., Williamson, C. D., Wong, D. S. & Colberg-Poley, A. M. Trafficking of UL37 proteins into mitochondrion-associated membranes during permissive human cytomegalovirus infection. J. Virol. 84, 7898–7903 (2010).
Kaarbo, M. et al. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 11, 935–945 (2011).
Roumier, T. et al. HIV-1 protease inhibitors and cytomegalovirus vMIA induce mitochondrial fragmentation without triggering apoptosis. Cell Death Differ. 13, 348–351 (2006).
McCormick, A. L., Smith, V. L., Chow, D. & Mocarski, E. S. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 77, 631–641 (2003).
Poncet, D. et al. Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vMIA. J. Cell Biol. 174, 985–996 (2006).
Norris, K. L. & Youle, R. J. Cytomegalovirus proteins vMIA andm38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 82, 6232–6243 (2008).
Castanier, C., Garcin, D., Vazquez, A. & Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 11, 133–138 (2010).
Munger, J., Bajad, S. U., Coller, H. A., Shenk, T. & Rabinowitz, J. D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2, e132 (2006).
Chambers, J. W., Maguire, T. G. & Alwine, J. C. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 84, 1867–1873 (2010).
Horner, S. M., Park, H. S. & Gale, M. Jr. Control of innate immune signaling and membrane targeting by the Hepatitis C virus NS3/4A protease are governed by the NS3 helix alpha0. J. Virol. 86, 3112–3120 (2012).
Schwer, B. et al. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 78, 7958–7968 (2004).
Piccoli, C. et al. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 46, 58–65 (2007).
Li, Y., Boehning, D. F., Qian, T., Popov, V. L. & Weinman, S. A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 21, 2474–2485 (2007).
Gong, G., Waris, G., Tanveer, R. & Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 98, 9599–9604 (2001).
Hu, W. S. & Hughes, S. H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2, a006882 (2012).
Stewart, T. L., Wasilenko, S. T. & Barry, M. Vaccinia virus F1L protein is a tail-anchored protein that functions at the mitochondria to inhibit apoptosis. J. Virol. 79, 1084–1098 (2005).
Everett, H. et al. M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 191, 1487–1498 (2000).
Chen, C. P. et al. Modulating the activity of the channel-forming segment of Vpr protein from HIV-1. Eur. Biophys. J. 39, 1089–1095 (2010).
Fang, H. Y. et al. ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell. Sci. 123(Pt 7), 1171–1180 (2010).
Huang, C. Y., Chiang, S. F., Lin, T. Y., Chiou, S. H. & Chow, K. C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE 7, e33657 (2012).
Chatel-Chaix, L. et al. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 20, 342–356 (2016).
Welsch, S. et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5, 365–375 (2009).
Aguirre, S. et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 8, e1002934 (2012).
Chan, Y. K. & Gack, M. U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 17, 523–530 (2016).
Giorgi, C., Wieckowski, M. R., Pandolfi, P. P. & Pinton, P. Mitochondria associated membranes (MAMs) as critical hubs for apoptosis. Commun. Integr. Biol. 4, 334–335 (2011).
Escoll, P., Rolando, M. & Buchrieser, C. MAMs are attractive targets for bacterial repurposing of the host cell: MAM-functions might be key for undermining an infected cell. Bioessays 39 (2017).
Stone, S. J. & Vance, J. E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 275, 34534–34540 (2000).
Garsin, D. A. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat. Rev. Microbiol. 8, 290–295 (2010).
Thiennimitr, P. et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. USA 108, 17480–17485 (2011).
Mellin, J. R. et al. Riboswitches. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science 345, 940–943 (2014).
Hayashi, T., Rizzuto, R., Hajnoczky, G. & Su, T. P. MAM: more than just a housekeeper. Trends Cell. Biol. 19, 81–88 (2009).
Gilk, S. D. Role of lipids in Coxiella burnetii infection. Adv. Exp. Med. Biol. 984, 199–213 (2012).
Danese, A. et al. Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta 1858, 615–627 (2017).
TranVan Nhieu, G., Clair, C., Grompone, G. & Sansonetti, P. Calcium signalling during cell interactions with bacterial pathogens. Biol. Cell 96, 93–101 (2004).
Rimessi, A. et al. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 6, 6201 (2015).
Asmat, T. M., Tenenbaum, T., Jonsson, A. B., Schwerk, C. & Schroten, H. Impact of calcium signaling during infection of Neisseria meningitidis to human brain microvascular endothelial cells. PLoS ONE 9, e114474 (2014).
Hamasaki, M. et al. Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 (2013).
Rolando, M. et al. Legionella pneumophila S1P-lyase targets host sphingolipid metabolism and restrains autophagy. Proc. Natl. Acad. Sci. USA 113, 1901–1906 (2016).
Krysko, D. V. et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164 (2011).
Bronner, D. N. et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 43, 451–462 (2015).
Oslowski, C. M. et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 16, 265–273 (2012).
Mori, T., Hayashi, T., Hayashi, E. & Su, T. P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 8, e76941 (2013).
Martinon, F., Chen, X., Lee, A. H. & Glimcher, L. H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418 (2010).
Bischof, L. J. et al. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 4, e1000176 (2008).
Pillich, H., Loose, M., Zimmer, K. P. & Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell. Microbiol. 14, 949–964 (2012).
Alvarez, J. I. et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731 (2011).
Ferrari, C. C. et al. Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am. J. Pathol. 165, 1827–1837 (2004).
Fogal, B., Li, J., Lobner, D., McCullough, L. D. & Hewett, S. J. System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J. Neurosci. 27, 10094–10105 (2007).
Tramutola, A., Lanzillotta, C., Perluigi, M. & Butterfield, D. A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 133, 88–96 (2017).
Del Prete, D. et al. Localization and processing of the amyloid-beta protein precursor in mitochondria-associated membranes. J. Alzheimers Dis. 55, 1549–1570 (2017).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865 (2008).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 (2013).
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M. & Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life. Sci. 65, 1272–1284 (2008).
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G. & Anandatheerthavarada, H. K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100 (2008).
Guardia-Laguarta, C. et al. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259 (2014).
Gustin, A. et al. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS ONE 10, e0130624 (2015).
Codolo, G. et al. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375 (2013).
Yan, Y. et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015).
Rutter, G. A. & Pinton, P. Mitochondria-associated endoplasmic reticulum membranes in insulin signaling. Diabetes 63, 3163–3165 (2014).
Tubbs, E. et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294 (2014).
Sebastian, D. et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 109, 5523–5528 (2012).
Tseng, H. H., Vong, C. T., Kwan, Y. W., Lee, S. M. & Hoi, M. P. TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci. Rep. 6, 35016 (2016).
Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 (2011).
Bassani, R. A., Bassani, J. W. & Bers, D. M. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J. Physiol. 453, 591–608 (1992).
Robert, V. et al. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 20, 4998–5007 (2001).
Dorn, G. W. 2nd, Song, M. & Walsh, K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J. Mol. Cell. Cardiol. 78, 123–128 (2015).
Papanicolaou, K. N. et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 31, 1309–1328 (2011).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Kirii, H. et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 656–660 (2003).
Mallat, Z. et al. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ. Res. 89, E41–E45 (2001).
Elhage, R. et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 59, 234–240 (2003).
Menu, P. et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2, e137 (2011).
Pomerantz, B. J., Reznikov, L. L., Harken, A. H. & Dinarello, C. A. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc. Natl. Acad. Sci. USA 98, 2871–2876 (2001).
Kawaguchi, M. et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123, 594–604 (2011).
Le Page, S. et al. Increase in cardiac ischemia-reperfusion injuries in Opa1/− mouse model. PLoS ONE 11, e0164066 (2016).
Horner, S. M., Wilkins, C., Badil, S., Iskarpatyoti, J. & Gale, M. Jr. Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 10, e0117963 (2015).
Acknowledgements
This work was supported by the Italian Association for Cancer Research (AIRC), the Italian Ministry of Health, and Cariplo to C.G. S.P. was supported by a research fellowship FISM, cod. 2014/B/3. M.R.W. was supported by the Polish National Science Centre grant (UMO-2014/15/B/NZ1/00490).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Missiroli, S., Patergnani, S., Caroccia, N. et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis 9, 329 (2018). https://doi.org/10.1038/s41419-017-0027-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-017-0027-2
This article is cited by
-
IP3R1-mediated MAMs formation contributes to mechanical trauma-induced hepatic injury and the protective effect of melatonin
Cellular & Molecular Biology Letters (2024)
-
The mechanism underlying correlation of particulate matter-induced ferroptosis with inflammasome activation and iron accumulation in macrophages
Cell Death Discovery (2024)
-
Mitochondria-associated endoplasmic reticulum membrane (MAM): a dark horse for diabetic cardiomyopathy treatment
Cell Death Discovery (2024)
-
Endoplasmic reticulum stress and the unfolded protein response: emerging regulators in progression of traumatic brain injury
Cell Death & Disease (2024)
-
CGI1746 targets σ1R to modulate ferroptosis through mitochondria-associated membranes
Nature Chemical Biology (2024)
