
Abstract
Extracellular chromatin, for example in the form of neutrophil extracellular traps (NETs), is an important element that propels the pathological progression of a plethora of diseases. DNA drives the interferon system, serves as autoantigen, and forms the extracellular scaffold for proteins of the innate immune system. An insufficient clearance of extruded chromatin after the release of DNA from the nucleus into the extracellular milieu can perform a secret task of moonlighting in immune-inflammatory and occlusive disorders. Here, we discuss (I) the cellular events involved in the extracellular release of chromatin and NET formation, (II) the devastating consequence of a dysregulated NET formation, and (III) the imbalance between NET formation and clearance. We include the role of NET formation in the occlusion of vessels and ducts, in lung disease, in autoimmune diseases, in chronic oral disorders, in cancer, in the formation of adhesions, and in traumatic spinal cord injury. To develop effective therapies, it is of utmost importance to target pathways that cause decondensation of chromatin during exaggerated NET formation and aggregation. Alternatively, therapies that support the clearance of extracellular chromatin are conceivable.

Similar content being viewed by others
Facts
-
DNA has two jobs. DNA is (I) responsible for heredity, and (II) can moonlight to orchestrate certain pathways of the immune response.
-
The pro-inflammatory/pro-thrombotic activity of extracellular DNA can be driven by (aggregated) neutrophil extracellular traps (NETs).
-
Rupture of the nuclear envelope, chromatin decondensation, loading of the chromatin with granular and cytoplasmic proteins, and plasma membrane breakdown are key cellular events for the release of chromatin during NET formation.
-
Reactive oxygen species (ROS) and an increase of intracellular calcium levels activate several downstream effectors that are crucial for NET formation.
-
Myeloperoxidase (MPO), neutrophil elastase (NE), peptidyl arginine deiminase 4 (PAD4), and a plethora of further intracellular proteins determine the functional capabilities of NETs.
-
Extracellular chromatin’s moonlighting tasks foster various pathological conditions and drive inflammatory diseases such as spinal cord trauma, cancer, sepsis, immunothrombosis, periodontitis, obstruction of exocrine ducts and glands, and formation of stones or adhesions.
Open questions
-
How can unbalanced actions of extranuclear DNA-protein complexes be rebalanced?
-
What therapies can effectively block pathways leading to aberrant NET formation?
-
What therapies can support the clearance of NETs and their aggregates?
-
How can the protective role of extracellular chromatin be preserved?
Introduction
DNA is a polymeric macromolecule that displays distinct molecular and functional properties depending on its location [1,2,3]. Inside the nucleus, DNA serves as the essential molecule of heredity, encoding information for gene structure and regulation. Nuclear DNA is bound to histones in the form of nucleosomes, constituting a material known as chromatin [4, 5]. Once outside the cell, DNA can expand in space and display other functional activities to drive inflammation and thrombosis [6]. Moonlighting is an extra activity or occupation, sometimes performed in secret. If heredity and gene regulation are DNA’s main functions in the nucleus, immunity is DNA’s main function in the extracellular space, whether tissue or blood.
The structural bases of the intracellular and extracellular activities of DNA differ. The nuclear functions of DNA result from gene sequences and base modifications, while the extracellular functions result primarily from the charged phosphodiester backbone and its extended polymeric structure. The structures of DNA, both sequence and backbone, facilitate the binding of proteins and provide the basis for a multitude of intermolecular interactions. The translocation of DNA from the inside to the outside of the cell is the key mechanism that reveals the full diversity of DNA’s biological activities [5].
As demonstrated in many model systems, the translocation of DNA outside the cell can occur with cell death, stress and injury, with cell death being the predominant source of extracellular DNA [7,8,9]. With cell death, DNA is a byproduct that is often considered as debris. This DNA is subject to rapid removal. With persistence and heightened levels, however, DNA can become noxious or “dangerous” as it can enter cells and interact with nucleic acid sensors; these sensors are part of an internal host defense system which can be triggered by foreign DNA from bacteria or viruses as well as self-DNA arising from cell stress or impaired nuclease activity [10, 11].
In addition to inadvertent or programmed cell death, extracellular DNA and chromatin can occur in the context of neutrophil extracellular trap (NET) formation [12]. NET formation is an elaborate program of polymorphonuclear granulocytes that involves the movement of DNA from the nucleus to the cytoplasm where it mixes with the contents of granules to form NETs. The latter play diverse and important roles in inflammation. The principal components of NETs, DNA and histones, are ancient and can even be found in archaea. From the point of view of evolution, it is noteworthy that extracellular chromatin decorated with histones and other antimicrobial proteins also occurs in invertebrates such as crabs, mussels and sea anemones [13], fish [14, 15], birds [16], as well as protozoans and plants [17].
Also, in mammals, extracellular DNA traps can originate not only from neutrophils [18] but also from other immune cells (eosinophils, dendritic cells, monocytes, macrophages, mast cells, basophils, T cells, and B cells); DNA traps can also arise from non-immune cells (endothelial cells, platelets, and cardiomyocytes) [19]. The evolutionary conservation of DNA traps suggests that the evolution of DNA has involved both hereditary and gene regulation as well as the potential weaponization against invading pathogens [20].
Depending on whether extracellular DNA or chromatin arises from cell death or NET formation, the molecular properties of the DNA, as well as the identity of associated macromolecules (e.g., histones, enzymes) will differ. The release of DNA from dead and dying cells can be studied in both in vitro as well as in vivo models, although in vivo models allow better assessment of potential mechanisms of clearance and degradation as well as interplay of dead cells with phagocytic cells [21,22,23,24]. In vivo models to study DNA translocation can involve the transfer of dead and dying cells to a recipient animal or the in vivo induction of apoptosis or necrosis. Other models involve infection or the stimulation of inflammation that can lead to cell death as well as NET formation. In models tested thus far, extracellular DNA shows a major peak of approximately 166 bases – the size of a mononucleosome – no matter whether induction of death was by apoptosis or necroptosis [21, 22]. This size range is the same as that observed in studies on the molecular properties of DNA in the blood [25].
As demonstrated in in vivo models, the translocation of DNA into the blood depends on macrophages and can be modulated by glucocorticoids as well as sex hormones [22, 26,27,28]. Thus, the occurrence of DNA in the blood is the culmination of complex processes that are subject to strict regulation, including nucleolytic digestion. As these processes proceed, the size of extracellular DNA changes since DNA, when released during NET formation, for example, can show very high molecular weight (thousands to tens of thousands of bases) while, in the blood, most of the DNA is less than 200 bases [25].
In addition to a soluble form, extracellular DNA can exist as a particle [29]. This particulate form of DNA resides in microparticles which are released from cells during apoptosis and likely correspond to blebs on the cell surfaces [30, 31]. This DNA is accessible and antigenically active and can be bound by monoclonal anti-DNA antibodies as well as sera from patients with systemic lupus erythematosus (SLE) to form large immune complexes [32,33,34,35,36]. Mitochondria represent a further source for extracellular DNA in a particle form that can bind anti-DNA antibodies [37]. Recognition of the various physicochemical forms of DNA circulating in the blood is important since their detection may differ depending on the use of sera or plasma as well as the conditions for isolation and analysis.
In this conceptualization, extracellular DNA or chromatin is an ensemble of molecules that vary in their origin from different cell populations; mechanisms of translocation (e.g., apoptosis, necrosis, NET formation); different physicochemical forms (i.e., high vs low molecular weight, soluble vs particulate); and the array of associated macromolecules. Rather than debris or a simple byproduct of cell death, extracellular DNA represents a multifunctional complex that displays activities to drive the pathogenesis of many diseases. Importantly, extracellular DNA and chromatin can provide a structure to organize and promote the activity of other mediators and thereby intensify inflammation and drive thrombosis.
The pro-inflammatory and pro-thrombotic activity of extracellular DNA occurs prominently during the process of NET formation, a unique element in host defense based on the elaboration of extracellular DNA in a high molecular weight form that can serve as a scaffold decorated by other intracellular molecules. This review will focus on the extracellular release of DNA during the process of NET formation and the many roles that NETs can play in disease.
The process of NET formation and degradation
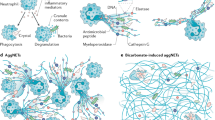
Depending on the cellular viability, NET formation has been classified as suicidal or vital NET formation [38,39,40,41,42]. Since the nuclear genome (~3.2 billion bp) is 200,000 times larger than the mitochondrial genome (16,569 bp) [38], nuclear DNA can indisputably form the backbone of the structure of NETs in suicidal NET formation (Fig. 1) [43]. However, mitochondrial DNA and over 20 other components are also associated with NETs [44, 45]. The nucleus is the source for extracellular DNA NETs in suicidal NET formation. In the nucleus, chromatin is enclosed by the nuclear envelope, which consists of outer and inner nuclear lipid membranes (ONM and INM) and the underneath nuclear lamina [38, 43, 46]. The latter is a filamentous structure consisting of A-type (A, C) or B-type (B1, B2) lamins [46]. A-type lamins are assembled as thick filament bundles, which affect the mechanical properties of the nuclei. In contrast, B-type lamins are assembled as a thin but highly organized meshwork which is crucial to the integrity and elasticity of the nuclear envelope [38, 43, 46].

Suicidal NET formation: a Chromatin decondensation is mediated by PAD4 and/or NE. b Nuclear envelope rupture is modulated by nuclear translocation of PKCα or CDK4/6 which mediate nuclear lamina disassembly (electron microscopy images of b1 well organized nuclear lamina, or b2 disassembled nuclear lamina [256]). c Rupture of the plasma membrane is achieved by disassembly of cortical cytoskeleton (c1, electron microscopy image of actin cortex [257]). (α, β) Representative confocal microscopy images of an untreated neutrophil (α) and a PMA-treated neutrophil with ruptured nuclear envelope and extracellular NETs in which nuclear DNA forms the backbone of NETs that are decorated with the disassembled lamin B (β), stainings of lamin B and DNA with fluorescent-labeled anti-lamin B1 and DAPI. Vital NET formation: Vital NET formation has been described as either derived through nuclear blebbing, or released from mitochondria.
The nuclear envelope is the first physical barrier for chromatin extranuclear extrusion. Nuclear envelope rupture occurs when the nuclear lamina is either cleaved proteolytically or disassembled by phosphorylation [38] (Fig. 1b). Lamin B is proteolytically cleaved by caspase-3 during apoptosis [43]. However, NET formation is a caspase-independent process [43, 47, 48], during which caspase-3 remains inactive [43]. Recent studies indicate that protein kinase C-α (PKC-α)-mediated lamin B phosphorylation and disassembly is responsible for nuclear envelope rupture [43, 49]. In addition, cyclin dependent kinase 4/6 (CDK4/6) controls NET formation through modulation of lamin A/C phosphorylation, resulting in nuclear envelope rupture [38, 50]. Mice with deficiency of CDK4/6 or PKCα [50, 51], or overexpression of lamin B [43], display impaired NET formation in vivo. Thus, kinase-mediated nuclear lamina phosphorylation-disassembly [43, 50], but not proteolytic cleavage [38], is responsible for nuclear envelope rupture during NET formation. PKCα and CDK4/6 are located in the cytoplasm of resting neutrophils, and their nuclear translocation requires a functional actin cytoskeleton in the early stage of neutrophil activation [38, 49, 52]. Genetic [49, 53] or pharmacologic [54] inhibition of actin assembly or its upstream regulatory molecules, Rho kinase [49] or Wiskott–Aldrich syndrome protein [53], impair NET formation. This argues for a crucial role of the actin cytoskeleton in NET formation [38, 52].
Nuclear DNA is tightly packaged as chromatin by histones. The extranuclear extrusion of chromatin requires its decondensation, which is mediated through histone citrullination by peptidyl arginine deiminase 4 (PAD4) [55] and/or histone cleavage by neutrophil elastase (NE) [39, 47] (Fig. 1a). In resting neutrophils, both PAD4 and NE are located in cytoplasmic granules [56, 57]. PAD4 has a nuclear localization sequence (NLS) which mediates nuclear translocation of cytoplasmic PAD4 [38]. Since NE does not have a NLS, it is unclear how NE is imported into the nucleus [38], however, the actin cytoskeleton might be involved in nuclear translocation of NE [58]. Also, CDK4/6- or PKCα-mediated nuclear envelope rupture may contribute to NE nuclear translocation which can be blocked by inhibition of these kinases [38, 50]. Furthermore, gasdermin D (GSDMD) pores may be involved in NE release from granules and its nuclear translocation. NE in turn may also process GSDMD for its maturation and pore formation in nuclear, granular, and plasma membranes [59, 60].
The plasma membrane is the second physical barrier for extracellular release of nuclear DNA. The cortical actin cytoskeleton is attached underneath the plasma membrane and strengthens its integrity [61, 62]. A recent study found that dynamic changes of actin polymerization in early stage, and actin depolymerization in late-stage, are accompanied by corresponding changes of Rho kinase activities [49]. The aforementioned dynamic changes explain the role of the actin cytoskeleton in the early-stage nuclear translocation of lamin kinase PKCα and CDK4/6 [38, 49, 52], and involvement of actin depolymerization in plasma membrane rupture in later stages of NET formation (Fig. 1c) [49, 62]. Disassembly of cortical cytoskeleton weakens the plasma membrane. This together with expanding forces from chromatin swelling [54], contributes to plasma membrane rupture and extracellular NET release.
Based on emerging evidence [38, 43, 49, 50, 54, 62], rupture of the nuclear envelope, nuclear chromatin decondensation, and the plasma membrane breakdown, are the key and necessary cellular events for nuclear chromatin extracellular release in suicidal NET formation (Fig. 1). The signaling pathways that regulate key cellular morphological changes might be candidate targets for therapeutics in NET-related diseases. Since NET formation has been described [63], and detailed in seminal experiments [18], the involvement of various signaling pathways has been reported [12, 38, 39]. Reactive oxygen species (ROS) are crucial for NET formation as they activate several downstream effectors. ROS modulate the release of granule myeloperoxidase (MPO) and NE [39, 47], and regulate cytoskeletal dynamics [53], which is involved in NET formation [49, 53, 54, 62]. Activated neutrophils generate ROS through activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 (NOX2) or mitochondrial dysfunction [39, 47, 48, 64]. Depending on the stimuli, NOX-dependent, and -independent pathways have been reported to drive NET formation [39, 47, 48, 64].
In NOX-dependent pathways, stimuli (like, PMA, LPS, PAF) activate NOX2 that drives NET formation through ROS generation [39, 47, 48, 65], while genetic mutation or pharmacological inhibition of NOX2 attenuates NET formation [39, 47, 48]. However, requirements for NOX are stimulus dependent, and NOX activity is not required for calcium ionophore-induced NET formation [66]. Calcium ionophores induce calcium influx, which activates the mitochondrial SK3 channel, resulting in mitochondrial ROS production for NOX-independent NET formation [64]. A recent study found that calcium ionophores activate calpain, which may with involvement of calcium-dependent PAD4 mediate interdomain proteolysis of the nuclear lamina and high mobility group box 1 protein (HMGB1); the latter an architectural chromatin binding protein [67]. The collective activity of PAD4 and calpain may contribute to the destruction of the nuclear lamina and, thus enable chromatin decondensation in calcium-mediated NET formation [67]. However, more detailed studies are needed for understanding the role of calpain in nuclear lamina disintegration. Intracellular calcium mobilization is also involved in regulation of actin cytoskeleton dynamics. These are important in nuclear translocation of PKCα and CDK4/6 for nuclear lamina disassembly [52] and NE for chromatin decondensation [58].
In contrast to suicidal NET formation, “vital NET formation” has also been reported as extracellular release of either mitochondrial [40] or nuclear [41] DNA, without loss of plasma membrane integrity (Fig. 1). Vital NET formation was initially described in neutrophils that were first primed by GM-CSF and then consequently stimulated with LPS/C5a, resulting in rapid release of NETs which DNA is solely from mitochondria [40]. An intact cytoskeleton is required for vital NET formation with involvement of ROS [68]. Extracellular release of mitochondrial DNA from viable cells has been observed not only in granulocytes [68], but also in lymphocytes [69] and amoebae [70]. This phenomenon has been considered an intrinsic innate immune response by either directly killing bacteria [70], or indirectly by inducing anti-viral interferons [68, 69]. Interestingly, mitochondrial DNA release has also been observed in viable fibroblasts [71] or chondrocytes [72], which might be an acute-phase response to mitochondrial stress or dysfunction. The latter findings, however, raise the question if extracellular release of mitochondrial DNA from viable cells is a broader phenomenon not limited to immune cells [73]. More studies are needed to understand this important and interesting phenomenon comparing extracellular release of mitochondrial DNA from viable immune cells vs non-immune cells.
In addition to mitochondrial DNA release, another study reported that exposure of neutrophils to Staphylococcus aureus can induce rapid NET formation without cell membrane breakdown [41]. Upon stimulation, the multilobular nucleus rapidly became rounded, followed by nuclear blebbing of chromatin containing vesicles, which deliver and release their contents into the extracellular space for NET formation [41]. The entire process occurs in 5–60 min in a ROS-independent manner [41] and requires chromatin decondensation [74]. Although the mechanism that regulates the nuclear blebbing in vital NET formation is unclear, histone modification during chromatin decondensation may contribute to nuclear blebbing, known to be determined by alteration of chromatin compaction and histone modification [75]. The budded nuclear vesicles may rupture over time and release their enclosed DNA into the cytoplasm, and eventually extrude into the extracellular space as described for suicidal NET formation [41]. Two studies found that parasites may induce rapid vital NET formation at 10–30 mins of neutrophil-parasite interaction, and suicidal NET formation when they are co-incubated for a longer time. The latter condition results in increased total NET formation [74, 76]. One may speculate that vital/rapid NET formation might be the early event of suicidal NET formation before the neutrophils lose their viability. More studies are needed to address the relationship between vital and suicidal NET formation with extracellular release of chromatin.
All in all, the coexistence between the suicidal lytic and vital NET formation remains uncertain [38, 45]. Most importantly, the diversity of NET formation signaling pathways makes it difficult to identify unified targets for therapeutic purposes of NET-related diseases. To control NET formation, there is a need for identifying the pathways in different clinical settings to allow specific targeting. Another option is to target and improve the degradation of NETs for a fine-tuning of the balance between NET formation and degradation. NETs are reportedly degraded by macrophages; preprocessing of NETs with DNase1 and opsonization with C1q facilitates this process [77]. Macrophages take up NETs through micropinocytosis, but how exactly the degradation is achieved warrants further research [77, 78]. Dendritic cells (DCs) are able to take up NETs, albeit to a much lesser extent than macrophages, and to secrete DNase1L3 for extracellular digestion of NETs [78]. Recently, it was also described that 13-series (T-series) resolvins reduce NET formation by enhancing NET uptake by human macrophages in a phospho–AMP-activated protein kinase (AMPK)-dependent manner [79]. One should be cautious in enhancing NET degradation since degraded NETs reportedly foster the growth of Actinobacillus pleuropneumoniae, causing severe porcine pneumonia;[80] this might also be true for further pathogens. The degradation of NETs by circulating DNases leads to the release of so-called NET degradation products (NDPs) such as cell-free DNA (complexed with MPO or NE) and histones. These NDPs themselves have toxic effects like fixation of complement (cell-free DNA) [81], induction of oxidative tissue damage (MPO) [82], promotion of thrombosis by local proteolysis of the tissue factor pathway inhibitor [83], or activation of platelets and cytotoxicity for epithelial cells (histones) [84,85,86,87].
The composition of NETs
NET-borne enzymes
During the process of NET formation or cell death, chromatin escapes the nuclear control with an abundance of various proteins. In a first assessment of the neutrophil proteome, a total of 251 major cellular proteins in different compartments were identified by gel-LC-MS/MS [88]. This proteome included the azurophilic granule proteins NE and MPO as well as PAD4, which catalyzes the deimination of arginine to citrulline and mediates chromatin decondensation [89, 90, 55]. In a recent proteome analysis by Petretto et al., 330 NET-associated proteins were identified; many with posttranslational modifications [91]. Of these, 74 were detected in all NETs but others differed dependent on the inducer of NET formation. Interestingly, the cellular origin of these NET-associated proteins seemed independent of the respective inducers with most proteins originating from the cytoplasm/cytoskeleton, followed by organelle- and membrane-derived proteins. The identification of only four to six different proteins associated with NETs from patients with SLE compared to rheumatoid arthritis (RA) determined that the nature of the stimulant is more important for the NET proteome composition than the underlying disease profile [92].
Aggregation of NETs
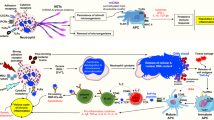
The binding of NE and other antimicrobial proteins to extruded chromatin of NETs mediate the digestion and elimination of microbial pathogens at the site of insult. At these areas of high cell densities, neutrophils tend to aggregate and form enzymatically stable clumps called aggregated NETs or aggNETs [93]. NETs and aggNETs immobilize, neutralize and/or kill bacteria [18], fungi [94], viruses [95], parasites [96], and inhibit their dissemination [97,98,99]. In addition, aggNETs also contribute to the resolution of inflammation. The externalized chromatin fibers are decorated with a plethora of cytoplasmic and granular proteases. Consequently, aggNETs can not only scavenge inflammatory mediators, but also degrade these molecules [100]. The proteolysis of toxic molecules like highly cationic histones, which had entered the extracellular space during NET formation, protects surrounding tissues from chronic damage and allows re-establishment of tissue homeostasis [101]. AggNETs also sequester and degrade inflammatory cytokines and chemokines. This process prevents further recruitment of neutrophils and supports the resolution of inflammation [93]. Despite possessing beneficial effects, NETs are also involved in the induction of pro-thrombotic events. NET-bound histones interact directly with T-cells resulting in Th17 differentiation [102], and activate platelets, thereby stimulating thrombogenesis [103]. Impaired NET aggregation and clearance can drive the development of autoimmunity and become detrimental. Therefore, a dysregulated immune response and an imbalance between NET formation and degradation can lead to devastating diseases as summarized in Fig. 2 and in the following paragraphs.

1. Neutrophils, via a trans-endothelial mechanism, are recruited to the site of an incident within hours. There they form NETs by releasing nuclear chromatin or mitochondrial DNA decorated with potent antimicrobial granular proteins. 2. Monocyte and activated macrophages secrete neutrophil chemoattractants that lead to a rapid influx of a large number of neutrophils. At these high densities, neutrophils aggregate, forming so-called aggregated NETs (aggNETs). These foster the resolution of inflammation by degrading small soluble mediators of inflammation. 3. Within days to weeks, after NETs enabled a successful entrapment of pathogens and restricted their dissemination, intracellular DNases break down the DNA backbone of NETs. These remnants can then be cleared by phagocytic cells (e.g., macrophages) to promote clearance and thereby restore homeostasis. 4. Ineffective clearance of NETs leads to their extended and prolonged ripening by forming stable aggNET-fibrin co-aggregates, in which fibrin polymerizes in the scaffold formed by NETs. In these aggregates fibrin can be citrullinated and, consequently, resists degradation by plasmin. In addition, the NETs incorporated in these aggregates were protected from DNases. A reduced clearance then may lead to long-term secondary inflammation and formation of stones and tophi.
Methods to detect NETs in tissue samples
The detection of tissue-borne NETs is crucial to identify dysregulated NET formation and clearance in infections, sepsis, autoimmune diseases, thrombosis, metabolic disorders, and cancer. Currently, the most general technique to visualize NETs in tissues is immunostaining of paraffin-embedded tissues followed by immunofluorescence microscopy. After deparaffinization of tissue sections, heat-induced epitope retrieval (HIER) buffer is used for rehydration, breaking the methylene bridges and making epitopes such as NE, MPO, and citrullinated histone H3 accessible for binding of antibodies. The antibody-stained tissues are counterstained with DNA intercalating dyes that have detected extended NETs in many tissue sections with reliable signal intensity [104]. Recently, a protocol for multiplex staining demonstrated the detection of NETs in paraffin-embedded human biopsies of phlegmonous appendicitis, lung abscess and non-small cell lung cancer [105]. Over the past two decades, numerous technological advances have been made in immunofluorescence microscopy. Recent achievements allow greater insights into the morphology and high-resolution analysis to detect subtle changes in the tissues. High resolution stimulated emission depletion (STED) microscopy was used to detect citrullinated NETs frequently recurring in tissue biopsies from patients with colon cancer. Using anti-DNA antibodies directed to extracellular chromatin, the authors distinguished between condensed and compacted chromatin inside neutrophils with a healthy appearance and ejected decondensed DNA displaying the typical characteristic of NETs [106].
Label-free emission recorded in immunofluorescence microscopy of tissue biopsies from patients with COVID-19 revealed widespread immune thrombosis in a devastated pulmonary vasculature due to native endogenous fluorophores. Contrary to standard detection of NETs via immunofluorescence using antibodies, a recently published article reported the detection of NETs in inflammatory diseases using a fluorogenic peptide. The authors developed a highly specific, triple-quenched, tri-branched fluorescent human neutrophil elastase sensor. This sensor offers a more than 20-fold increase in fluorescence intensity upon enzymatic cleavage and extends the current detection methods from antibodies to the first molecular probe detectors [107].
Additionally, several live-cell imaging techniques directly monitor the neutrophils’ release of NETs at the cellular level in various complex tissue and organs affected by infection and autoimmunity. Two-photon microscopy enables real-time detection of NETs in Aspergillus fumigatus infected murine lung lobules using the SYTOX dye without organ fixation [108]. Intravital microscopy showed activated platelets resulting in the formation of NETs in liver sinusoids [109], and using SYTOX dye, spinning disk confocal intravital microscopy visualized the formation of NETs, over two hours on murine skin infected with Staphylococcus aureus [110,111,112]. Cell-permeable and impermeable DNA dyes allow the direct visualization of intact neutrophils and of NETs in various organs, respectively [112,113,114]. Intravital microscopy with a laser scanning microscope characterized NETs in blood vessels of different organs [110, 115].
To date, classical antibody-based immunofluorescence methods are still the most commonly used to detect neutrophils and NETs in tissue sections. However, many promising new methods like live-cell imaging and STED microscopy have recently been developed to directly monitor neutrophils and NETs in vivo and with ultrahigh resolution, respectively.
Extracellular chromatin moonlighting diseases
Immunothrombosis
In the last decade, it has become increasingly clear that neutrophils and especially NETs are intertwined into the processes of thrombus formation and maturation in diverse pathological settings [116]. The term immunothrombosis has been coined to highlight the interaction of the cellular innate immune system with pathological thrombosis [117]. The evolutionary advantage of the activation of thrombosis by players of the innate immune system lies in physically trapping infectious agents in occluded vessels to limit spread via the circulation and thus contain an inflammatory focus [118]. Exaggerated immunothrombosis, however, is central in the exacerbation of several pathological settings including coagulopathy in sepsis [109], necroinflammation [119], and severe COVID-19 [120, 121]. As shown in pancreatitis [122], not only blood vessels, but also glandular ducts can be occluded by NETs. Apart from physical trapping of microbes, immunothrombosis also fulfils beneficial hemostatic tasks in the setting of mucosal damage in acute flares of ulcerative colitis [123]. Here, the absence of PAD4 is associated with increased mucosal blood loss. The interactions of neutrophils and components of NETs with classical players of thrombosis are numerous; these players include platelets, serine proteases of the coagulation cascade, fibrinolysis and the fibrin mesh itself. Neutrophils and NETs interact with these players via membrane-bound receptors, degranulated effector proteins, chromatin of NETs, including NET-bound nuclear and granular proteins [124] and extracellular vesicles [125]. Activated platelets can induce formation of NETs [126]. More specifically, deletion of HMGB1 in platelets reduced NET formation and associated organ damage in various experimental models [127]. Vice versa, platelets may bind to and aggregate on extracellular chromatin of NETs [103].
The aggNETs provide a scaffold for thrombus formation and are able to occlude vessels and ducts [103, 128]. The obstruction of the microvasculature in organs and a consequent inhibition of the blood flow in the capillaries, together with NET-driven endothelial dysfunction, may precipitate organ failure and mortality [129, 130]. Thereby, aggNETs can contribute to the pathogenesis of various diseases. As already mentioned above, NETs occlude the vessels in patients with COVID-19 [120]. Furthermore, NETs-associated occlusions have been reported for coronary vessels in acute myocardial infarction and artherosclerosis [131, 132] and for cerebral vessels in ischemic stroke [133].
In addition to cellular interactions which may foster NET formation and pathological thrombosis, soluble mediators are studied. PAD4 becomes a focus of attention since it reportedly links inflammation and thrombosis. Injection of recombinant human PAD4 in vivo induced the formation of von Willebrand factor (vWF)-platelet strings in mesenteric venules. These strings are naturally degraded by ADAMTS13, a metalloproteinase, but citrullination of ADAMTS13 dramatically reduces the endogenous enzymatic activity [134]. In line with these findings, studies have shown that a class of serpins with an arginine residue in the P1 position (including antithrombin, C1INH, 1-antiplasmin, PAI1/PAI2) is inhibited by citrullination [135, 136], thus unleashing the proteolytic power of the serine proteases thrombin, plasmin and tissue plasminogen activator in the thrombo-inflammatory microenvironment. PAD4-guided thrombin activation may then further facilitate thrombus maturation by FXIII-mediated cross-linking [123]. Citrullination has also recently been identified as a major posttranslational modifier that impacts proteolysis [67]. Future studies will unravel the translational potential of targeting immunothrombosis in clinical settings. These studies could explain the puzzling failure of classical anticoagulants to prevent thrombus formation under certain septic conditions and in disseminated intravascular coagulopathy. Pan-PAD inhibitors and PAD4-specific inhibitors are already central tools in the research of NETs and immunothrombosis and will surely be further studied as therapeutic options in general.
NETs in lung diseases
Chronic respiratory diseases affect the airways and other structures of the lung [137]. In 2017, 544.9 million people worldwide were affected by a chronic lung disease, such as asthma or chronic obstructive pulmonary disease (COPD), making them the third leading cause of death behind cardiovascular diseases and cancer [138]. Even though asthma was always considered to be an eosinophilic disease, recent reports also highlight the role of neutrophils in this disease [139,140,141]. Similar to this subset of patients with neutrophilic asthma, patients with COPD show high neutrophilic airway inflammation; higher levels of blood neutrophil counts have been correlated with mortality in these patients [142, 143]. In both lung diseases, NETs were found in the airways of patients and were associated with inflammation [144], and in the case of COPD, also with airflow limitation [145].
Next to chronic respiratory diseases, acute lung injury and acute respiratory distress syndrome (ARDS) are further major causes of morbidity and mortality, especially in the critically ill patients. In these disorders, acute lung inflammation, as indicated by excessive transepithelial neutrophil migration and the release of pro-inflammatory and cytotoxic mediators, disrupts the endothelial and epithelial barriers of the lungs [146, 147]. Increased plasma levels of NETs have been associated with ARDS severity and mortality and lower plasma levels of DNase1 were associated with the development of sepsis-induced ARDS. This indicates that a balance in NET formation and degradation is crucial to prevent lung injury [148]. In this context, disulfiram, an aldehyde dehydrogenase inhibitor, was recently shown to inhibit NET formation and to protect from acute lung injury in a mouse model [149].
Cystic fibrosis (CF) is characterized by impaired mucus hydration and clearance due to mutations in the CFTR gene leading to chronic pulmonary infection and (neutrophilic) inflammation [150]. The sputum of patients with CF is heavily loaded with NETs and NET-related proteins. The activity of NE and the presence of MPO are correlated with disease progression, severity and reduction in lung function [151,152,153,154]. Interestingly, it was shown that NE has a higher enzymatic activity within the extracellular DNA of sputum from patients with CF [155].
Despite the seemingly negative influence of neutrophils and NET formation on disease severity in the above-mentioned lung diseases, it is also becoming increasingly clear that it is not the NET formation per se that is responsible for worse disease outcomes but rather an imbalance in NET formation and degradation. It was, for example, shown in a murine model of pathogen-induced lung injury that a complete PAD4 deficiency reduced NET formation and, therefore, lung injury but was counterbalanced by an increased bacterial load and inflammation [148]. Additionally, neutrophils seem to be not only responsible for tissue disruption and early lung damage but also for orchestrating later repair. Here they promote epithelial proliferation and release proteases, needed for the processing of the collagen scar [156].
NETs in autoimmune diseases
Autoimmunity is defined as loss of self-tolerance, meaning that, cellular or humoral immunity or both, respond against endogenous macromolecules and cells. If this response injures cells or tissues, it is usually referred to as autoimmune disease [157]. Despite their importance in pathogen clearance, NETs contribute to the development and pathogenesis of various autoimmune diseases such as RA, SLE, Anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV), anti-phospholipid Syndrome (APS), psoriasis, and others [158, 159].
Disruption of the balance between NET formation and degradation by DNases in favor of the formation results in accumulation of the released chromatin and the associated proteins into the extracellular matrix, the interstitium and into the lumina of vessels and ducts. Here these released nuclear constituents can serve as sources of autoantigens that may drive the development of autoantibodies and immune complexes, especially if the material carries post-translational modifications like oxidation [160], citrullination [161], carbamylation [161], or neoepitopes generated after proteolytic cleavage [162]. These autoantibodies are directed against a plethora of highly variable disease-specific targets, like double-stranded (ds)DNA in SLE [163], citrullinated proteins in RA [164], anti-lysosome-associated membrane protein 2 and anti-MPO in AAV [165, 166], or phospholipids in APS [167]. These autoantibodies can also alter the persistence and immunogenic potential of NETs themselves. Binding of autoantibodies to the chromatin structures stabilizes NETs and prevents their degradation by DNases [168, 169]. Anti-dsDNA-NET complexes of patients with SLE stimulate type I interferon secretion by mononuclear phagocytes, and NF-κB activity in endothelial cells in an Fc-gamma dependent manner. Thus, they enhance inflammatory immune responses and foster the progression of disease and autoimmunity [170]. NET formation in capillaries and aggNET formation in larger vessels activate endothelial cells and the coagulation cascade, and promote platelet aggregation. Together with the autoantibodies, NETs build immune complexes and foster thrombogenesis. The occlusion of vessels, especially those of the microvascular bed, can precipitate organ damage, and can even be fatal [171].
NETs additionally modulate immune responses through interaction with other immune cells and humoral components; NET-associated proteins, especially histones, serve as damage-associated molecular patterns [161]. NETs activate the inflammasome [172], the complement system via classical, alternative and lectin pathways [81, 165], and the coagulation cascade [171]. The formation of NETs by splenic neutrophils induces immunoglobulin class switch and, thus, can shape B cell responses [173]. In SLE, specific LL37-DNA complexes trigger self-reactive memory B cells for autoantibody production [174]. NETs lower the activation threshold of T cells [175], and directly activate production of type I interferons by plasmacytoid dendritic cells (pDCs), the hallmark cytokines of SLE [176].
A self-reinforcing loop of dysregulated NET formation and inflammation is created, as some NET-mediated responses drive further neutrophil attraction and NET formation [158]. In AAV with microscopic polyangiitis and SLE, this loop is exacerbated by reduced DNase1 activities and the consecutive accumulation and aggregation of NET remnants [177]. Low DNase activities can occur by genetic deficiency [178], consumption of the enzyme, circulating inhibitors [168] [179] or autoantibodies impairing the activity of DNase1L3 as observed in patients with sporadic SLE [180]. Hence, prevention of NET formation by inhibitors or supporting NET degradation by addition of DNases represent therapeutic approaches for the treatment of NET-driven chronic autoimmune diseases [171, 181].
Obstruction of exocrine ducts and stone diseases
The original defensive function of NETs can contribute to the development of obstructive and subsequently inflammatory diseases when ducts of exocrine glands are affected. Neutrophils physiologically patrol the ducts of exocrine organs [122, 182, 183]. The factors that trigger NET and aggNET formation within the ducts are various, ranging from changes in ion concentrations or pH, to crystal precipitations, bacteria or foreign bodies [93, 122, 182, 184, 185].
Irrespective of the cause, aggNET formation results in reduction of the excretory flow and further accumulation of occlusive material within the NETs. Secretory stasis, which develops gradually, can in turn create an environment in which calculi can form. This process initiates a vicious circle of further obstruction of the ducts, flow rate reduction and inflammation of the adjacent gland, as reported for the pancreas [122, 186], the gall bladder [182], tooth-supporting tissues [187,188,189], ocular [190, 191] and salivary glands [185].
By incorporating crystals, pathogens, cellular debris and viable immune cells, aggNETs serve as a glue that increasingly condenses the material to facilitate tophus and calculus formation, as observed in gouty arthritis [161, 192], and stone diseases like cholelithiasis [12], and sialolithiasis [185], and meibomian gland disorders.
Gouty arthritis develops as uric acid precipitates in the form of monosodium urate (MSU) crystals in the joints, causing acute inflammation [93, 193]. The crystals are taken up by resident macrophages [194], followed by NALP3 inflammasome activation [195, 196], pro-inflammatory cytokine secretion, and abundant neutrophil recruitment [197, 198]. The latter bind to the crystals and induce NET formation [199]. During this process, the neutrophils release pro-inflammatory mediators, like tumor necrosis factor α (TNFα) [200] and interleukin-6 (IL-6), and the neutrophil attractant CXCL8 as well as the elicitor of neutrophil extravasation, CCL3, and CXCL10, which plays a critical role in oxidative stress induced inflammation [192, 201, 202]. In the presence of high neutrophil counts, the NETs are not sufficiently degraded by DNases. The NETs tend to co-aggregate with the crystals and form tophi that may reach several cm in size. When the pro-inflammatory boost has terminated, the tophus-borne proteases facilitate the resolution of inflammation by degrading certain cytokines and chemokines [93, 197]. In vitro, MSU crystals also induce ROS-dependent NET formation [193]. Cholesterol crystals activate the complement system, and generate C3a and C5a that facilitate further neutrophil influx [203]. In contact with the crystals NETs are generated and promote growth of gall stones. Various kinds of crystals, among other sterile stimuli, are potent inducers of neutrophil activation and NET formation [204].
Likewise, sialoliths are formed in patients with sialadenitis. The presence of leucocytes in saliva, along with high concentrations of bicarbonate ions and calcium-based crystals, trigger NET formation, a step that contributes to the development of sialoliths [185]. The aggregation of NETs then leads to its growth, a common final path in sialolithogenesis. Sialoliths reflect the mechanism of lithogenesis by their layered structure of alternating organic and inorganic components, resulting in an appositional growth of a stone. Once a macroscopic sialolith is formed, salivary gland ducts may be occluded and lose their function [185]; chronic inflammations and autoimmunity may occur. Further candidates for NET-driven pathologies are stones in kidney, pancreas, prostate, as well as calcinosis cutis.
NETs in the periodontal crevice precipitate periodontitis
The first encounter between neutrophils and dental biofilms occurs within the space delineated by gingival and oral tooth surfaces. This space, referred to as gingival crevice, is filled with a transudate from blood plasma referred to as periodontal crevicular fluid. The latter is characterized by excessive NET formation in periodontitis [187, 189]. The crevice contains abundant outer membrane vesicles originating from the dental biofilm that are endocytosed by crevicular neutrophils. These vesicles orchestrate bacterial colonisation, delivery of virulence factors, and pathogenesis. Bacteria use outer membrane vesicles to modulate the host’s immune response, which eventually allows the bacteria to evade the immunity of the host [205].
When the pre-activated neutrophils that infiltrate the gingival epithelium enter the periodontal crevice [206], they encounter and endocytose a multitude of lipopolysaccharide-filled outer membrane vesicles [205, 207]. These vesicles, containing components of the bacteria’s outer membrane, had been translocated from the early endosomal compartments into the neutrophils’ cytosols, the caspase-4/11/GSDMD signaling pathway is activated and NETs are formed [208,209,210,211]. Indeed, caspase-4/11-deficient neutrophils form fewer NETs when compared with wild type controls [208]. Surprisingly, the toll-like receptors (TLR) 2, 3, 4, 7, and 9 are not required for NET formation by neutrophils stimulated with outer membrane vesicles from oral pathogens in vitro [212]. TLR4 is not necessary for caspase-4/11-mediated NET formation [211]. Chloroquine (inhibitor of TLR3, 7, and 9) and oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (inhibitor of TLR2 and 4) did not affect NET formation when activated with supernatant of oral pathogens in vitro [212]. Virtually all crevicular neutrophils appear to be in some stage of NET formation [187]. Despite the ability of caspase 4/11 to induce NETs independently of NE, MPO and PAD4 [59], NE translocation and H3 citrullination was observed in virtually all crevicular neutrophils [187] and suggests their involvement in crevicular NET formation.
NET formation in ischemic disease
Many steps of NET formation depend on a sufficient supply with oxygen in tissues [213]. Also, changes of pH in the microenvironment influence neutrophil capabilities of NET formation [184]. Tissue alkalosis favors the release of NETs whereas more acidic conditions lead to reduced NET formation [184]. Commonly, tissue hypoxia is accompanied by acidification of the microenvironment due to metabolic changes.
In human tissues, hypoxia-inducible factor-1α (HIF-1α) regulates cellular responses to low oxygen [214, 215]. Under hypoxia, HIF-1α is stabilized and translocated to the nucleus where it induces the transcription of hypoxia-regulated genes [216]. In neutrophils, translocated nuclear HIF-1α upregulates the transcription of NF-kB, prolongs their viability [216], and promotes degranulation and chemotaxis during hypoxia [217, 218].
In human acute myocardial infarction, an ischemic disease, infiltrating neutrophils show high nuclear HIF-1α content and remain primarily viable [219]. In contrast, neutrophils with low nuclear HIF-1α protein levels form NETs [219]. By staying viable in hypoxic tissue neutrophils remain capable of phagocytosis and clear cell debris as an important step of wound healing [220].
The role of NETs in cancer
The increased occurrence of NETs in tumor indicates a worse prognosis for cancer patients. NETs are associated with a high histopathological tumor grade, disease progression, metastasis and reduced disease-free and cancer-related survival in various cancer entities [106, 221,222,223]. The presence of NETs in cancer patients is often indirectly detected through high serum levels of MPO-DNA complexes [222, 224, 225] and to a lesser extent directly in the tumor tissues [105, 106]. A recent study showed that citrullinated NETs in human colon cancer tissues were correlated to the stages 3/4 [106].
The functions of NETs in tumorigenesis have been extensively evaluated in murine models. Interestingly, surgical stress and increased LPS levels after postoperative infections were found to induce NET formation concomitantly with an increased occurrence of metastases [222, 225]. NETs may directly support metastasis formation by trapping tumor cells at the distant site through the coiled-coil domain containing protein 25 (CCDC25), which can bind to NETs and is expressed on certain cancer cells (colorectal, breast, prostate, liver) [223]. Moreover, many key processes involved in cancerogenesis and metastasis are co-regulated by NETs. These processes include the establishment of an immune evasive micromilieu, the activation of dormant tumor cells, tumor cell extravasation, angiogenesis and vascular permeability [226, 227]. Moreover, NETs promote a mesenchymal, pro-metastatic phenotype in breast [228], colorectal [106], gastric [229], and pancreatic cancer [230] cell lines by inducing epithelial-mesenchymal transition (EMT) associated with an increased migration and invasion of the tumor cells. Specifically, the protein content of NETs seems to be necessary for the EMT induction [106]. Altogether, the present knowledge indicates that NETs may be active components in the progression of cancer and putative targets of therapy and prevention.
NETs in ocular diseases
NETs serve important functions in ocular antimicrobial responses [231, 232]. However, NETs are also involved in pathologies of the eye. Severe cases of chronic Dry Eye Disease (DED) in graft-versus-host disease have been associated with NETs [233] and hyperosmolar stress is thought to induce NET formation on the ocular surface of patients with DED [234]. NETs are involved in molecular pathological alterations in patients with corneal injuries [232, 235]. The choroidal and retinal compartments can also be affected by NETs. Patients with Behcet’s disease, a subtype of non-infectious uveitis, showed an increased formation of NETs possibly responsible for the extended vasculitis in this disease [236]. In vivo and in vitro data of diabetic retinopathy, a major reason for irreversible blindness worldwide, suggest that high blood glucose levels can induce NET formation [237, 238].
Perspective
In addition to the aforementioned diseases, further moonlighting tasks of extracellular chromatin in the form of NETs come to light. There is increasing evidence that NETs contribute to peritoneal adhesions and traumatic spinal cord injury as discussed shortly in the following paragraphs.
NETs in adhesions
Peritoneal adhesions are a common consequence of serosal repair after almost all abdominal interventions. Adhesions are associated with serious complications such as intestinal obstruction, pelvic pain, and infertility [239]. As a result, the quality of life of millions of patients throughout the world is affected. In the US, complications from adhesions cost more than two billion dollar per year and are responsible for more than 5% of hospital re-admissions in surgery [240].
Recently, it has been reported that formation and aggregation of NETs worsened primary and secondary intention wound healing. NETs intensified and prolonged the inflammatory phase [241]. Activated neutrophils have been found in burn patients even months after the initiating thermal injury [242]. DNases reportedly accelerated dermal wound healing in mice and in diabetic patients [238, 241]. Contrary to the notion that inflammation is essential for wound healing, areas with low levels of neutrophils, macrophages and T cells, like oral wounds, healed faster with almost no scarring [243].
Despite its clinical impact, the pathomechanisms of adhesions remain poorly understood. Adhesion formation is a form of peritoneal healing which consists of hemostasis, angiogenesis, and tissue remodeling [244]. The most important factor appears to be the inflammation orchestrated by the innate immune system [245]. Within hours, neutrophils are recruited to sites of bacterial infection or sterile tissue injury where they start phagocytosis, degranulation, and NET formation [18, 244].
A recent preprint describes that abundant fibrin-associated NET deposits form murine as well as human adhesions. The digestion of extracellular DNA with DNases abolished the formation of adhesions induced after surgical procedures. These data suggest that NETs form the first scaffold and that fibrin attachment stabilizes the primary structure to eventually form mechanically robust adhesions [246]. Mice with a targeted deletion of PAD4 (Padi4-/-) or mice treated with DNases showed significantly reduced adhesions.
NETs in spinal trauma
Traumatic spinal cord injury (tSCI) can result in permanent paralysis of patients. Beside individual sequelae and psychosocial trauma, the socioeconomic burden is substantial [89]. A recent multicenter cohort study showed that tSCI patients are among the most resource intense patient group of $11.193 per admission in Canada [247]. Treatment strategies include surgical intervention, type and timing of anticoagulation, as well as the use of corticosteroids. The long-term use of the latter has a number of adverse effects. Evidence-based strategies and novel pharmacologic treatment options are lacking [248,249,250].
The pathophysiology of tSCI consists of two distinct phases. A primary mechanical injury disrupts axons, neuronal cells, surrounding glia cells, and the blood-brain barrier. Neutrophils accumulate locally within minutes after spinal injury and initiate a phase of secondary damage via the release of NETs, aggravating cell damage and severity of neurological deficits after the initial trauma [251]. Secondary injuries include vascular damage, disturbed hemostasis, edema formation, and particularly inflammation [248].
A murine model showed that a decrease of neutrophil infiltration by selective inhibition of phosphodiesterase 4 (PDE4-I 0.5 or 1.0 mg/kg s.c. bolus; IC486051) decreased MPO, key markers of oxidative stress, and leukocyte infiltration. This resulted in cellular protection, locomotor improvements (by the Basso-Beattie-Bresnahan scale (BBBS)), and reduced neuropathic pain [252]. MPO, a hallmark enzyme of NETs, increased the levels of the pro-inflammatory cytokines IL-6, IL-1ß and TNFα [253]. Indeed, in a murine model of spinal tSCI, the lack of MPO reduced neutrophil infiltration, pro-inflammatory cytokine expression and apoptosis. Consequently, the motor recovery of the MPO knockout mice was improved (by BBBS). The findings indicate that MPO precipitates secondary injury and exacerbates tissue damage after tSCI, mainly via MPO-derived HOCl mediated apoptotic cell death [254].
Interestingly, intravenous DNase1 treatment (5 mg/kg) of rats one hour after tSCI decreased pro-inflammatory cytokine levels in favor of the anti-inflammatory IL-10. The treatment attenuated the NET-induced neuroinflammation and the tSCI-associated edema; it reduced glial and fibrotic scarring as quantified 28 days after injury [251].
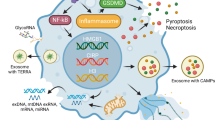
Considering these findings, anti-NET-therapies (PAD4 or DNase1) should be evaluated in further mechanistic studies (Fig. 3). The aim of these studies is to transfer in vitro data and the pre-clinical findings of the animal models from bench-to-bedside into patients with tSCI. Topical applications of DNases could be greatly beneficial during surgical decompression of the spine, minimizing secondary injury and scarring. Pharmacological stunning of neutrophils during the extended surgical window or treatment of patients with heparin, which has been shown to dismantle NETs and to limit NET formation [103, 255], are further treatment possibilities.

Neutrophils infiltrate the lesion core within hours and release NETs which lead to local tissue damage disrupting the blood-spinal cord barrier (BSCB). Resulting tissue hypoxia promotes neuronal apoptosis. Inhibition of peptidylarginine deiminase 4 (PAD4) or degradation of NETs via DNase1 could alleviate damage and promote functional recovery after tSCI.
Conclusion
Chromatin extrusion and the formation of NETs have been the subject of intense investigation since its discovery in 2004. The characteristic feature of these DNA traps is to limit the spread of invading pathogens, kill or suppress them and preserve tissue integrity. NET formation is evolutionarily preserved and must be considered advantageous (Graphical Abstract). Generally, NETs are beneficial. However, there is also a downside to NETs. In their fight against invaders, infiltrating neutrophils and excessive NET formation trigger many pathological processes. NETs can be considered offenders in immunothrombosis, autoimmune diseases, gout, obstruction of exocrine ducts and stone formation, periodontitis, adhesions, spinal trauma, cancer and ocular disorders (Graphical Abstract). Given the involvement of NETs in various pathologies, several state-of-the-art technologies have identified moonlighting extranuclear DNA-protein complexes. As a result, many interventions, especially regarding different forms of DNase and heparin, have been shown to accelerate the disruption and clearance of NETs. However, effective therapies are required for the future that can block pathways leading to the aberrant formation of NETs and preserve the ejected DNA’s protective role.
References
Duvvuri B, Lood C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. Front Immunol. 2019;10:502.
Soni C, Reizis B. Self-DNA at the epicenter of SLE: immunogenic forms, regulation, and effects. Front Immunol. 2019;10:1601.
Pisetsky DS. The origin and properties of extracellular DNA: from PAMP to DAMP. Clin Immunol. 2012;144:32–40.
Onufriev AV, Schiessel H. The nucleosome: from structure to function through physics. Curr Opin Struct Biol. 2019;56:119–30.
Pisetsky DS. Mechanisms of chromatin remodeling and repurposing during extracellular translocation. Adv Protein Chem Struct Biol. 2017;106:113–37.
Fernandez-Dominguez IJ, Manzo-Merino J, Taja-Chayeb L, Duenas-Gonzalez A, Perez-Cardenas E, Trejo-Becerril C. The role of extracellular DNA (exDNA) in cellular processes. Cancer Biol Ther. 2021;22:267–78.
Beyer C, Pisetsky DS. Modeling nuclear molecule release during in vitro cell death. Autoimmunity. 2013;46:298–301.
Mazlo A, Jenei V, Burai S, Molnar T, Bacsi A, Koncz G. Types of necroinflammation, the effect of cell death modalities on sterile inflammation. Cell Death Dis. 2022;13:423.
Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis. 2021;26:152–62.
Pisetsky DS. The central role of nucleic acids in the pathogenesis of systemic lupus erythematosus [version 1; peer review: 3 approved]. F1000Res. 2019;8.
Okude H, Ori D, Kawai T. Signaling through nucleic acid sensors and their roles in inflammatory diseases. Front Immunol. 2020;11:625833.
Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019;26:395–408.
Robb CT, Dyrynda EA, Gray RD, Rossi AG, Smith VJ. Invertebrate extracellular phagocyte traps show that chromatin is an ancient defence weapon. Nat Commun. 2014;5:4627.
Brogden G, von Kockritz-Blickwede M, Adamek M, Reuner F, Jung-Schroers V, Naim HY, et al. beta-Glucan protects neutrophil extracellular traps against degradation by Aeromonas hydrophila in carp (Cyprinus carpio). Fish Shellfish Immunol. 2012;33:1060–4.
Zhao J, Chen W, Zhang Y, Liu Q, Yang D, Wang Z. Bacterial infection induces pyroptotic signaling-mediated neutrophil extracellular traps (NETs) formation in turbot (Scophthalmus maximus). Fish Shellfish Immunol. 2022;127:982–90.
Chuammitri P, Ostojic J, Andreasen CB, Redmond SB, Lamont SJ, Palic D. Chicken heterophil extracellular traps (HETs): novel defense mechanism of chicken heterophils. Vet Immunol Immunopathol. 2009;129:126–31.
Ramos-Martinez E, Hernandez-Gonzalez L, Ramos-Martinez I, Perez-Campos Mayoral L, Lopez-Cortes GI, Perez-Campos E, et al. Multiple Origins of Extracellular DNA Traps. Front Immunol. 2021;12:621311.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5.
Mamtimin M, Pinarci A, Han C, Braun A, Anders HJ, Gudermann T, et al. Extracellular DNA Traps: origin, function and implications for anti-cancer therapies. Front Oncol. 2022;12:869706.
Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198:773–83.
Jiang N, Reich CF 3rd, Monestier M, Pisetsky DS. The expression of plasma nucleosomes in mice undergoing in vivo apoptosis. Clin Immunol. 2003;106:139–47.
Jiang N, Reich CF 3rd, Pisetsky DS. Role of macrophages in the generation of circulating blood nucleosomes from dead and dying cells. Blood. 2003;102:2243–50.
Choi JJ, Reich CF 3rd, Pisetsky DS. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand J Immunol. 2004;60:159–66.
Beyer C, Stearns NA, Giessl A, Distler JH, Schett G, Pisetsky DS. The extracellular release of DNA and HMGB1 from Jurkat T cells during in vitro necrotic cell death. Innate Immun. 2012;18:727–37.
Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science. 2021;372
Pisetsky DS, Jiang N. The generation of extracellular DNA in SLE: the role of death and sex. Scand J Immunol. 2006;64:200–4.
Choi JJ, Reich CF 3rd, Pisetsky DS. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology. 2005;115:55–62.
Jiang N, Pisetsky DS. The effect of dexamethasone on the generation of plasma DNA from dead and dying cells. Am J Pathol. 2004;164:1751–9.
Malkin EZ, Bratman SV. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020;11:584.
Reich CF 3rd, Pisetsky DS. The content of DNA and RNA in microparticles released by Jurkat and HL-60 cells undergoing in vitro apoptosis. Exp Cell Res. 2009;315:760–8.
Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. 2008;15:183–91.
Mobarrez F, Fuzzi E, Gunnarsson I, Larsson A, Eketjall S, Pisetsky DS, et al. Microparticles in the blood of patients with SLE: Size, content of mitochondria and role in circulating immune complexes. J Autoimmun. 2019;102:142–9.
Mobarrez F, Vikerfors A, Gustafsson JT, Gunnarsson I, Zickert A, Larsson A, et al. Microparticles in the blood of patients with systemic lupus erythematosus (SLE): phenotypic characterization and clinical associations. Sci Rep. 2016;6:36025.
Spencer DM, Gauley J, Pisetsky DS. The properties of microparticles from RAW 264.7 macrophage cells undergoing in vitro activation or apoptosis. Innate Immun. 2014;20:239–48.
Soop A, Hallstrom L, Frostell C, Wallen H, Mobarrez F, Pisetsky DS. Effect of lipopolysaccharide administration on the number, phenotype and content of nuclear molecules in blood microparticles of normal human subjects. Scand J Immunol. 2013;78:205–13.
Ullal AJ, Reich CF 3rd, Clowse M, Criscione-Schreiber LG, Tochacek M, Monestier M, et al. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J Autoimmun. 2011;36:173–80.
Pisetsky DS, Spencer DM, Mobarrez F, Fuzzi E, Gunnarsson I, Svenungsson E. The binding of SLE autoantibodies to mitochondria. Clin Immunol. 2020;212:108349.
Liu ML, Lyu X, Werth VP. Recent progress in the mechanistic understanding of NET formation in neutrophils. FEBS J. 2021;289:3954–66.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nature reviews Immunology. 2018;18:134–47.
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44.
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–25.
Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol. 2011;127:1260–6.
Li Y, Li M, Weigel B, Werth VP, Mall M, Liu ML. Nuclear envelope rupture and NET formation is driven by PKCα‐mediated lamin B disassembly. EMBO Rep. 2020;21.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53.
Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New insights into neutrophil extracellular traps: mechanisms of formation and role in inflammation. Front Immunol. 2016;7:302.
Goldberg MW, Huttenlauch I, Hutchison CJ, Stick R. Filaments made from A- and B-type lamins differ in structure and organization. J Cell Sci. 2008;1212:215–25.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41.
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304.
Li M, Lyu X, Liao J, Werth VP, Liu ML. Rho Kinase regulates neutrophil NET formation that is involved in UVB-induced skin inflammation. Theranostics. 2022;12:2133–49.
Amulic B, Knackstedt SL, Abu Abed U, Deigendesch N, Harbort CJ, Caffrey BE, et al. Cell-cycle proteins control production of neutrophil extracellular traps. Dev Cell. 2017;43:449–62.
Li Y, Travers JB, Werth VP, Liu ML. Pkcα Deficiency Protected mice from UVB Induced-Skin Inflammation through Attenuation of Neutrophil Netosis. 2018 ACR/ARHP Annual Meeting; 22.10.20218; Chicago: Arthritis & Rheumatology; 2018.
Liu ML. Functional actin cytoskeleton is required in early stage of NETosis induction. Proc Natl Acad Sci USA. 2020;117:22653–4.
Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol. 2017;216:4073–90.
Neubert E, Meyer D, Rocca F, Gunay G, Kwaczala-Tessmann A, Grandke J, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9:3767.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–13.
Zhou Y, An LL, Chaerkady R, Mittereder N, Clarke L, Cohen TS, et al. Evidence for a direct link between PAD4-mediated citrullination and the oxidative burst in human neutrophils. Sci Rep. 2018;8:15228.
Jones JE, Causey CP, Knuckley B, Slack-Noyes JL, Thompson PR. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr Opin Drug Disco Devel. 2009;12:616–27.
Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8:883–96.
Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3.
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3.
Koster DV, Mayor S. Cortical actin and the plasma membrane: inextricably intertwined. Curr Opin Cell Biol. 2016;38:81–9.
Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci USA. 2020;117:7326–37.
Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. 1996;59:229–40.
Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. 2015;112:2817–22.
Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur J Clin Invest. 2018;48:e12951.
Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. 2012;92:841–9.
Gosswein S, Lindemann A, Mahajan A, Maueroder C, Martini E, Patankar J, et al. Citrullination licenses calpain to decondense nuclei in neutrophil extracellular trap formation. Front Immunol. 2019;10:2481.
Yousefi S, Simon D, Stojkov D, Karsonova A, Karaulov A, Simon HU. In vivo evidence for extracellular DNA trap formation. Cell Death Dis. 2020;11:300.
Ingelsson B, Soderberg D, Strid T, Soderberg A, Bergh AC, Loitto V, et al. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc Natl Acad Sci USA. 2018;115:E478–E87.
Zhang X, Zhuchenko O, Kuspa A, Soldati T. Social amoebae trap and kill bacteria by casting DNA nets. Nat Commun. 2016;7:10938.
Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, et al. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196:1571–81.
Keller L, Casey J, Delco M, Mitochondrial DNA. is released by viable chondrocytes after induction of mitochondrial dysfunction. Osteoarthr Cartil. 2019;27:S192–S3.
Valenti D, Vacca RA, Moro L, Atlante A. Mitochondria Can Cross Cell Boundaries: An overview of the biological relevance, pathophysiological implications and therapeutic perspectives of intercellular mitochondrial transfer. Int J Mol Sci. 2021;22:8312.
Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, et al. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci Rep. 2015;5:18302.
Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, Adam SA, et al. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell. 2018;29:220–33.
Zhou E, Silva LMR, Conejeros I, Velasquez ZD, Hirz M, Gartner U, et al. Besnoitia besnoiti bradyzoite stages induce suicidal- and rapid vital-NETosis. Parasitology. 2020;147:401–9.
Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol. 2013;191:2647–56.
Lazzaretto B, Fadeel B. Intra- and extracellular degradation of neutrophil extracellular traps by macrophages and dendritic cells. J Immunol. 2019;203:2276–90.
Chiang N, Sakuma M, Rodriguez AR, Spur BW, Irimia D, Serhan CN. Resolvin T-series reduce neutrophil extracellular traps. Blood. 2022;139:1222–33.
de Buhr N, Bonilla MC, Pfeiffer J, Akhdar S, Schwennen C, Kahl BC, et al. Degraded neutrophil extracellular traps promote the growth of Actinobacillus pleuropneumoniae. Cell Death Dis. 2019;10:657.
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522–31.
Hawkins CL, Davies MJ. Role of myeloperoxidase and oxidant formation in the extracellular environment in inflammation-induced tissue damage. Free Radic Biol Med. 2021;172:633–51.
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–96.
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–61.
Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708–14.
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21.
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7:e32366.
Tomazella GG, da Silva I, Laure HJ, Rosa JC, Chammas R, Wiker HG, et al. Proteomic analysis of total cellular proteins of human neutrophils. Proteome Sci. 2009;7:32.
Malekzadeh H, Golpayegani M, Ghodsi Z, Sadeghi-Naini M, Asgardoon M, Baigi V, et al. Direct cost of illness for spinal cord injury: a systematic review. Global Spine J. 2022;12:1267–81.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91.
Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS One. 2019;14:e0218946.
Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, et al. Caught in a Trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front Immunol. 2019;10:423.
Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20:511–7.
Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006;8:668–76.
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12:109–16.
Abi Abdallah DS, Lin C, Ball CJ, King MR, Duhamel GE, Denkers EY. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect Immun. 2012;80:768–77.
Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007;13:981–5.
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017–25.
Dainichi T, Nakajima S, Iwata M, Kabashima K. Net effects of NETs: new concepts. J Invest Dermatol. 2020;140:939–41.
Hahn J, Schauer C, Czegley C, Kling L, Petru L, Schmid B, et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019;33:1401–14.
Knopf J, Leppkes M, Schett G, Herrmann M, Munoz LE. Aggregated NETs sequester and detoxify extracellular histones. Front Immunol. 2019;10:2176.
Wilson AS, Randall KL, Pettitt JA, Ellyard JI, Blumenthal A, Enders A, et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun. 2022;13:528.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–5.
Brinkmann V, Abu Abed U, Goosmann C, Zychlinsky A. Immunodetection of NETs in paraffin-embedded tissue. Front Immunol. 2016;7:513.
de Andrea CE, Ochoa MC, Villalba-Esparza M, Teijeira A, Schalper KA, Abengozar-Muela M, et al. Heterogenous presence of neutrophil extracellular traps in human solid tumours is partially dependent on IL-8. J Pathol. 2021;255:190–201.
Stehr AM, Wang G, Demmler R, Stemmler MP, Krug J, Tripal P, et al. Neutrophil extracellular traps drive epithelial-mesenchymal transition of human colon cancer. J Pathol. 2022;256:455–67.
Rios MR, Garoffolo G, Rinaldi G, Megia-Fernandez A, Ferrari S, Robb CT, et al. A fluorogenic peptide-based smartprobe for the detection of neutrophil extracellular traps and inflammation. Chem Commun (Camb). 2021;57:97–100.
Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6:e1000873.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9.
de Buhr N, von Kockritz-Blickwede M. How neutrophil extracellular traps become visible. J Immunol Res. 2016;2016:4604713.
Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122:2784–94.
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–93.
Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123:3446–58.
Alasmari SZ. In vivo imaging of neutrophil extracellular traps (NETs): visualization methods and outcomes. Biomed Res Int. 2020;2020:4192745.
Tanaka K, Koike Y, Shimura T, Okigami M, Ide S, Toiyama Y, et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS One. 2014;9:e111888.
Thalin C, Hisada Y, Lundstrom S, Mackman N, Wallen H. Neutrophil extracellular traps: villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol. 2019;39:1724–38.
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35.
Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45.
Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol. 2017;28:1753–68.
Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine. 2020;58:102925.
Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21:319–29.
Leppkes M, Maueroder C, Hirth S, Nowecki S, Gunther C, Billmeier U, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun. 2016;7:10973.
Leppkes M, Lindemann A, Gosswein S, Paulus S, Roth D, Hartung A, et al. Neutrophils prevent rectal bleeding in ulcerative colitis by peptidyl-arginine deiminase-4-dependent immunothrombosis. Gut. 2022;71:2414–29.
Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–76.
Caillon A, Trimaille A, Favre J, Jesel L, Morel O, Kauffenstein G. Role of neutrophils, platelets, and extracellular vesicles and their interactions in COVID-19-associated thrombopathy. J Thromb Haemost. 2022;20:17–31.
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074–88.
Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. 2015;125:4638–54.
Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: back in the thrombosis spotlight. Blood 2019;133:2186–97.
Shi H, Zuo Y, Navaz S, Harbaugh A, Hoy CK, Gandhi AA, et al. Endothelial cell-activating antibodies in COVID-19. Arthritis Rheumatol. 2022;74:1132–8.
Yaykasli KO, Schauer C, Munoz LE, Mahajan A, Knopf J, Schett G, et al. Neutrophil extracellular trap-driven occlusive diseases. Cells. 2021;10:2208.
de Boer OJ, Li X, Teeling P, Mackaay C, Ploegmakers HJ, van der Loos CM, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost. 2013;109:290–7.
Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. 2014;114:947–56.
Laridan E, Denorme F, Desender L, Francois O, Andersson T, Deckmyn H, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. 2017;82:223–32.
Sorvillo N, Mizurini DM, Coxon C, Martinod K, Tilvawala R, Cherpokova D, et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ Res. 2019;125:507–19.
Tilvawala R, Nguyen SH, Maurais AJ, Nemmara VV, Nagar M, Salinger AJ, et al. The rheumatoid arthritis-associated citrullinome. Cell Chem Biol. 2018;25:691–704.e6.
Tilvawala R, Nemmara VV, Reyes AC, Sorvillo N, Salinger AJ, Cherpokova D, et al. The role of SERPIN citrullination in thrombosis. Cell. Chem Biol. 2021;28:1728–39.e5.
World Health Organization. Chronic respiratory diseases 2022 https://www.who.int/health-topics/chronic-respiratory-diseases#tab=tab_1.
Collaborators GBDCRD. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8:585–96.
Yang X, Li H, Ma Q, Zhang Q, Wang C. Neutrophilic asthma is associated with increased airway bacterial burden and disordered community composition. Biomed Res Int. 2018;2018:9230234.
Pabreja K, Gibson P, Lochrin AJ, Wood L, Baines KJ, Simpson JL. Sputum colour can identify patients with neutrophilic inflammation in asthma. BMJ Open Respir Res. 2017;4:e000236.
Halwani R, Sultana A, Vazquez-Tello A, Jamhawi A, Al-Masri AA, Al-Muhsen S. Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J Asthma. 2017;54:893–904.
Moermans C, Heinen V, Nguyen M, Henket M, Sele J, Manise M, et al. Local and systemic cellular inflammation and cytokine release in chronic obstructive pulmonary disease. Cytokine. 2011;56:298–304.
Lonergan M, Dicker AJ, Crichton ML, Keir HR, Van Dyke MK, Mullerova H, et al. Blood neutrophil counts are associated with exacerbation frequency and mortality in COPD. Respir Res. 2020;21:166.
Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology. 2016;21:467–75.
Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner WD, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. 2015;16:59.
Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23:243–52.
Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307.
Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3.
Adrover JM, Carrau L, Dassler-Plenker J, Bram Y, Chandar V, Houghton S, et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight. 2022;7.
Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet. 2021;397:2195–211.
Manzenreiter R, Kienberger F, Marcos V, Schilcher K, Krautgartner WD, Obermayer A, et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J Cyst Fibros. 2012;11:84–92.
Kim JS, Okamoto K, Rubin BK. Pulmonary function is negatively correlated with sputum inflammatory markers and cough clearability in subjects with cystic fibrosis but not those with chronic bronchitis. Chest. 2006;129:1148–54.
Dittrich AS, Kuhbandner I, Gehrig S, Rickert-Zacharias V, Twigg M, Wege S, et al. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur Respir J. 2018;51.
Keir HR, Chalmers JD. Neutrophil extracellular traps in chronic lung disease: implications for pathogenesis and therapy. Eur Respir Rev. 2022;31
Guerra M, Halls VS, Schatterny J, Hagner M, Mall MA, Schultz C. Protease FRET Reporters Targeting Neutrophil Extracellular Traps. J Am Chem Soc. 2020;142:20299–305.
Blazquez-Prieto J, Lopez-Alonso I, Huidobro C, Albaiceta GM. The emerging role of neutrophils in repair after acute lung injury. Am J Respir Cell Mol Biol. 2018;59:289–94.
Lleo A, Invernizzi P, Gao B, Podda M, Gershwin ME. Definition of human autoimmunity–autoantibodies versus autoimmune disease. Autoimmun Rev. 2010;9:A259–66.
Fousert E, Toes R, Desai J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells. 2020;9:915.
Podolska MJ, Mahajan A, Knopf J, Hahn J, Boeltz S, Munoz L, et al. Autoimmune, rheumatic, chronic inflammatory diseases: Neutrophil extracellular traps on parade. Autoimmunity. 2018;51:281–7.
Kurien BT, Scofield RH. Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev. 2008;7:567–73.
Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. 2016;46:223–9.
Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6:6–12.
Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201.
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5:178ra40.
Tang S, Zhang Y, Yin SW, Gao XJ, Shi WW, Wang Y, et al. Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin Exp Immunol. 2015;180:408–18.
O’Sullivan KM, Lo CY, Summers SA, Elgass KD, McMillan PJ, Longano A, et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015;88:1030–46.
Bertolaccini ML, Amengual O, Andreoli L, Atsumi T, Chighizola CB, Forastiero R, et al. 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmun Rev. 2014;13:917–30.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–8.
Zuo Y, Yalavarthi S, Navaz SA, Hoy CK, Harbaugh A, Gockman K, et al. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. JCI Insight. 2021;6.
Lou H, Wojciak-Stothard B, Ruseva MM, Cook HT, Kelleher P, Pickering MC, et al. Autoantibody-dependent amplification of inflammation in SLE. Cell Death Dis. 2020;11:729.
Grayson PC, Kaplan MJ. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. J Leukoc Biol. 2016;99:253–64.
Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190:1217–26.
Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2011;13:170–80.
Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, et al. Netting neutrophils activate autoreactive B cells in lupus. J Immunol. 2018;200:3364–71.
Tillack K, Breiden P, Martin R, Sospedra M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J Immunol. 2012;188:3150–9.
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20.
Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol. 2014;25:990–7.
Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43:1186–8.
Sudakov NP, Klimenkov IV, Byvaltsev VA, Nikiforov SB, Konstantinov YM. Extracellular actin in health and disease. Biochem (Mosc). 2017;82:1–12.
Hartl J, Serpas L, Wang Y, Rashidfarrokhi A, Perez OA, Sally B, et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J Exp Med. 2021;218.
Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8:361ra138.
Munoz LE, Boeltz S, Bilyy R, Schauer C, Mahajan A, Widulin N, et al. Neutrophil extracellular traps initiate gallstone formation. Immunity. 2019;51:443–50.e4.
Bilyy R, Bila G, Vishchur O, Vovk V, Herrmann M. Neutrophils as main players of immune response towards nondegradable nanoparticles. Nanomaterials (Basel). 2020;10:1273.
Maueroder C, Mahajan A, Paulus S, Gosswein S, Hahn J, Kienhofer D, et al. Menage-a-Trois: The Ratio of Bicarbonate to CO2 and the pH regulate the capacity of neutrophils to form NETs. Front Immunol. 2016;7:583.
Schapher M, Koch M, Weidner D, Scholz M, Wirtz S, Mahajan A, et al. Neutrophil extracellular traps promote the development and growth of human salivary stones. Cells. 2020;9:2139.
Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renstrom E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. 2015;149:1920–31.e8.
Vitkov L, Minnich B, Knopf J, Schauer C, Hannig M, Herrmann M. NETs are double-edged swords with the potential to aggravate or resolve periodontal inflammation. Cells. 2020;9:2614.
Wang J, Zhou Y, Ren B, Zou L, He B, Li M. The role of neutrophil extracellular traps in periodontitis. Front Cell Infect Microbiol. 2021;11:639144.
Vitkov L, Munoz LE, Schoen J, Knopf J, Schauer C, Minnich B, et al. Neutrophils orchestrate the periodontal pocket. Front Immunol. 2021;12:788766.
Mahajan A, Hasikova L, Hampel U, Gruneboom A, Shan X, Herrmann I, et al. Aggregated neutrophil extracellular traps occlude Meibomian glands during ocular surface inflammation. Ocul Surf. 2021;20:1–12.
Estua-Acosta GA, Zamora-Ortiz R, Buentello-Volante B, Garcia-Mejia M, Garfias Y. Neutrophil extracellular traps: current perspectives in the eye. Cells. 2019;8:979.
Maueroder C, Kienhofer D, Hahn J, Schauer C, Manger B, Schett G, et al. How neutrophil extracellular traps orchestrate the local immune response in gout. J Mol Med (Berl). 2015;93:727–34.
Schorn C, Janko C, Krenn V, Zhao Y, Munoz LE, Schett G, et al. Bonding the foe—NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front Immunol. 2012;3:376.
Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61.
Munoz LE, Leppkes M, Fuchs TA, Hoffmann M, Herrmann M. Missing in action-The meaning of cell death in tissue damage and inflammation. Immunol Rev. 2017;280:26–40.
Schorn C, Frey B, Lauber K, Janko C, Strysio M, Keppeler H, et al. Sodium overload and water influx activate the NALP3 inflammasome. J Biol Chem. 2011;286:35–41.
Popa-Nita O, Naccache PH. Crystal-induced neutrophil activation. Immunol Cell Biol. 2010;88:32–40.
Gresnigt MS, Joosten LA, Verschueren I, van der Meer JW, Netea MG, Dinarello CA, et al. Neutrophil-mediated inhibition of proinflammatory cytokine responses. J Immunol. 2012;189:4806–15.
Reichel CA, Puhr-Westerheide D, Zuchtriegel G, Uhl B, Berberich N, Zahler S, et al. C-C motif chemokine CCL3 and canonical neutrophil attractants promote neutrophil extravasation through common and distinct mechanisms. Blood. 2012;120:880–90.
Michalec L, Choudhury BK, Postlethwait E, Wild JS, Alam R, Lett-Brown M, et al. CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammation. J Immunol. 2002;168:846–52.
Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. 2014;192:2837–45.
Desai J, Foresto-Neto O, Honarpisheh M, Steiger S, Nakazawa D, Popper B, et al. Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap-like chromatin. Sci Rep. 2017;7:15003.
Tiku V, Tan MW. Host immunity and cellular responses to bacterial outer membrane vesicles. Trends Immunol. 2021;42:1024–36.
Vitkov L, Munoz LE, Knopf J, Schauer C, Oberthaler H, Minnich B, et al. Connection between periodontitis-induced lowgrade endotoxemia and systemic diseases: neutrophils as protagonists and targets. Int J Mol Sci. 2021;22:4647.
Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and Caspase-11 activation. Cell. 2016;165:1106–19.
Burgener SS, Schroder K. Neutrophil extracellular traps in host defense. Cold Spring Harb Perspect Biol. 2020;12.
Perlee D, de Beer R, Florquin S, van der Poll T, van ‘t Veer, de Vos C, et al. Caspase-11 contributes to pulmonary host defense against Klebsiella pneumoniae and local activation of coagulation. Am J Physiol Lung Cell Mol Physiol. 2020;319:L105–L14.
Chen KW, Demarco B, Broz P. Beyond inflammasomes: emerging function of gasdermins during apoptosis and NETosis. EMBO J. 2020;39:e103397.
Rathinam VAK, Zhao Y, Shao F. Innate immunity to intracellular LPS. Nat Immunol. 2019;20:527–33.
Hirschfeld J, White PC, Milward MR, Cooper PR, Chapple ILC. Modulation of neutrophil extracellular trap and reactive oxygen species release by periodontal bacteria. Infect Immun. 2017;85.
Nishinaka Y, Arai T, Adachi S, Takaori-Kondo A, Yamashita K. Singlet oxygen is essential for neutrophil extracellular trap formation. Biochem Biophys Res Commun. 2011;413:75–9.
Sadiku P, Walmsley SR. Hypoxia and the regulation of myeloid cell metabolic imprinting: consequences for the inflammatory response. EMBO Rep. 2019;20.
Borella R, De Biasi S, Paolini A, Boraldi F, Lo Tartaro D, Mattioli M, et al. Metabolic reprograming shapes neutrophil functions in severe COVID-19. Eur J Immunol. 2022;52:484–502.
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201:105–15.
McGovern NN, Cowburn AS, Porter L, Walmsley SR, Summers C, Thompson AAR, et al. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J Immunol. 2011;186:453–63.
Lodge KM, Cowburn AS, Li W, Condliffe AM. The impact of hypoxia on neutrophil degranulation and consequences for the host. Int J Mol Sci. 2020;21:1183.
Dolling M, Eckstein M, Singh J, Schauer C, Schoen J, Shan X, et al. Hypoxia promotes neutrophil survival after acute myocardial infarction. Front Immunol. 2022;13:726153.
Walmsley SR, Cowburn AS, Clatworthy MR, Morrell NW, Roper EC, Singleton V, et al. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108:3176–8.
Berger-Achituv S, Brinkmann V, Abed UA, Kuhn LI, Ben-Ezra J, Elhasid R, et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48.
Wang WW, Wu L, Lu W, Chen W, Yan W, Qi C, et al. Lipopolysaccharides increase the risk of colorectal cancer recurrence and metastasis due to the induction of neutrophil extracellular traps after curative resection. J Cancer Res Clin Oncol. 2021;147:2609–19.
Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583:133–8.
Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. 2019;79:5626–39.
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76:1367–80.
Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52:856–71.e8.
Yang L, Liu L, Zhang R, Hong J, Wang Y, Wang J, et al. IL-8 mediates a positive loop connecting increased neutrophil extracellular traps (NETs) and colorectal cancer liver metastasis. J Cancer. 2020;11:4384–96.
Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, Konig S, et al. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers (Basel). 2020;12:1542.
Zhu T, Zou X, Yang C, Li L, Wang B, Li R, et al. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelialmesenchymal transition. Int J Mol Med. 2021;48.
Jin W, Yin H, Li H, Yu XJ, Xu HX, Liu L. Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J Cell Mol Med. 2021;25:5443–56.
Mahajan A, Gruneboom A, Petru L, Podolska MJ, Kling L, Maueroder C, et al. Frontline Science: Aggregated neutrophil extracellular traps prevent inflammation on the neutrophil-rich ocular surface. J Leukoc Biol. 2019;105:1087–98.
Zhu B, Zhang L, Yuan K, Huang X, Hu R, Jin X. Neutrophil extracellular traps may have a dual role in Pseudomonas aeruginosa keratitis. Eur J Clin Microbiol Infect Dis. 2021;40:169–80.
An S, Raju I, Surenkhuu B, Kwon JE, Gulati S, Karaman M, et al. Neutrophil extracellular traps (NETs) contribute to pathological changes of ocular graft-vs.-host disease (oGVHD) dry eye: Implications for novel biomarkers and therapeutic strategies. Ocul Surf. 2019;17:589–614.
Tibrewal S, Ivanir Y, Sarkar J, Nayeb-Hashemi N, Bouchard CS, Kim E, et al. Hyperosmolar stress induces neutrophil extracellular trap formation: implications for dry eye disease. Invest Ophthalmol Vis Sci. 2014;55:7961–9.
Navas A, Magana-Guerrero FS, Dominguez-Lopez A, Chavez-Garcia C, Partido G, Graue-Hernandez EO, et al. Anti-inflammatory and anti-fibrotic effects of human amniotic membrane mesenchymal stem cells and their potential in corneal repair. Stem Cells Transl Med. 2018;7:906–17.
Safi R, Kallas R, Bardawil T, Mehanna CJ, Abbas O, Hamam R, et al. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behcet’s disease. J Dermatol Sci. 2018;92:143–50.
Wang L, Zhou X, Yin Y, Mai Y, Wang D, Zhang X. Hyperglycemia induces neutrophil extracellular traps formation through an NADPH Oxidase-dependent pathway in diabetic retinopathy. Front Immunol. 2018;9:3076.
Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–9.
ten Broek RP, Strik C, Issa Y, Bleichrodt RP, van Goor H. Adhesiolysis-related morbidity in abdominal surgery. Ann Surg. 2013;258:98–106.
Fritjofsson A, Lingardh G, Odelberg-Johnson O, Vikterlof KJ. Intraurethral irradiation of prostatic cancer using afterloading technique. Prog Clin Biol Res. 1976;6:255–66.
Heuer A, Stiel C, Elrod J, Konigs I, Vincent D, Schlegel P, et al. Therapeutic targeting of neutrophil extracellular traps improves primary and secondary intention wound healing in mice. Front Immunol. 2021;12:614347.
Singh V, Devgan L, Bhat S, Milner SM. The pathogenesis of burn wound conversion. Ann Plast Surg. 2007;59:109–15.
Szpaderska AM, DiPietro LA. Inflammation in surgical wound healing: friend or foe? Surgery. 2005;137:571–3.
Stommel MW, Strik C, van Goor H. Response to pathological processes in the peritoneal cavity–sepsis, tumours, adhesions, and ascites. Semin Pediatr Surg. 2014;23:331–5.
Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. 2018;19:327–41.
Boettcher M, Schönfeld L, Heuer A, Elrod J, Stiel C, Raluy LP et al. Neutrophil extracellular traps orchestrate formation of peritoneal adhesions. 2021.
Porgo TV, Moore L, Truchon C, Berthelot S, Stelfox HT, Cameron PA, et al. Patient-level resource use for injury admissions in Canada: A multicentre retrospective cohort study. Injury. 2019;50:1192–201.
Zhang Y, Al Mamun A, Yuan Y, Lu Q, Xiong J, Yang S, et al. Acute spinal cord injury: Pathophysiology and pharmacological intervention (Review). Mol Med Rep. 2021;23:417.
Fehlings MG, Tetreault LA, Wilson JR, Kwon BK, Burns AS, Martin AR, et al. A clinical practice guideline for the management of acute spinal cord injury: introduction, rationale, and scope. Glob Spine J. 2017;7:84S–94S.
Fehlings MG, Wilson JR, Harrop JS, Kwon BK, Tetreault LA, Arnold PM, et al. Efficacy and safety of methylprednisolone sodium succinate in acute spinal cord injury: a systematic review. Glob Spine J. 2017;7:116S–37S.
Feng Z, Min L, Liang L, Chen B, Chen H, Zhou Y, et al. Neutrophil extracellular traps exacerbate secondary injury via promoting neuroinflammation and blood-spinal cord barrier disruption in spinal cord injury. Front Immunol. 2021;12:698249.
Bao F, Fleming JC, Golshani R, Pearse DD, Kasabov L, Brown A, et al. A selective phosphodiesterase-4 inhibitor reduces leukocyte infiltration, oxidative processes, and tissue damage after spinal cord injury. J Neurotrauma. 2011;28:1035–49.
Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–78.
Kubota K, Saiwai H, Kumamaru H, Maeda T, Ohkawa Y, Aratani Y, et al. Myeloperoxidase exacerbates secondary injury by generating highly reactive oxygen species and mediating neutrophil recruitment in experimental spinal cord injury. Spine (Philos Pa 1976). 2012;37:1363–9.
Manfredi AA, Rovere-Querini P, D’Angelo A, Maugeri N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharm Res. 2017;123:146–56.
Aebi U, Cohn J, Buhle L, Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323:560–4.
Mukherjee K, Gu C, Collins A, Mettlen M, Samelko B, Altintas MM, et al. Simultaneous stabilization of actin cytoskeleton in multiple nephron-specific cells protects the kidney from diverse injury. Nat Commun. 2022;13:2422.
Funding
This work was partially supported by Medical Research Service, Veterans administration and NIH grant R01 AR073935 to DSP; by the Deutsche Forschungsgemeinschaft (DFG) 446358093 to MB; SCHA 2040/1-1 to CS, STU 238/10-1 to MS, TRR241 subproject B04 to MH and ML and subproject A06 to MS, FOR 2438 (subproject 2 to EN and MS), TRR 305 subproject B08 to EN, FOR 2886 PANDORA subproject B3 to MH, FOR 2886 PANDORA to GS, CRC1181 to GS, CRC1181 subproject C03 to MH; by the Interdisciplinary Center for Clinical Research (IZKF) of the Clinical Center Erlangen to MS; by the German Bundeswirtschaftsministerium ZF4010106MD9 to ML; by the German Bundesministerium für Bildung und Forschung project MASCARA to GS; by H2020-FETOPEN-2018-2019-2020-01; 861878 “NeutroCure” to MH and by European Research Council Synergy grant 4D Nanoscope to GS; by the Volkswagen-Stiftung (Grant 97744) to MH; by the Programm des Bayerischen Wissenschaftsministeriums zur Corona-Forschung to MH and ML; and by Lupus Research Alliance 416805 to MLL and NIH R21AI144838 to MLL. Open Access funding enabled and organized by Projekt DEAL.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, J., Boettcher, M., Dölling, M. et al. Moonlighting chromatin: when DNA escapes nuclear control. Cell Death Differ 30, 861–875 (2023). https://doi.org/10.1038/s41418-023-01124-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-023-01124-1
